

Abstract
Bacteriophage T4 initiates origin-dependent replication via an R-loop mechanism in vivo. During in vitro reactions, the phage-encoded gp59 stimulates loading of the replicative helicase, gp41, onto branched intermediates, including origin R-loops. However, although gp59 is essential for recombination-dependent replication from D-loops, it does not appear to be required for origin-dependent replication in vivo. In this study, we have analyzed the origin-replicative intermediates formed during infections that are deficient in gp59 and other phage replication proteins. During infections lacking gp59, the initial replication forks from two different T4 origins actively replicated both leading- and lagging-strands. However, the retrograde replication forks from both origins were abnormal in the gp59-deficient infections. The lagging-strand from the initial fork was elongated as a new leading-strand in the retrograde direction without lagging-strand synthesis, whereas in the wild-type, leading- and lagging-strand synthesis appeared to be coupled. These results imply that gp59 inhibits the polymerase holoenzyme in vivo until the helicase-primase (gp41-gp61) complex is loaded, and we thereby refer to gp59 as a gatekeeper. We also found that all origin-replicative intermediates were absent in infections deficient in the helicase gp41 or the single-strand-binding protein gp32, regardless of whether gp59 was present or absent. These results argue that replication from the origin in vivo is dependent on both the helicase and single-strand-binding protein and demonstrate that the strong replication defect of gene 41 and 32 single mutants is not caused by gp59 inhibition of the polymerase.
The initiation of DNA replication at origin sequences generally involves localized unwinding within an AT-rich region of the origin, promoted by either an initiator protein or an origin transcript (for review, see Ref. 1). The unwound region provides an assembly site for the replication apparatus, beginning with the replicative helicase that is responsible for more extensive unwinding of the parental strands. In the well studied Escherichia coli system, DnaA binds to specific sequences within the origin of replication, oriC, and promotes localized unwinding of the DNA and recruitment of the replicative helicase, DnaB. DnaB catalyzes further unwinding of the region, and DnaG primase then synthesizes RNA primers for both leading- and lagging-strand synthesis (1).
In the ColE1 replicon of certain plasmids and also in mitochondrial DNA, RNA polymerase generates an origin transcript that forms a persistent R-loop. The origin R-loop thereby provides an unwound region that serves as assembly site for the replicative helicase, in addition to providing the primer for leading-strand synthesis (2-4).
Bacteriophage T4 uses two major replication initiation mechanisms that are regulated to occur during certain phases of the infective cycle (5-7). At early times, origin-dependent replication proceeds from several origins, including the well characterized ori(uvsY) and ori(34) (8-11). The structure of both origins consists of a promoter just upstream of a DNA unwinding element (Fig. 1B, top). In vivo studies measuring permanganate hypersensitivity demonstrated that the origin transcript forms a persistent R-loop within the DNA unwinding element, supporting an R-loop initiation mechanism (12). As the T4 infection progresses, origin-dependent replication is repressed and recombination-dependent replication predominates. Recombination-dependent replication is dependent upon T4 recombination proteins and involves initiation from D-loops formed by strand invasion (13) (for review, see Ref. 6). T4 recombination-dependent replication overcomes the problem of replicating the ends of the infecting linear T4 DNA, which are left partially single-stranded after origin-dependent replication. Because T4 DNA ends are terminally redundant and circularly permuted, these unreplicated 3′-ends can undergo strand invasion into a homologous internal region of another molecule or the opposite end of the same molecule. Strand invasion results in the formation of a D-loop with a displaced single strand that serves as an assembly site for the replication machinery. As with the R-loop mechanism described above, the invading 3′-end (DNA, in this case) serves as a primer for leading-strand synthesis (5-7).
Fig. 1.

Model of replication initiation at ori(uvsY). A, the major steps in the R-loop replication mechanism are shown: (i) the initial replication fork is activated by polymerase extension of the RNA within the R-loop (leading-strand synthesis) and assembly of the helicase; (ii) the initial fork exits the restriction fragment of interest on the right side; (iii) the retrograde replication fork is initiated; (iv) the retrograde fork exits the restriction fragment on the left side. B, a schematic of a simple Y-arc with the characteristic comet arc detected from N/N two-dimensional gel electrophoresis of restriction fragments containing ori(uvsY). The replicative intermediates that comprise the comet arc consist of molecules like those after step ii, panel A, as shown in the diagram in panel B, right (see the Introduction for discussion of the different branch point locations). The overall origin structure is shown above the intermediates in panel B, with P indicating the origin promoter and DUE indicating the DNA unwinding element.
We have previously analyzed the mechanism of T4 origin firing in vivo using neutral-neutral (N/N)1 two-dimensional gel analyses. In this technique, restriction fragments are separated on the basis of mass in the first dimension and mass and shape in the second dimension (14). Branched DNA molecules thereby migrate more slowly than linear DNA molecules in the second dimension. Families of Y-shaped and θ-form replicative intermediates form distinctive arc patterns that deviate from the diagonal of linear DNA molecules. A restriction fragment containing the T4 chromosomal ori(uvsY) resulted in a novel arc referred to as the comet arc, rather than the characteristic bubble arc that is generally seen with replication origins (15) (see Fig. 1). The replicative intermediates that comprise the comet arc consist of Y-molecules with the branch points within the origin transcription unit. The structure of these molecules provides strong evidence that the RNA within the origin R-loop is used to prime leading-strand synthesis in vivo (15). The heaviest region of the comet, referred to as the comet nucleus, contains intermediates with branch points very near the 5′-end of the transcript. The intermediates within the comet arc result from the delay between the initiation of the two different replication forks during the two-stage bidirectional replication pathway; these are molecules in which only the first fork has initiated and already exited the restriction fragment (see Fig. 1B).
Why do some molecules have a branch point downstream of the transcription start site? We infer that the 5′-end of the RNA is processed to varying extents in these molecules and the parental DNA strands rewind as the RNA is degraded, prior to synthesis of the first Okazaki fragment. This would generate Y-shaped molecules with branch points throughout the transcription unit, i.e. the molecules within the comet tail (see Fig. 1B). Evidence supporting this model includes the formation of a comet with only a nucleus in DNA isolated from an RNaseH-deficient infection or from a mutant origin with a very short origin transcript (15).
Recent in vitro studies with a circular synthetic R-loop substrate confirmed that the T4 replication machinery efficiently uses R-loops to initiate replication (16). Unit-length nascent leading-strands and Okazaki fragments were synthesized in the presence of the T4 DNA polymerase (gp43), clamp and clamp loader (gp45 and gp44/62), replicative helicase (gp41), helicase loader (gp59), primase (gp61), single-stranded DNA-binding protein (gp32), and topoisomerase (gp39/52/60). The further addition of T4 RNaseH and DNA ligase allowed generation of completely intact replicated products. In reactions using a synthetic R-loop comprised of a radiolabeled transcript in the absence of RNaseH, the unit-length products were radiolabeled, demonstrating that the origin transcript served as the primer for leading-strand synthesis (16).
These in vivo and in vitro results argue that T4 initiates bidirectional replication in a two-step process. In the initial replication fork, the origin transcript is used to prime leading-strand synthesis, and in the delayed retrograde replication fork, the 3′-end of the first Okazaki fragment from the initial fork is presumably used as primer for leading-strand synthesis. In the in vitro system, the leftward fork was not detected. In this case, the initial replication fork travels around the relatively small (5.7-kb) circular substrate rapidly, leaving only a short window of time that is apparently insufficient for assembly of the retrograde fork. The loading of the replicative helicase is presumably quite different for the initial and retrograde replication forks. The displaced strand of the R-loop provides an attractive target for loading the helicase on the initial fork. However, a single-stranded region presumably must be generated near the branch point to allow loading of the helicase for the retrograde replication fork, and this could explain the delay in the retrograde fork.
Another key factor in helicase loading is the phage-encoded gp59 protein (17-19). gp59 greatly stimulates gp41 loading in vitro, particularly when single-stranded DNA is coated with gp32 (18, 20-24). gp59 binds preferentially to fork DNA, which may allow targeting of gp59/41 to both R-loops and D-loops (16, 19, 25). In addition, gp59 has been shown to bind tightly to gp32, which might also help target gp59/41 to these initiating DNA structures (23, 24, 26-28). The gp59-mediated assembly of gp41 onto gp32-coated single-stranded DNA most likely involves a ternary complex between these three proteins (23, 24, 29). Recent in vitro studies indicate that gp59 travels with the replication fork, perhaps playing a role in the coupling of leading- and lagging-strand synthesis (30, 31).
As judged by in vivo plasmid model systems, gp59 is essential for the process of recombination-dependent replication but dispensable for origin-dependent replication (9, 32, 33). In addition, gp59-deficient mutants replicate some phage chromosomal DNA early, implying successful origin-dependent replication (34-37). In this report, we have found that chromosomal origin replication does indeed occur in gp59-deficient infections; however, the mechanism of initiation in the absence of gp59 is altered. In particular, the leading-strand for the retrograde replication fork is significantly extended without lagging-strand synthesis, arguing that gp59 normally holds the polymerase in check until the replicative helicase is loaded. These in vivo results are consistent with recent in vitro results that revealed an inhibitory effect of gp59 on T4 DNA polymerase (see “Discussion”).
EXPERIMENTAL PROCEDURES
Materials—Restriction enzymes were purchased from New England Biolabs, Taq DNA polymerase from Invitrogen, radiolabeled nucleotides from PerkinElmer Life Sciences, and Nytran membranes from Schleicher & Schuell. Oligonucleotides were synthesized by Sigma-Genosys Biotechnologies, Inc. Luria broth contained bacto-tryptone (Difco Laboratories) at 10 g/liter, yeast extract (Difco Laboratories) at 5 g/liter, and NaCl at 10 g/liter.
E. coli and Bacteriophage T4 Strains—E. coli strains include CR63 (K12, supD λr) (38) and AB1 (araD139 Δ(ara-leu)7697 ΔlacX74 galU galK hsdR rpsL) (9). Bacteriophage T4 strains included K10 (amB262 (gene 38) amS29 (gene 51) nd28 (denA) rIIPT8 (denB-rII deletion)), which is considered as the wild-type for these studies, K10-59am (as K10, with gene 59 amber mutation amHL628) (9, 32), K10-41am (as K10, with gene 41 amber mutation amN81) (39), and K10-32am (as K10, with gene 32 amber mutation amA453) (39). K10-59am 41am and K10-59am 32am were generated by crossing the appropriate single mutant K10 strains and identifying the double mutant progeny. K10-59am-GC was created by crossing K10-GC (15) with K10-59am and identifying the double mutant progeny.
T4 strain KD1 was constructed using the T4 insertion/substitution system (40, 41). Briefly, triple overlapping SwaI restriction sites were inserted into the phage genome within the intergenic region between gene 34 and rnh (map position 150773, all map positions in this report are from the T4 genome data base 3/2003 release; insert sequence 5′-TTTAAATTTAAATTTAAAT-3′ oriented counterclockwise with respect to the T4 map). Triple overlapping sites were used to facilitate complete digestion of T4-modified DNA for the two-dimensional gel analyses. After the recombination steps of the insertion/substitution system (40, 41), a phage strain containing the SwaI insert in its chromosome was identified by PCR followed by SwaI restriction analysis and was verified by DNA sequencing. Phage strain KD1-59am was constructed by crossing phage strains KD1 and K10-59am and identifying the double mutant (SwaI insert and 59am) progeny.
Two-dimensional Neutral-Neutral Agarose Gel Analyses—E. coli AB1 cells were grown in Luria broth to an A560 of 0.5 (∼4 × 108 cells/ml) at 37 °C and then infected with the appropriate phage at a multiplicity of 6 plaque-forming units/cell. At the indicated time points, 1.5-ml aliquots were removed, cell pellets were collected by centrifugation and frozen quickly in dry ice/ethanol bath, and total nucleic acids were cross-linked with 5-methyl-psoralen (trioxsalen) and purified as previously described (42). The first dimension of the N/N two-dimensional gels was run in 0.5× TBE buffer (1× = 89 mm Tris base, 89 mm boric acid, 2 mm Na3EDTA) at 1 V/cm for 29 h. The second-dimension gel contained ethidium bromide at 0.3 μg/ml; electrophoresis was at 4 °C in 0.5× TBE buffer containing ethidium bromide (0.3 μg/ml) at 4.5 V/cm for 16 h (with buffer recirculation). Gels were analyzed by Southern blot hybridization with a probe prepared using the Random Primed kit (Roche Applied Science). Southern blots were visualized by exposures to x-ray film.
For the PacI-SwaI digests of ori(uvsY) (4.5-kb fragment), DNA samples were digested with PacI for 12–14 h at 37 °C, followed by an additional 12–14-h digestion at 25 °C with SwaI. The first-dimension gel contained 0.4% agarose, and the second-dimension gel contained 1% agarose. The probe consisted of a radioactive 1.4-kb HindIII T4 DNA fragment corresponding to T4 map coordinates 114,420–115,771 bp.
For the PsiI digest of ori(uvsY) (2.1-kb fragment), DNA samples were digested with PsiI for 12–14 h at 37 °C. The first-dimension gel contained 0.6% agarose, and the second-dimension gel contained 1.2% agarose. The probe consisted of a radioactive 0.815-kb PCR fragment corresponding to T4 map coordinates 115,004–115,819 bp.
For the SwaI digest of ori(34) (5.4-kb fragment), DNA samples were digested with SwaI for 12–14 h at 25 °C. The first-dimension gel contained 0.4% agarose, and the second-dimension gel contained 1% agarose. The probe consisted of a radioactive 2.2-kb PCR fragment corresponding to T4 map coordinates 150,782–152,975 bp.
Two-dimensional Neutral-Alkaline Agarose Gel Analyses—The conditions for the first-dimension (neutral) gel are as described above for the PacI-SwaI ori(uvsY) N/N agarose gel. The appropriate gel slice was soaked in 50 mm NaOH and 1 mm Na3EDTA at room temperature for 15 min twice with shaking. The second-dimension gel contained 1% agarose and was run at 4 °C in 50 mm NaOH and 1 mm Na3EDTA at ∼1 V/cm for 42 h with recirculation. Gels were analyzed by Southern blot hybridization using radioactive RNA transcripts (see below) as probe. After analysis of the nascent leading-strand, the probe was removed from the blot and the blot reprobed with an RNA transcript that hybridizes to the nascent lagging-strand. The Southern blots were analyzed by exposure to a Phosphorimager screen (Amersham Biosciences).
Generation of RNA Probes—The template for in vitro transcription reactions consisted of a PCR product from ori(uvsY) DNA (position +462 to +1004, with respect to the ori(uvsY) promoter transcription) (10) with a T7 promoter at one end and an SP6 promoter at the other. Transcription from the T7 promoter generated an RNA transcript that hybridized to the nascent leading-strand, whereas the transcript generated from the SP6 promoter hybridized to the nascent lagging-strand. PCR reactions used T4 DNA as the template. The transcript was labeled by incorporation of [α-32P]UTP residues, using the MAXIscript SP6/T7 kit (Ambion Co.).
RESULTS
Replication from ori(uvsY) Occurs in the Absence of gp59—A major function of gp59 in vitro is the efficient loading of gp41, but previous in vivo experiments indicated that gp59 is not required for origin-dependent replication (see the Introduction). We therefore began by determining whether origin-replicative intermediates are formed during a gene 59 amber mutant infection of a non-suppressing E. coli strain (an rpsL mutation was included to reduce the leakiness of an amber mutation; see Refs. 44 and 43). Using N/N two-dimensional gel analyses, replicative intermediates were indeed detected on ori(uvsY)-containing restriction fragments generated from the gp59-deficient infection (Fig. 2B). The intermediates migrated along the simple Y-arc and were similar, but not identical, to those in the comet arc from the wild-type infection (compare Fig. 2A and 2B). As a control, we also analyzed DNA isolated from a gene 41 amber mutant infection, deficient in the replicative helicase. In this case, only the diagonal line of linear DNA was detected, with no significant accumulation of replicative intermediates (Fig. 2C). Therefore, replication can initiate from ori(uvsY) in the absence of the helicase loader gp59, but not in the absence of the replicative helicase gp41. The initiation of replication in the absence of gp59 strongly suggests that gp41 can be loaded at the origin without gp59. However, an alternate interpretation is that gp41 is not required for origin replication and that gp59 inhibits gp41-independent replication; this possibility is explored below.
Fig. 2.
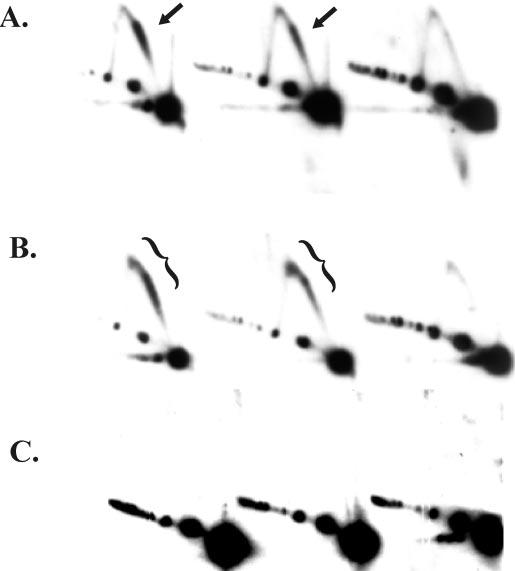
Replicative intermediates formed during a 59 amber mutant infection migrate as an extended comet arc in two-dimensional N/N gels. Cells lacking an amber suppressor were infected with either wild-type T4 (A) (strain K10; see “Experimental Procedures”), a gene 59 amber mutant (B), or a gene 41 amber mutant (C). After a 3-min attachment period, the infected cells were incubated for 4, 6, and 8 min (left to right), and DNA was harvested and purified as described under “Experimental Procedures.” The DNA was cleaved with PacI and SwaI, separated by N/N two-dimensional gel electrophoresis, and visualized by Southern hybridization. The comet arc is indicated by arrows, and the extended comet by brackets.
The replicative intermediates detected during a time course of wild-type and gp59-deficient infections were distinct. In both cases, the branched replicative intermediates were present at the early 4-min time point, reduced at the 6-min time point, and absent at the 8-min time point (Fig. 2, A and B). We have previously found that the UvsW helicase, which is expressed as a late protein, is responsible for turning origin replication off at this time by unwinding the R-loop intermediate (45). Although the comets produced by the 59 mutant and wild-type were quite similar at the 4-min time point, there was a definite lengthening of the comet beyond the apex of the simple Y-arc in the 59 mutant infection. This trend was much more pronounced with the 6-min sample, where a large fraction of the intermediates from the 59 mutant infection were near the apex.
We conclude that the replicative intermediates formed in the absence of gp59 are structurally unique. The branch points of the wild-type ori(uvsY) intermediates in the comet nucleus are very near the origin promoter (see the Introduction). Therefore, molecules in the extended comet of the 59 mutant infection presumably have branch points upstream of the origin promoter, suggesting that retrograde replication had occurred in some of these replicative intermediates.
Replication in the Absence of gp59 Is Origin-dependent—An alternative explanation of the extended comet is that replication forks initiated from outside the restriction fragment get blocked or stalled upstream of ori(uvsY) in the 59 mutant. To distinguish between these models, we examined DNA from a 59 mutant phage that also carried a GC-rich insertion mutation in ori(uvsY). This GC-rich insertion abolishes replication from ori(uvsY) but does not affect transcription from the origin promoter (15). As shown in Fig. 3, there were no detectable replicative intermediates in the double mutant phage infection, providing strong evidence that the intermediates in the extended comet from gp59-deficient infections originate from an active ori(uvsY).
Fig. 3.
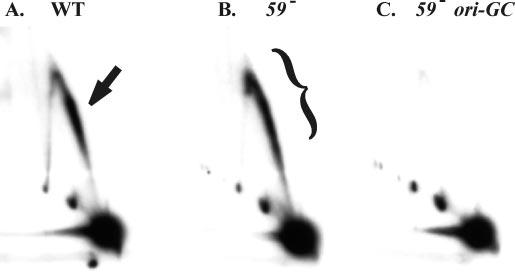
The extended comet arc is dependent upon active ori(uvsY) initiation. Cells lacking an amber suppressor were infected with either wild-type T4 (A), a gene 59 amber mutant (B), or a gene 59 amber mutant carrying the GC-rich insertion in ori(uvsY)(C). DNA was isolated 4 min after the attachment period and analyzed as described in the legend to Fig. 1. The comet arc is indicated by the arrow and the extended comet by a bracket.
The 59 Mutant Produces Extended Comets at Another T4 Replication Origin—Another well characterized T4 replication origin is ori(34), which, like ori(uvsY), consists of a promoter just upstream of a DNA unwinding element. N/N two-dimensional gel analyses of a restriction fragment containing ori(34) revealed both a comet structure and a bubble arc commonly seen for θ-form replication (11). We do not understand why both branched and θ molecules were detected at ori(34) and not at ori(uvsY). One possibility is that the initial replication fork from ori(34) needed to traverse a longer distance to exit the restriction fragment than in the case of ori(uvsY), which might have facilitated detection of θ intermediates.
Particularly with that concern in mind, we wanted to compare ori(34) to ori(uvsY) using more similar restriction fragments. However, T4 DNA is heavily modified, and therefore only a handful of restriction enzymes can be used. Because there were no natural sites for an enzyme that cleaves T4 DNA and generates an appropriate fragment, we inserted a SwaI restriction site into the phage genome downstream of ori(34). The genome of this new phage strain, KD1, has a 5.4-kb SwaI fragment containing ori(34) just to the right of center (see “Experimental Procedures”). N/N two-dimensional gel analysis of the ori(34)-containing SwaI fragment from a KD1 phage infection revealed a pattern very similar to that of ori(uvsY)-containing fragments, with a comet just to the right of the apex (Fig. 4A). In the gp59-deficient infection, the comet was again extended into the apex region, although in this case it appeared to extend slightly outside of the Y-arc region (in the upper left direction; Fig. 4B). Therefore, an aberrant form of replication also occurs at ori(34) in the absence of gp59.
Fig. 4.

Extended comet arc at ori(34) in a 59 amber mutant infection. Cells lacking an amber suppressor were infected with either wild-type T4 (A) or a gene 59 amber mutant (B). DNA was isolated 4, 6, and 8 min after the attachment period and analyzed by N/N two-dimensional gel electrophoresis after digestion with SwaI. The replicative forms were visualized by Southern hybridization with an ori(34) probe. Schematics of the patterns are shown on the right. The comet arc is indicated by arrows and the extended comet by brackets.
This SwaI digest also revealed a strong arc of single-stranded DNA in both the wild-type and gp59-deficient infections (Fig. 4). We have also detected this arc from digests with the restriction enzyme PsiI. We found that both SwaI and PsiI fail to cleave single-stranded M13mp18 DNA, but PacI does (data not shown). This explains the absence of the single-stranded DNA arc in the two-dimensional gels shown above (Figs. 2 and 3). We confirmed that the nucleic acid within this arc is single-stranded DNA by sensitivity to nuclease S1 and resistance to RNaseA (data not shown). The single-stranded DNA arc was present in both the wild-type and gp59-deficient infections, with a similar shape in each case (data not shown). In addition, the arc was detected when the gels were probed with a non-origin region of the T4 genome, and so these molecules are not specific to origin regions.
The extended comet arc from the SwaI digest of ori(34) (Fig. 4) looked slightly different from that of the PacI-SwaI digest of ori(uvsY) (Figs. 2B and 3B), extending slightly in the upper left direction away from the Y-arc. This difference could be a function of PacI digestion of single-stranded DNA, and we therefore repeated the two-dimensional analysis of ori(uvsY) with enzyme PsiI, which generates a 2.2-kb fragment with ori(uvsY) near the middle. Branched replicative intermediates that correspond to the comet or extended comet were again detected in both wild-type and gp59-deficient infections (Fig. 5; note that the Y-arc is more compact because of the smaller size of this restriction fragment). As with the ori(34) intermediates, the PsiI-cleaved intermediates of ori(uvsY) also extend to the upper left away from the Y-arc in DNA from the 59 mutant infection (Fig. 5B). The extension of the comet away from the Y-arc may therefore relate to some single-stranded character of the replicative intermediates in the 59 mutant infection, but this characteristic seems to be shared by both origins.
Fig. 5.
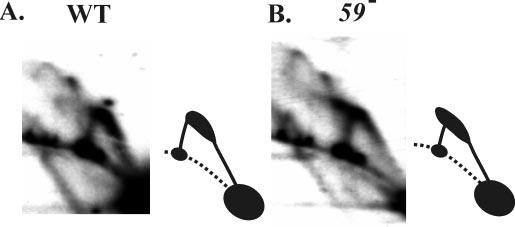
Comet arcs with a small restriction fragment containing ori(uvsY). Cells lacking an amber suppressor were infected with either wild-type T4 (A) or a gene 59 amber mutant (B). DNA was isolated 4 min after the attachment period and analyzed by N/N two-dimensional gel electrophoresis after digestion with PsiI. The replicative forms were visualized by Southern hybridization with an ori(uvsY) probe.
Structural Analysis of the Branched Intermediates Comprising the Extended Comet—Two general models might explain the extended comet molecules in the gp59-deficient samples. In the first model, the initial replication fork consists of leading-strand-only synthesis because the absence of gp59 precludes gp41 (and hence the gp61 primase) loading. This would result in replicative intermediates consisting of branched Y-molecules containing one duplex and one single-stranded arm (Fig. 6A). Depending on the restriction enzyme used, the single-stranded arm of the replication fork could be cleaved at the appropriate restriction site or could be generally intact (perhaps randomly broken at various distances from the origin; Fig. 6A, iii and iv). It is not clear how such partially single-stranded molecules would migrate in a two-dimensional gel, but it seems at least plausible that the difference in the extended arcs described above could be explained by these two types of structures. In the second model, the initial replication fork is the same in the gp59-deficient and wild-type infection, but the retrograde replication fork is altered (Fig. 6B). The gp59-deficient infection might support unusually slow leading- and lagging-strand synthesis in the retrograde direction (Fig. 6B, iv and v). Alternatively, the gp59-deficient infection might support slow leading-strand-only replication in the retrograde direction (Fig. 6B, vi; branch migration might generate molecules like those in panel vii).
Fig. 6.

Models depicting possible replicative intermediates generated during a 59 amber mutant infection. A, intermediates are shown that could be formed during leading-strand-only replication of the initial fork. B, top, intermediates formed from both leading- and lagging-strand synthesis of the initial fork are shown. Bottom left (intermediates iv and v), coupled leading- and lagging-strand synthesis of the retrograde fork is shown. Bottom right, leading-strand-only replication of the retrograde fork (inter-mediates vi and vii) is shown.
To directly address the nature of the nascent strands synthesized during wild-type and 59 mutant infections, we performed neutral-alkaline (N/A) two-dimensional gel electrophoresis. We used first-dimension (neutral) conditions identical to those in the neutral-neutral gel experiments above (Figs. 2 and 3), followed by second-dimension alkaline conditions that would separate nascent and parental strands. Strand-specific probes were then used in the Southern blot to distinguish leading- and lagging-strand products. We also included DNA from a negative control infection with a phage deficient in the replicative helicase (gp41), which produce no detectable replicative intermediates in the neutral-neutral two-dimensional gels above (Fig. 2C).
The top panel of Fig. 7A displays a schematic of the above-characterized neutral-neutral two-dimensional patterns, just above a depiction of the same molecules collapsed in the first dimension. The N/A gels start with the same first dimension conditions but utilize denaturing conditions in the second dimension. Nascent leading-strands were detected from wild-type and gp59-deficient infections, but only a trace was detected in the gp41-deficient infection (Fig. 7B). The wild-type nascent leading-strands ran within a very condensed horizontal area, corresponding to the migration of the replicative intermediates in the nucleus of the comet in the N/N gels. The nascent leading-strands from the gp59-deficient infection displayed a much wider horizontal area that corresponded to the migration of the extended comet in the first (neutral) dimension. As judged by the second dimension migration (vertical), the lengths of the nascent leading-strands were essentially the same from both the wild-type and gp59-deficient infections (Fig. 7B). Furthermore, the nascent leading-strands from the gp59-deficient infections were of similar size throughout the region of the extended comet (i.e. the DNA ran as a vertical arc rather than a diagonal arc). These results imply that the nascent leading-strands of the initial fork were not extended by ligation to nascent lagging-strands of a retrograde fork, even though the comet is markedly extended in the gp59-deficient infections.
Fig. 7.
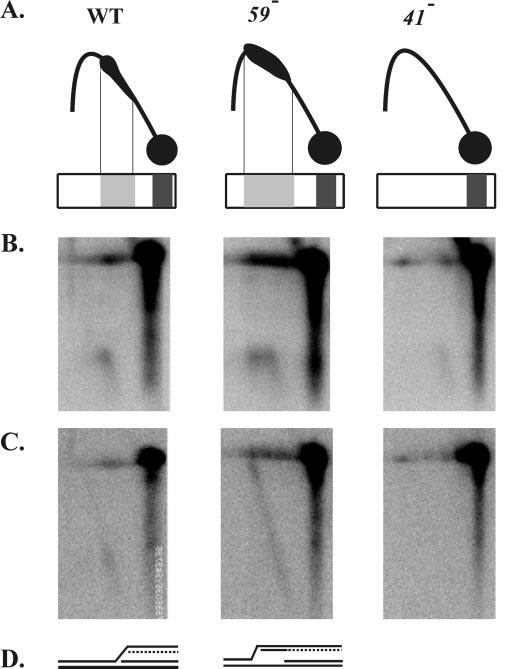
The lagging-strand of the initial fork is elongated in the retrograde direction in a 59 amber mutant infection. A, schematic drawings of the above characterized N/N two-dimensional patterns of wild-type and 59 and 41 amber mutant infections. Immediately below the two-dimensional patterns are representations of the intermediates as they would be collapsed in the first-dimension gel. The first-dimension conditions are the same for the N/N two-dimensional gels shown above and the N/A gels shown in panels B and C. B, N/A two-dimensional gel electrophoresis of DNA collected 4 min after the attachment period and digested with PacI and SwaI. DNA was visualized by Southern blotting with a strand-specific probe that hybridizes to the nascent leading-strand products of the initial fork (wild-type on the left, 59 amber mutant in the center, and 41 amber mutant on the right). C, the blot was washed and reprobed with a strand-specific probe that hybridizes to the nascent lagging-strand products of the initial fork. D, diagram summarizing the structures inferred from the N/A two-dimensional gel analysis.
In the wild-type infection, the nascent lagging-strands of the initial fork migrated similarly to the nascent leading-strands, with only a faint extension in the upper left direction (Fig. 7C). In contrast, the nascent lagging-strands from the gp59-deficient infections were extended diagonally up toward the line of linear DNA (Fig. 7C). These extended nascent strands represent initiation of retrograde DNA synthesis, that is, the first Okazaki fragment of the initial fork was extended as leading-strand synthesis in the retrograde direction. Because only this strand was extended, the detected intermediates underwent leading-strand-only synthesis in the retrograde direction. As expected, the gp41-deficient control infections did not generate detectable nascent lagging-strands.
These results strongly support the model for the extended comet of 59 mutant infections shown in Fig. 6B (molecules vi and/or vii). The DNA polymerase molecule(s) that extends the new leading-strand in the retrograde direction could be, in principle, either the same DNA polymerase that completed the first Okazaki fragment on the initial fork or another molecule that is recruited after both polymerases from the initial fork have left the scene. We favor the latter interpretation, particularly because the extension of the comet in the gp59-deficient infection is much more dramatic at the second time point than at the first (see Fig. 2). Another issue worth commenting on is that retrograde leading-strands of nearly full length are detected in the N/A gel (Fig. 7), and yet the extended comet region is up near the apex of the Y-arc and does not approach the line of linears at 2N (Figs. 2-5). The simplest explanation is that Y-branched DNA with an extended single-stranded region (on the retrograde lagging-strand template) behaves differently than totally duplex Y-branched DNA. For example, formation of molecules like those depicted in Fig. 6B (molecule vii) could readily explain migration in the apex region of the Y-arc. Overall, these results imply that during a wild-type infection, gp59 prevents T4 DNA polymerase from initiating the retrograde fork until gp41 is successfully loaded (see “Discussion”).
Does gp59 Prevent Replication in gp41- or gp32-deficient Infections?—The absence of replicative intermediates in gp41-deficient infections (Fig. 2C) could be due to either a dependence of replication on gp41 or an inhibition of DNA polymerase by gp59 when gp41 is absent. If it is because of inhibition by gp59, then removing gp59 should alleviate the block and result in the production of replicative intermediates. We therefore generated a 41/59 double amber mutant phage to test this hypothesis. Replicative intermediates of ori(uvsY) were detected in a N/N two-dimensional gel from both the wild-type and gp59-deficient infections (Fig. 8, A and B), but not from either the 41 single mutant or the 41/59 double mutant infections (Fig. 8, C and D). Therefore, gp41 is required for generation of replicative intermediates in the absence of gp59, even though gp59 is a loading factor for gp41 (see “Discussion”).
Fig. 8.
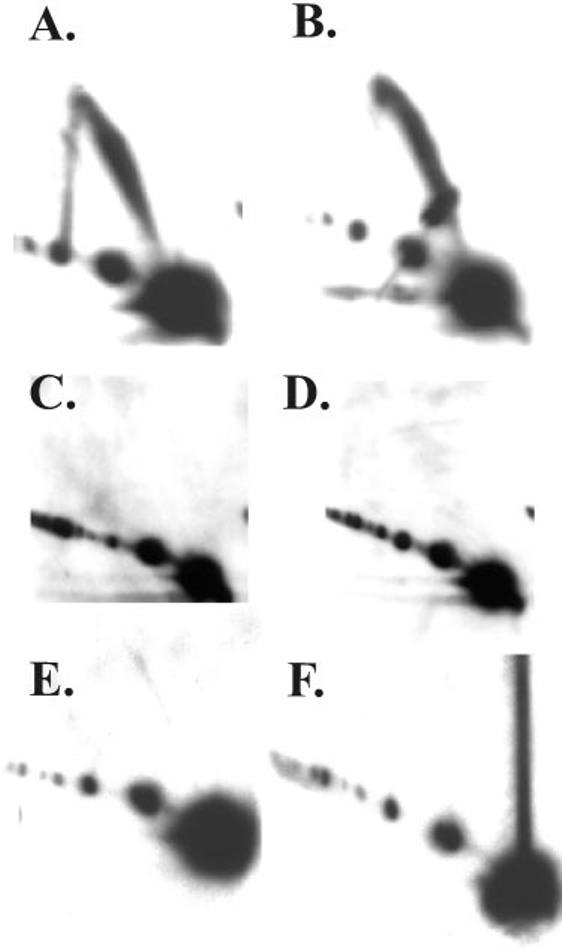
Replication from ori(uvsY) requires gp41 and gp32. DNA was isolated 4 min after the attachment period and analyzed by N/N two-dimensional gel electrophoresis as described in the legend to Fig. 2. The infecting phage was wild-type (A) or carried the following amber mutations: 59 (B), 41 (C), 41 and 59 (D), 32 (E), 32 and 59 (F).
Recently, Jones et al. (24) demonstrated that the T4 single-stranded DNA-binding protein, gp32, is required for helicase-dependent leading-strand synthesis in vitro only when the helicase is loaded by gp59. These results suggested that gp32 might be required for generating replicative intermediates in an otherwise wild-type infection but dispensable in a gp59-deficient infection where gp41 loads by the gp59-independent pathway. Put another way, the presence of gp59 would inhibit generation of replicative intermediates in a gp32-deficient infection.
We first analyzed replicative intermediates of ori(uvsY) from a 32 mutant infection and found no detectable intermediates in the N/N two-dimensional gel (Fig. 8E). The absence of replicative intermediates could be explained by an absolute requirement for gp32 or by gp59-mediated inhibition when gp32 is absent. To distinguish these possibilities, we constructed a 32/59 double mutant phage. Replicative intermediates were also absent in the double mutant samples, indicating an absolute requirement for gp32 during origin-dependent replication in vivo (Fig. 8F).
DISCUSSION
Origin-replicative Intermediates in the Absence of gp59—Although gp59 functions to load the T4 replicative helicase gp41 during in vitro reactions, the protein appears dispensable for origin-dependent replication in vivo (9, 32, 34-37). The two-dimensional gel analyses described here directly verify that two T4 origins, ori(uvsY) and ori(34), generate replicative intermediates during gp59-deficient infections. The migration of these replicative intermediates on N/N two-dimensional gels was similar, but not identical, to those formed during a wild-type infection. Although they migrated on or very near the arc of simple Y-branched DNA, the intermediates from the gp59-deficient infection formed an extended comet consistent with some retrograde replication. Analysis of N/A two-dimensional gels with strand-specific probes revealed that the branched molecules from both wild-type and gp59-deficient infections contained nascent leading- and lagging-strands on the initial replication fork (Fig. 9, initial fork). However, the N/A gels also revealed that the nascent lagging-strand of the initial replication fork had been extended in the retrograde direction to form the new nascent leading-strand of the retrograde fork in DNA from the gp59-deficient infections (Fig. 9, retrograde fork). This leading-strand-only replication in the retrograde direction apparently proceeds slowly, allowing accumulation of intermediates. These data argue that gp59 inhibits retrograde leading-strand-only synthesis during a wild-type infection until gp41 loads to allow retrograde replication that rapidly exits the restriction fragment.
Fig. 9.

Models for initial and retrograde replication fork function during wild-type and gp59-deficient infections. The RNA within the R-loop is indicated by the squiggly line, the leading-strand products by solid lines, and the lagging-strand products by dashed lines.
Origin-dependent replicative intermediates were not detected in 41 or 41 59 mutant infections, indicating the gp41 is essential for origin-dependent replication even in the absence of gp59. How does gp41 load without gp59? The displaced strand of the origin R-loop presents a likely loading site for the initial fork, because gp41 can load onto single-stranded DNA in vitro in the absence of gp59 (17, 19, 23, 24). However, the loading of gp41 for the retrograde fork in the absence of gp59 may be more difficult, because the lagging-strand template for that fork starts out duplex (see Fig. 9B). Perhaps 5′-exonuclease action is necessary to degrade the RNA and/or DNA from the nascent leading-strand of the initial fork, or helicase action might be needed to unwind the parental helix (Fig. 9B). With regard to the possibility of helicase action being important, a previous report suggests that the T4-encoded helicase Dda is involved in gp41 loading at T4 origins (46). However, we saw very similar extended comets in a 59-dda (deletion) double mutant as in the 59 single, arguing that Dda is not necessary (data not shown).
Production of the replicative intermediates in the comet is abolished when the T4-encoded helicase UvsW is expressed from its late promoter and unwinds the R-loop substrate for initiation (45). Complete disappearance of the comet itself would then require either retrograde synthesis past the next restriction site and/or degradation of the replication intermediate (e.g. by branch cleavage). We note that the intermediates in the extended comet of the gp59-deficient infection disappear with similar kinetics as seen in the wild-type infection. Disappearance of these molecules could occur when the leading-strand-only synthesis extends past the next restriction site. Alternatively or in addition, the helicase-primase complex may be loaded once sufficient single-stranded DNA is generated on the lagging-strand template arm, allowing rapid synthesis of both leading- and lagging-strands in the retrograde fork (see Fig. 9B).
What Does the Intensity of the Comet Arcs Reveal about Mutant Infections?—The intensities of the comet arc from wild-type and gp59-deficient infections appeared roughly similar. Although this might suggest that the frequency of initiation at each origin is unchanged by the 59 mutation, this conclusion is premature. The comet arc consists of molecules in which the initial fork has fired and exited the restriction fragment, whereas the retrograde fork has not yet fired (or is still within the restriction fragment, as in the gp59-deficient infections). Therefore, the efficiencies of both initial and retrograde fork function can affect comet intensity. It is possible that both the initial and retrograde forks are initiated with full efficiency in the absence of gp59; alternatively, the efficiency of both forks could have been either increased or decreased by similar amounts in the mutant infection.
The Gatekeeper gp59 Coordinates Leading- and Lagging-strand Replication—The data presented here imply that gp59 prevents premature initiation of the retrograde replication fork in wild-type infections, thus acting as a gatekeeper and prohibiting leading-strand-only synthesis. These results suggest a model in which gp59 holds the holoenzyme in check at the branch point left behind by the initial fork until the primosome is correctly assembled and coupled leading- and lagging-strand synthesis becomes possible (Fig. 9).
gp59 inhibition of T4 holoenzyme has been demonstrated in a number of biochemical experiments. In vitro replication assays using the synthetic R-loop substrate first suggested that gp59 blocks the progression of DNA polymerase in the absence of gp41 (16). This conclusion was confirmed by additional studies using either singly primed M13 DNA (extended by holoenzyme in the first stage of a two-stage reaction) or a nicked substrate that is replicated by a rolling circle mechanism (23, 24). More recently, Xi et al. (47) have presented a detailed study of gp59-mediated inhibition of T4 DNA polymerase, showing that the inhibition involves a direct protein-protein interaction (also see below).
We suspect that gp59 also inhibits DNA polymerase extension of the RNA primer within the R-loop during initiation of the initial replication fork, ensuring coupled leading- and lagging-strand synthesis for that fork. However, the two-dimensional gel results did not prove to be informative on this issue. If the suspicion is true, gp59-deficient (but not wild-type) infections should generate R-loops in which the RNA has been extended by DNA polymerase without any lagging-strand synthesis. However, such molecules have never been characterized by two-dimensional gel electrophoresis; they would presumably be very labile because origin R-loops are only stable in negatively supercoiled DNA in vitro (45, 48, 49).
Several in vitro studies indicate that the synthesis of leading- and lagging-strands is coupled during the elongation phase of DNA replication (50-53). Coupling ensures that most of the DNA remains double-stranded as the fork progresses, even if one of the two polymerases encounters a blocking lesion (54). gp59 could also play an active role in maintaining coupling during elongation, as it does during initiation. Ishmael et al. (30) have demonstrated an interaction between gp59 and the holoenzyme by cross-linking and fluorescence resonance energy transfer analyses, and the interaction was evident in both static and actively replicating forks. Furthermore, gp59 was localized to an elongating replication fork by an elegant approach involving electron microscopy (31). Although these experiments place gp59 at the elongating replication fork, they do not clarify whether gp59 in the elongating fork plays the role of coordinating leading- and lagging-strand synthesis, for example, when the fork encounters a blocking lesion. More experiments will be necessary to test this possibility.
Role of gp59 in Recombination-dependent Replication—gp59-deficient mutants have a “DNA-delay” phenotype, indicating that this protein is essential for recombination-dependent replication (34-37). Furthermore, a 59 mutation strongly blocks replication in several different plasmid model systems for recombination-dependent replication (32, 33, 44, 55). Recombination-dependent replication in T4 depends on the generation of a D-loop by the combined action of UvsX, UvsY, and gp32, and these proteins remain bound to the resulting D-loop (50, 56). The conversion of this D-loop into a functional replication fork is the key step in recombination-dependent replication. gp41 loading and subsequent DNA replication from a D-loop is dependent upon gp59, which repositions the recombination proteins to efficiently load the helicase (56). The simplest explanation for why gp59 is strictly required for this mode of replication and not for origin replication is that gp41 requires assistance to load onto the D-loop coated with recombination proteins. It is also possible that the timing of the infective cycle plays some role. For example, the concentration of gp32 may be significantly different in the very early stages of the infection, when origin replication occurs, or other differences in the protein composition may be critical in the early versus late stages of the infective cycle.
Acknowledgment
We thank Karyn Belanger for preliminary two-dimensional gel analyses of the 59 and 41 mutant infections.
Footnotes
This work was supported by National Institutes of Health Grant GM34622. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: N/N neutral-neutral; N/A, neutral-alkaline.
REFERENCES
- 1.Kornberg A, Baker T. DNA Replication. W. H. Freeman and Company; New York: 1992. [Google Scholar]
- 2.Kues U, Stahl U. Microbiol. Rev. 1989;53:491–516. doi: 10.1128/mr.53.4.491-516.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee DG, Clayton D. J. Biol. Chem. 1996;271:24262–24269. doi: 10.1074/jbc.271.39.24262. [DOI] [PubMed] [Google Scholar]
- 4.Shadel G, Clayton D. Annu. Rev. Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- 5.Luder A, Mosig G. Proc. Natl. Acad. Sci. U. S. A. 1982;79:1101–1105. doi: 10.1073/pnas.79.4.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kreuzer KN, Morrical SW. In: Molecular Biology of Bacteriophage T4. Karam JD, editor. American Society for Microbiology; Washington, D. C.: 1994. pp. 28–42. [Google Scholar]
- 7.Mosig G, Colowick ME, Gruidl A, Chang A, Harvey AJ. FEMS Microbiol. Rev. 1995;17:83–98. doi: 10.1111/j.1574-6976.1995.tb00190.x. [DOI] [PubMed] [Google Scholar]
- 8.Kreuzer KN, Alberts BM. J. Mol. Biol. 1986;188:185–198. doi: 10.1016/0022-2836(86)90303-7. [DOI] [PubMed] [Google Scholar]
- 9.Kreuzer KN, Engman HW, Yap WY. J. Biol. Chem. 1988;263:11348–11357. [PubMed] [Google Scholar]
- 10.Menkens AE, Kreuzer KN. J. Biol. Chem. 1988;263:11358–11365. [PubMed] [Google Scholar]
- 11.Doan PL, Belanger KG, Kreuzer KN. Genetics. 2001;157:1077–1087. doi: 10.1093/genetics/157.3.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carles-Kinch K, Kreuzer KN. J. Mol. Biol. 1997;266:915–926. doi: 10.1006/jmbi.1996.0844. [DOI] [PubMed] [Google Scholar]
- 13.Formosa T, Alberts BM. Cell. 1986;47:793–806. doi: 10.1016/0092-8674(86)90522-2. [DOI] [PubMed] [Google Scholar]
- 14.Brewer B, Fangman W. Cell. 1987;51:463–471. doi: 10.1016/0092-8674(87)90642-8. [DOI] [PubMed] [Google Scholar]
- 15.Belanger KG, Kreuzer KN. Mol. Cell. 1998;2:693–701. doi: 10.1016/s1097-2765(00)80167-7. [DOI] [PubMed] [Google Scholar]
- 16.Nossal NG, Dudas KC, Kreuzer KN. Mol. Cell. 2001;7:31–41. doi: 10.1016/s1097-2765(01)00152-6. [DOI] [PubMed] [Google Scholar]
- 17.Barry J, Alberts BM. J. Biol. Chem. 1994;9:33049–33062. [PubMed] [Google Scholar]
- 18.Morrical SW, Beernick HTH, Dash A, Hempstead K. J. Biol. Chem. 1996;271:20198–20207. doi: 10.1074/jbc.271.33.20198. [DOI] [PubMed] [Google Scholar]
- 19.Jones CE, Mueser TC, Nossal NG. J. Biol. Chem. 2000;275:27145–27154. doi: 10.1074/jbc.M003808200. [DOI] [PubMed] [Google Scholar]
- 20.Hurley JM, Chervitz SA, Jarvis TC, Singer BS, Gold L. J. Mol. Biol. 1993;229:398–418. doi: 10.1006/jmbi.1993.1042. [DOI] [PubMed] [Google Scholar]
- 21.Raney KD, Carver TE, Benkovic SJ. J. Biol. Chem. 1996;271:14074–14081. doi: 10.1074/jbc.271.24.14074. [DOI] [PubMed] [Google Scholar]
- 22.Lefebvre SD, Wong ML, Morrical SW. J. Biol. Chem. 1999;274:22830–22838. doi: 10.1074/jbc.274.32.22830. [DOI] [PubMed] [Google Scholar]
- 23.Ma Y, Wang T, Villemain JL, Giedroc DP, Morrical SW. J. Biol. Chem. 2004;279:19035–19045. doi: 10.1074/jbc.M311738200. [DOI] [PubMed] [Google Scholar]
- 24.Jones CE, Green EM, Stephens JA, Muesser TC, Nossal NG. J. Biol. Chem. 2004;24:25721–25728. doi: 10.1074/jbc.M402128200. [DOI] [PubMed] [Google Scholar]
- 25.Mueser TC, Jones CE, Nossal NG, Hyde CC. J. Mol. Biol. 1999;296:597–612. doi: 10.1006/jmbi.1999.3438. [DOI] [PubMed] [Google Scholar]
- 26.Xu H, Wang Y, Bleuit JS, Morrical SW. Biochemistry. 2001;26:7651–7661. doi: 10.1021/bi010116n. [DOI] [PubMed] [Google Scholar]
- 27.Yonesaki T. J. Biol. Chem. 1994;269:1284–1289. [PubMed] [Google Scholar]
- 28.Morrical SW, Hempstead K, Morrical MD. J. Biol. Chem. 1994;269:33069–33081. [PubMed] [Google Scholar]
- 29.Ishmael FT, Alley SC, Benkovic SJ. J. Biol. Chem. 2002;277:20555–20562. doi: 10.1074/jbc.M111951200. [DOI] [PubMed] [Google Scholar]
- 30.Ishmael FT, Trakselis MA, Benkovic SJ. J. Biol. Chem. 2003;278:3145–3152. doi: 10.1074/jbc.M209858200. [DOI] [PubMed] [Google Scholar]
- 31.Chastain PD, II, Makov AM, Nossal NG, Griffith J. J. Biol. Chem. 2003;23:21276–21285. doi: 10.1074/jbc.M301573200. [DOI] [PubMed] [Google Scholar]
- 32.Kreuzer KN, Yap WY, Menkens AE, Engman HW. J. Biol. Chem. 1988;263:11366–11373. [PubMed] [Google Scholar]
- 33.Kreuzer KN, Saunders M, Weislo LJ, Kreuzer HW. J. Bacteriol. 1995;177:6844–6853. doi: 10.1128/jb.177.23.6844-6853.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu R, Ma F-J, Yeh Y-C. Virology. 1972;47:147–156. doi: 10.1016/0042-6822(72)90248-6. [DOI] [PubMed] [Google Scholar]
- 35.Wu R, Yeh YC. Virology. 1974;59:108–122. doi: 10.1016/0042-6822(74)90209-8. [DOI] [PubMed] [Google Scholar]
- 36.Cunningham R, Berger H. Virology. 1977;80:67–82. doi: 10.1016/0042-6822(77)90381-6. [DOI] [PubMed] [Google Scholar]
- 37.Wu JR, Yeh YC. J. Virol. 1978;27:103–117. doi: 10.1128/jvi.27.1.103-117.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edgar R, Denhardt G, Epstein R. Genetics. 1964;49:635–648. doi: 10.1093/genetics/49.4.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benson KH, Kreuzer KN. J. Virol. 1992;66:6960–6968. doi: 10.1128/jvi.66.12.6960-6968.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Selick HE, Kreuzer KN, Alberts BM. J. Biol. Chem. 1988;263:11336–11347. [PubMed] [Google Scholar]
- 41.Kreuzer KN, Selick HE. In: Molecular Biology of Bacteriophage T4. Karam JD, editor. American Society for Microbiology; Washington, D. C.: 1994. pp. 452–454. [Google Scholar]
- 42.Belanger KG, Mirzayan C, Kreuzer HE, Alberts BM, Kreuzer KN. Nucleic Acids Res. 1996;24:2166–2175. doi: 10.1093/nar/24.11.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karam JD, O'Donnell PV. J. Virol. 1973;11:933–945. doi: 10.1128/jvi.11.6.933-945.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.George JW, Kreuzer KN. Genetics. 1996;143:1507–1520. doi: 10.1093/genetics/143.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dudas KC, Kreuzer KN. Mol. Cell. Biol. 2001;21:2706–2715. doi: 10.1128/MCB.21.8.2706-2715.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gauss PK, Park K, Spencer TE, Hacker KJ. J. Bacteriol. 1994;176:1667–1672. doi: 10.1128/jb.176.6.1667-1672.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xi J, Zhuang Z, Zhang Z, Selzer T, Spiering MM, Hammes GG, Benkovic SJ. Biochemistry. 2005;44:2305–2318. doi: 10.1021/bi0479508. [DOI] [PubMed] [Google Scholar]
- 48.Kaback DB, Angerer LM, Davidson N. Nucleic Acids Res. 1979;6:2499–2517. doi: 10.1093/nar/6.7.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landgraf R, Chen C, Sigman D. Nucleic Acids Res. 1995;23:3516–3523. doi: 10.1093/nar/23.17.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salinas F, Benkovic SJ. Proc. Natl. Acad. Sci. U. S. A. 2000;97:7196–7201. doi: 10.1073/pnas.97.13.7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kadyrov FA, Drake JW. J. Biol. Chem. 2001;276:29559–29566. doi: 10.1074/jbc.M101310200. [DOI] [PubMed] [Google Scholar]
- 52.Trakselis MA, Mayer MU, Ishmael FT, Roccasecca RM, Benkovic SJ. Trends Biochem. Sci. 2001;26:566–572. doi: 10.1016/s0968-0004(01)01929-6. [DOI] [PubMed] [Google Scholar]
- 53.Yang J, Trakselis MA, Roccasecca RM, Benkovic SJ. J. Biol. Chem. 2003;278:49828–49838. doi: 10.1074/jbc.M307406200. [DOI] [PubMed] [Google Scholar]
- 54.Trakselis MA, Roccasecca RM, Yang J, Valentine AM, Benkovic SJ. J. Biol. Chem. 2003;278:49839–49849. doi: 10.1074/jbc.M307405200. [DOI] [PubMed] [Google Scholar]
- 55.Tomso DT, Kreuzer KN. Genetics. 2000;155:1493–1504. doi: 10.1093/genetics/155.4.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beernink HTH, Morrical SW. Trends Biochem. Sci. 1999;24:385–389. doi: 10.1016/s0968-0004(99)01451-6. [DOI] [PubMed] [Google Scholar]