

Abstract
Histone acetylation is implicated in the regulation of herpes simplex virus type 1 (HSV-1) latency. However, the role of histone acetylation in HSV-1 reactivation is less clear. In this study, the well established model system, quiescently-infected, neuronally-differentiated PC12 (QIF-PC12) cells, was used to address the participation of histone acetylation in HSV-1 reactivation. In this model, sodium butyrate and trichostatin A (TSA), two histone deacetylase inhibitors, stimulated production of infectious HSV-1 progeny from a quiescent state. To identify viral genes responsive to TSA, we analyzed representative α, β, and γ viral genes using quantitative real-time polymerase chain reaction. Only the latency-associated transcript (LAT) accumulated in response to TSA treatment, under culture conditions that restricted virus replication and spread. This led us to evaluate the importance of LAT expression on TSA-induced reactivation. In QIF-PC12 cells, the LAT deletion mutant virus dLAT2903 reactivated equivalently with its wild type parental strain (McKrae) after TSA treatment, as well as forskolin and heat stress treatment. Both viruses also reactivated equivalently from latently infected trigeminal ganglia explants from rabbits. In contrast, there was a marked reduction in the recovery of dLAT2903, as compared to wild type virus, from the eyes of latently infected rabbits following epinephrine iontophoresis. These combined in vitro, ex vivo and in vivo data suggest that LAT is not required for reactivation from latently infected neuronal cells per se, but may enhance processes that allow for the arrival of virus at, or close to, the site of original inoculation (i.e., recrudescence).
Keywords: herpes simplex virus, latency-associated transcript, quiescence, reactivation, histone deacetylase inhibitor, PC12 cells
Introduction
Herpes simplex virus type 1 (HSV-1) is a large alpha herpesvirus capable of two types of infection. The initial lytic infection occurs in epithelium and results in the ultimate destruction of infected cells. Invasion of local peripheral nerve endings and retrograde axonal transport (Johnson, 1964) produces an infection of regional sensory ganglionic neurons (Hill et al 1972; Stevens and Cook, 1971). Neurons that survive retain the virus in a latent state. During latency, HSV-1 DNA forms an episome (Rock and Fraser, 1983) and the majority of the genome is transcriptionally repressed (reviewed in Preston, 2000). The region(s) responsible for regulating viral latency and periodic reactivation of HSV-1 remain largely unknown.
Evidence is accumulating that histone acetylation and derepression of viral gene expression may contribute to the regulation of the lytic and latent life cycles of HSV-1 (Hobbs and DeLuca, 1999; Poon et al, 2003; Kubat et al, 2004). Histones regulate gene activity via modulation of their interactions with DNA (Grunstein, 1997). Histones are post-translationally modified by acetylation, phosphorylation, and ubiquitination (Hong et al, 1993). Acetylation of the histone tail regions by acetyltransferases results in altered chromatin structure, making the nucleosome accessible to the RNA polymerase II complex and transcription initiation (Galasinski et al, 2000; Grunstein, 1997; Reeves, 1984). Recent evidence indicates that when expression/processing of the infected cell polypeptide ICP0 is perturbed and HSV-1 DNA has the opportunity to be silent in the nucleus, the viral DNA is in chromatin-like structures amenable to modification by histone acetylation (Poon et al 2003). Further, inhibition of histone deacetylases by trichostatin A (TSA) results in activation of the HSV-1 ICP0 promoter in neonatal dorsal root neurons in vitro (Arthur et al, 2001). However, whether infectious progeny result from latently infected neurons after TSA treatment is not known.
The latency-associated transcript (LAT) promoter of latent HSV-1 DNA has been found to be associated with acetylated histone H3 (Kubat et al, 2004). This suggests that the LAT promoter is euchromatic and a transcriptionally active structure. However, it is not known if LAT responds to histone deacetylation inhibition. In addition, the role of LAT during the induction phase of reactivation within the neuron after TSA treatment is not known.
In the neurally-derived PC12 cell line, several HSV-1 promoters are responsive to the histone deacetylase inhibitor, sodium butyrate (Frazier et al, 1996). Since histone deacetylase inhibitors enhance HSV gene expression in cells displaying a neuronal phenotype, it was important to determine if these agents could activate a cryptic HSV-1 genome and produce viral progeny in neuronally-differentiated PC12 cells. Rat pheochromocytoma-derived PC12 cells, following neuronal differentiation, support HSV-1 in a quiescent state that is reminiscent of latency (Danaher et al, 1999a; Su et al, 1999). These cells are advantageous in allowing for the establishment of quiescence (i.e., infectious virus not detected in culture supernatants and lysates) with wild-type HSV strains (Danaher et al, 1999a), and the maintenance of quiescence without the requirement of antiviral agents (Danaher et al, 1999a; 1999b). Moreover, reactivation of HSV from quiescently infected, neuronally-differentiated (QIF)-PC12 cells occurs following exposure to diverse stimuli such as physical (heat), chemical (forskolin), and biological (pituitary adenylate cyclase activating peptide) agents (Danaher et al, 1999a; 1999b; 2001; 2003). In addition, the proportion of QIF-PC12 cells that support reactivation is similar to that observed in vivo.
In this report, we used in vitro (i.e., QIF-PC12 cells), ex vivo, and in vivo models of latency, the LAT deletion mutant dLAT2903, and inhibitors of histone deacetylation to further evaluate the participation of histone deacetylation and LAT in virus reactivation and recrudescence. Herein we report that 1) treatment of HSV-1 QIF-PC12 cells with inhibitors of histone deacetylases results in activation of the cryptic viral genome as indicated by the production of infectious progeny, 2) the major LAT accumulates in QIF-PC12 cells following TSA treatment, and 3) whereas the absence of LAT reduced measurable in vivo reactivation at the eye after epinephrine iontophoresis, LAT was not required for efficient reactivation of HSV-1 from explanted latently-infected trigeminal ganglia or from QIF-PC12 cells following induction with TSA and other known in vitro reactivation stimuli. These studies indicate that LAT is dispensable for reactivation, but enhances processes that allow for the arrival of virus at, or close to, the site of original inoculation (i.e., recrudescence).
Results
Histone deacetylase inhibitors induce HSV-1 reactivation from quiescence
The fact that the HSV-1 genome is maintained in association with nucleosomes during latency (Deshmane and Fraser, 1989), and the ICP0 promoter appears responsive to histone deacetylase inhibition in replication-defective HSV recombinants (Arthur et al, 2001), led us to determine whether inhibition of histone deacetylases can induce reactivation of replication-competent HSV-1 from a quiescent state. QIF-PC12 cells were used to examine the effect of histone deacetylase inhibitors on viral genomes harbored within neuronal cells per se, i.e. without the indirect effects due to glial cells, cells of the immune system, and other potential in vivo participants. QIF-PC12 cell cultures were established with HSV-1 strain 17+ and treated with sodium butyrate. Supernatants were monitored daily for the production of virus over the next 8 days. Treatment with sodium butyrate resulted in dramatic reactivation of HSV-1 (Fig. 1A). In contrast, virus did not reactivate significantly above background following treatment with a broad range of concentrations of 5′-azacytidine, an inhibitor of DNA methylation (data not shown). Sodium butyrate inhibits histone deacetylases (Candido et al, 1978), but also has pleiotropic activities (Birren et al, 1987; Kruh, 1982). To further support the involvement of inhibition of histone deacetylation in HSV-1 reactivation, QIF-PC12 cells were treated with TSA, a specific inhibitor of histone deacetylase (Van Lint et al, 1996; Yoshida et al, 1990). TSA treatment resulted in dose-dependent reactivation of the cryptic viral genome, with a maximal response observed at the highest concentration tested (400 ng/ml) (Fig. 1B). These findings suggest that inhibition of histone deacetylation results in altered cellular and/or viral gene expression that favors the production of infectious progeny from a cryptic HSV-1 genome.
Fig. 1.
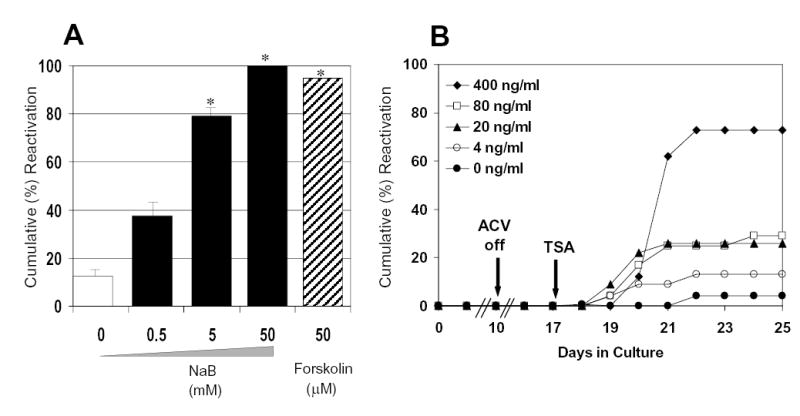
Effect of sodium butyrate (NaB) and TSA on HSV-1 strain 17+ reactivation from QIF-PC12 cells. Cells, grown in duplicate 12-well plates per treatment group, were neuronally differentiated with nerve growth factor (NGF) then infected with HSV-1 strain 17+ at an MOI of 5 in the presence of acycloguanosine (ACV). Seven days after the withdrawal of ACV from the medium (day 17 postinfection), culture medium was changed to medium containing (A) NaB (solid bars), forskolin (striped bar), or no drug (clear bar), or (B) TSA at the indicated concentrations, as well as no treatment, control medium. Reactivation was monitored for 8 days using culture supernatants titered onto Vero cells. Histogram shows results of cumulative wells positive by day 8 post-induction. * P < 0.05 (mock vs. treatment with agent). For all treatment points, the standard deviations were < 10%. Similar results were obtained in duplicate NaB and TSA experiments.
Induction pattern of α (immediate-early), β (early), and γ (late) gene transcription following TSA treatment
Based on the histone deacetylase inhibitor findings, it was important to identify viral genes responsive to TSA treatment. To investigate this, RNA isolated from QIF-PC12 cells established with strain 17+ was reverse transcribed, and the quantity of cDNA synthesized for select host and viral transcripts following TSA induction was determined by real-time PCR. The primers and probes used for the analyses of representative α, β and γ genes and the cellular control GAPDH are indicated in Table 1 and Figures 2A and 2B. In this experiment, the culture medium contained ACV from the day of infection until the day of harvest to maintain quiescence, limit viral gene expression dependent on DNA synthesis, and focus on those initial events that occur following induction without the complication of measuring subsequent events due to a spreading infection. In parallel, QIF-PC12 cells maintained in 12-well plates without ACV were analyzed and shown to reactivate HSV-1 as a control (Fig. 3A).
Table 1.
Real Time PCR Primers and Probes.
| Primer/Probe | Sequence | Location | Source |
|---|---|---|---|
| ICP27 F | CGCCAAGAAAATTTCATCGA
G |
114,766 → 114,786 | Cohrs et al 2000 |
| ICP27 R | ACATCTTGCACCACGCCAG | 114,811 ← 114,829 | |
| ICP27 Probe | CTGGCCTCCGCCGACGAGAC | 114,790 → 114,809 | |
| Major LAT F | CCCACGTACTCCAAGAAGGC | 119,721 → 119,740
6,631 ← 6,650 |
Cohrs et al 2000 |
| Major LAT R | AGACCCAAGCATAGAGAGCC
AG |
6,579 → 6,600
119,771 ← 119,792 |
|
| Major LAT Probe | CCCACCCCGCCTGTGTTTTTG
TG |
119,747 → 119,769
6,602 ← 6,624 |
|
| α4 F | CCGTCCCTGTCCTTTTTCC | 146,926 → 146,944
131,289 ← 131,307 |
* |
| α4 R | GTAGGTCACCTACGGACTCT
CGAT |
131,183 → 131,206
147,027 ← 147,050 |
|
| α4 Probe | CCGCGCTAGTTCCGCGTCGA | 146,964 → 146,983
131,250 ← 131,269 |
|
| Minor LAT F | CGCATGCGCTGTGGTTT | 119,287 → 119,303
7,068 ← 7,084 |
* |
| Minor LATR | AGAACAGGAAAGGCGATGG
A |
7,023 → 7,042
119,329 ← 119,348 |
|
| Minor LAT Probe | CCCGGCAGAACACCGAGGAA
AAA |
7,044 → 7,066
119,305 ← 119,327 |
|
| VP16 F | TCGACGACTTGGGCTTTAGC | 104,864 ← 104,883 | * |
| VP16 R | GAAAACAGATCCTCGTTCCA
GGTA |
104,811 → 104,834 | |
| VP16 Probe | CCCCGCGCTATGTACCATGC
TCG |
104,836 ← 104,858 | |
| UL39 F | ATAGCCAATCCATGACCCTG
TATG |
89,661 → 89,684 | Cohrs et al 2000 |
| UL39 R | GGGTGGAGGCTGGGAGG | 89,707 ← 89,723 | |
| UL39 Probe | CACGGAGAAGGCGGACGGG
A |
89,686 → 89,705 | |
| Spliced α0 F | CCCTCCAGCCGCATACG | 2,238 → 2,254
124,117 ← 124,133 |
* |
| Spliced α0 R | CCTCAGAGTCGCTGCTGTCC | 123,225 → 123,244
3,127 ← 3,146 |
|
| Spliced α0 Probe | CAGCGCGAGCCCGCCC | Probe spans intron | |
| GAPDHS | GAACATCATCCCTGCATCCA | Medhurst et al 2000 | |
| GAPDHA | CCAGTGAGCTTCCCGTTCA | ||
| GAPDH Probe | CTTGCCCACAGCCTTGGCAG
C |
Designed using Primer Express software (PE Applied Biosystems).
Fig. 2.

Location of primers used to assess LAT cDNA levels. (A) Top line represents the region spanning from nucleotide position 117,000 to 124,000 of the prototype viral genome. Locations of the α0 transcripts and 8.3-kb minor LAT and 2.0-kb major LAT are displayed. (B) Nucleotide positions of regions assessed by real-time PCR represent the flanking boundaries of PCR products. The expression of LATs was assessed at three locations. The minor 8.3-kb LAT was assessed at the region 5′ of the major LAT to evaluate changes in expression from the LAP1 promoter (Dobson et al, 1989; Zwaagstra et al, 1990), and also 3′ of the major 2.0-kb LAT to include changes occurring from the LAP2 promoter (Chen et al, 1995; Goins et al, 1994; Nicosia et al, 1993; Wechsler et al, 1988). The third site was within the 2.0-kb major LAT (119,721 to 117,792). Note that primers used to assess levels of α0 cDNA span the first intron of the α0 gene transcript. Transcription from LAP1 initiates at nucleotide 118,801.
Figure 3.
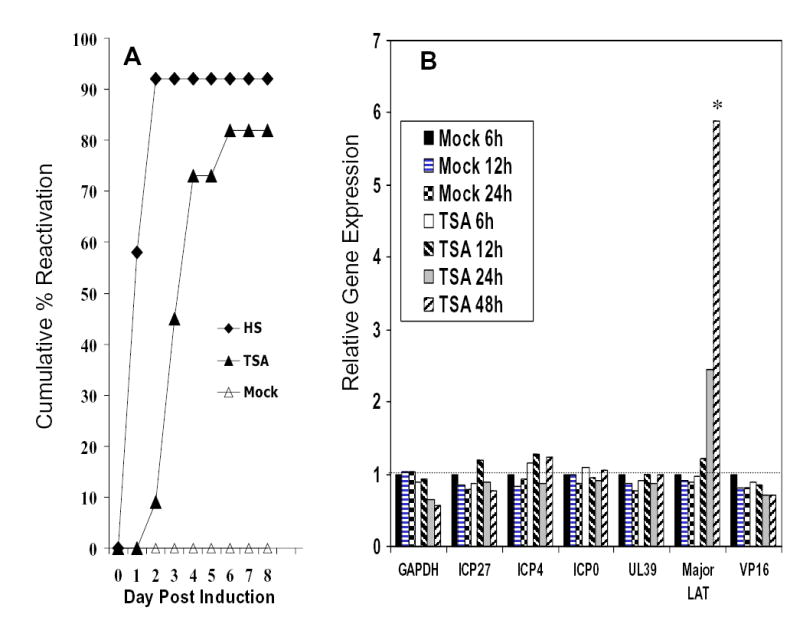
HSV-1 strain 17+ reactivates after TSA induction, and expression pattern of viral genes following TSA-induced reactivation. (A) QIF-PC12 cells were established and maintained in 12-well plates and assayed for reactivation as described in Figure 1, as a positive control for experiment shown in B. Ten days after ACV was removed, cultures were induced with TSA (400 ng/ml), heat stress (HS, 43°C x 3 h, positive control), or mock medium on day 17 postinfection. (B) QIF-PC12 cells, maintained in medium containing ACV, in parallel 6-well plates, were harvested from duplicate wells and RNA isolated at the times indicated following mock or TSA treatment. RNA was reverse transcribed and the quantity of cDNA synthesized for selected host and viral transcripts was determined by real-time PCR. The average numbers of cDNA copies were determined from triplicate reactions and are presented relative to the levels detected 6 h after mock treatment. The 6 h mock treatment was set at 1 for each transcript. Sufficient amounts of cDNA were used in all PCRs such that all representatives of α, β, and γ class genes examined were readily detected. Signal was rarely detected in controls lacking Rtase (-Rtase). When detected in –Rtase controls, the level was substantially below the levels detected in experimental samples. * P < 0.05 (LAT vs. mock treatment). For all cDNA results, the mean standard deviations were < 17%.
Figure 3B shows that the level of the cellular GAPDH transcript remained constant at 6, 12, and 24 h post-mock treatment and 6 and 12 h post-TSA treatment, then decreased by approximately 40 to 50% by 24 and 48 h post-TSA treatment. During this time frame of quiescence, expression of all viral transcripts examined was detected and decreased to varying degrees after mock treatment. Following TSA treatment on day 17 postinfection, the amount of the major LAT more than doubled by 24 h and increased nearly 5-fold by 48 h post treatment (P < 0.05 compared with mock treatment). Similar increases were observed in the minor LAT in most assays (data not shown). In contrast, increases were not detected in any of the representative α, β or γ genes.
LAT is not required for reactivation of a cryptic HSV-1 genome in neuronal cells
Because LAT accumulated in response to TSA treatment and the role of LAT during the induction phase of reactivation has not been characterized, we investigated whether LAT plays a direct role in reactivation. A correlative approach using in vitro, ex vivo, and in vivo models of latency was employed (Miller et al, 2003). This strategy allowed us to observe the role of LAT during HSV-1 reactivation from latency at the cellular and tissue levels and in the animal.
In the in vitro analysis, neuronally-differentiated PC12 cultures were infected with the HSV-1 LAT deletion mutant dLAT2903 or its parent strain McKrae at a multiplicity of infection (MOI) of 5 as described in the Materials and Methods. The quiescent phase was established and maintained in 99% of cultures until induction on day 17 pi. Activation of HSV-1 from QIF-PC12 cells was measured using daily aliquots of culture supernatants that were titered onto Vero cells. In replicate experiments, HSV-1 reactivated from dLAT2903-infected cultures after induction with TSA or forskolin (Fig. 4). For each treatment, the level and kinetics of reactivation from dLAT2903-infected QIF-PC12 cells was equivalent with that of cells infected with its parent McKrae strain. Similar results were observed following heat stress (data not shown).
Fig. 4.
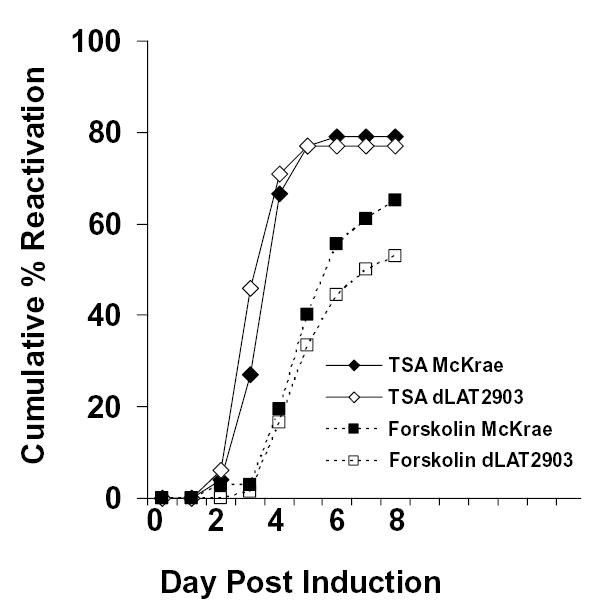
Reactivation of dLAT2903 and McKrae from QIF-PC12 cells is similar. Cells were grown in duplicate 12-well plates per treatment group, neuronally differentiated with NGF, and infected with the indicated strain at an MOI of 5. On day 17 postinfection, cultures were exposed to maintenance medium (mock), medium containing TSA (400 ng/ml) or forskolin (50 μM). Reactivation was monitored daily for 8 days using culture supernatants titered onto Vero cells. Data represents means from at least two independent experiments. Spontaneous reactivation from mock induced cultures was less than 12%. The mean standard deviations for each time point were less than 15%. Similar results were observed when TSA treatment was performed on day 31 postinfection.
To determine if viral genome copy number contributed to the observed response, the amount of viral DNA present in QIF-PC12 cells was measured from triplicate cultures by quantitative real-time PCR. The viral DNA copy was virtually equivalent for cultures infected with the two strains (McKrae = 3.85 ± 0.12 x 106 genome equivalents vs. dLAT2903 = 4.23 ± 0.4 x 106 genome equivalents per 100 ng PC12 DNA).
The explant and in vivo studies were performed by infecting the eyes of New Zealand white rabbits with HSV-1 McKrae, dLAT2903, and its rescue (dLAT2903R). These strains were selected because the absence of LAT in dLAT2903 has been well documented to reduce reactivation efficiency in vivo (Perng et al, 1994). The acute infection was monitored by slit lamp examination (SLE). No differences in the severity of corneal lesions were observed among infections by the three strains. After the ocular infections resolved, latency was established after 4 weeks post-infection in rabbit trigeminal ganglia (TG). With this model, we evaluated reactivation in vivo using detection of ocular shedding of HSV-1 induced by transcorneal adrenergic iontophoresis, and ex vivo using TGs that were dissected and cultured as explants in medium containing serum. Aliquots of supernatant explant medium were removed daily and tested for cytopathic effects on primary rabbit kidney (PRK) cells beginning on day 7 post-explant (PE) through day 30 PE. In the ex vivo model, dLAT2903 reactivated equivalently and within a similar time frame as the McKrae and rescue strain (Table 2). The findings indicate that LAT is not required for efficient reactivation of these strains ex vivo.
Table 2.
Reactivation of HSV-1 from explanted, latently-infected rabbit trigeminal ganglia.
| Virus | Time to Positive for Reactivation in Days (Range) | Explants Positive for Reactivationa/Total Explants (%) | P valuesb |
|---|---|---|---|
| McKrae | 15.50 (10–21) | 7/8 (88%) | |
| dLAT2903 | 14.25 (9–20) | 13/15 (87%) | 0.9549 |
| dLAT2903R | 15.33 (9–21) | 10/11 (91%) | 0.8111 |
Reactivation determined as described in the Material and methods.
Compared with McKrae.
In the in vivo latency model, which served as a control, reactivation induced by epinephrine iontophoresis was high in rabbits latently infected with McKrae and dLAT2903R (Table 3). In contrast, reactivation of the LAT deletion mutant dLAT2903 was significantly less as determined by the frequency of eyes and tear-film swabs positive for virus (i.e., recrudescence). To determine whether viral DNA copy number contributed to the observed outcome, the amount of HSV-1 DNA per TG was quantified by real-time PCR. The copy number per TG (9 or 10 TGs per virus group) for the LAT null virus dLAT2903 (2095 ± 545 [SEM]) was virtually equivalent to that of McKrae (1920 ± 517) and dLAT2903R (1808 ± 517).
Table 3.
Ocular reactivation in HSV-1 latently infected rabbits.
| Virus | Rabbits Positive for Reactivationa/Total Rabbits (%) | Eyes Positive for Reactivation/Total Eyes (%) | Eye Swabs Positive for Reactivation/Total Swabs (%) | P valuesb |
|---|---|---|---|---|
| McKrae | 8/8 (100%) | 15/16 (94%) | 47/112 (42%) | |
| dLAT2903 | 2/8 (25%) | 2/16 (13%) | 6/112 (5%) | <0.0001 |
| dLAT2903R | 7/8 (88%) | 14/16 (88%) | 41/112 (37%) | 0.4941 |
Determined by recrudescence in the eye.
P values are a statistical analysis comparing McKrae to dLAT2903 and dLAT2903R for swabs positive per total swabs.
Collectively, these data indicate that LAT is not required for efficient reactivation from the neuronal cells or TG explants. However, LAT appears to contribute to our ability to detect reactivation in vivo when measured as recrudescence at the eye. In addition, the observed difference in reactivation efficiency of the two strains in vivo was not attributable to different viral genome copy number in cells harboring cryptic HSV-1 genomes.
Discussion
In the present study, we used QIF-PC12 cells, TG explants, and the rabbit eye model of latency to examine the effect of histone deacetylase inhibitors and LAT expression on HSV-1 reactivation. The QIF-PC12 model was used because it provides a useful system similar to latency in vivo (Miller et al, 2003; Millhouse and Wigdahl, 2000), with the distinct advantages that all cells have a neuronal phenotype and can be infected with wild-type HSV-1 strains, and the predictable disruption of quiescence and the resulting activation of virus production following exposure to physical (heat), chemical (forskolin), and biological (pituitary adenylate cyclase activating peptide) stimuli (Danaher et al, 1999a; Danaher et al, 1999b; Danaher et al, 2001). This culture system allows one to analyze events that regulate the transition of a cryptic genome to an active genome without the complications of immunological processes that can limit one’s ability to distinguish between reactivation (a neuronal event) and recrudescence (the subsequent epithelial event) in vivo.
In the current study, treatment of QIF-PC12 cells with inhibitors of histone deacetylases resulted in the production of infectious HSV-1 progeny. Our findings regarding sodium butyrate and TSA are consistent with observations on herpesvirus DNA (Poon et al, 2003; Kubat et al, 2004) and the role of histone deacetylases in the regulation of gene expression (Van Lint et al, 1996). During latency, HSV-1 DNA has been detected in association with nucleosome-like structures (Deshmane and Fraser, 1989). Thus, a post-translational change in the histone state could alter the expression of herpesvirus genes. Arthur et al 2001 found that cytomegalovirus IE1 and HSV-1 ICP0 promoters are responsive to TSA during quiescence in neonatal dorsal root neurons. A product of the ICP0 gene appears to be operationally similar to inhibitors of histone deacetylases (Hobbs and DeLuca, 1999; Poon et al, 2003), and the LAT promoter has been shown to be associated with hyperacetylated H3 histones during latency (Kubat et al, 2004). In human γ-herpesviruses, a correlation between repression by histone deacetylases and maintenance of latency and, conversely, histone acetylation and viral reactivation has been demonstrated (Bryant and Farrell, 2002; Gwack et al, 2001; Jenkins et al, 2000). Thus, it is plausible that the HSV-1 genome is silenced during latency by a mechanism involving histones, and those processes that allow for acetylation of histones result in derepression and transcription of host and/or viral genes required of reactivation (Hsia and Shi, 2002; Poon et al, 2003). It is important to note that regulation of viral quiescence does not appear to involve demethylation, as the HSV-1 genome is reported to be unmethylated (Dressler et al, 1987; Low et al, 1969; Lundberg et al, 2003; Kubat et al, 2004) and virus production was not induced from QIF-PC12 cells following 5′-azacytidine treatment, over a broad range of concentrations tested (data not shown).
The accumulation of the major LAT following TSA induction, in the absence of obvious increased expression from representative genes of α (ICP0, 4, 27), β (UL39), and γ (VP16, a γ1 gene) classes, was unexpected. HSV-1 immediate early promoters are responsive to sodium butyrate or TSA in neonatal neurons (Arthur et al, 2001) and PC12 cells (Frazier et al, 1996), and infectious progeny are generally detectable in QIF-PC12 cultures by day 3 post-TSA treatment. Thus, if viral transcription levels increased, 48 h would have been sufficient time to detect such increases. One possible explanation is that viral transcription initiates in only a few cells in the latently infected population (Arthur et al, 2001; Danaher et al, 1999a; Sawtell and Thompson, 1992), and when assessed among the majority of cells that do not permit reactivation, the increase in transcription may be masked. The presence of ACV may also have restricted the occurrence of an event (i.e., DNA replication) critical for increased viral gene transcription in neurons (Kosz-Vnenchak et al, 1993; Nichol et al, 1996). Alternatively, the changes induced by TSA could act at a post-transcriptional level or on cellular genes important for reactivation.
Although LAT accumulates in QIF-PC12 cells after TSA treatment, the biological significance of this event is less clear. LAT is not required for HSV-1 reactivation in vitro (Fig. 4). Similarly, LAT is not required for reactivation from latently-infected ganglion explants of rabbits (Table 2) or BALB/c mice (Perng et al, 2001), or from outbred Swiss Webster mice following establishment with wild-type levels of viral latency (Thompson and Sawtell, 1997). In contrast, LAT is important for efficient reactivation in vivo (Bloom et al, 1997; Hill et al, 1997; Hill et al, 1990; Thompson and Sawtell, 1997; Trousdale et al, 1991). Evidence supports the fact that in vivo neuroprotection (Thompson and Sawtell, 2001) and down regulation of productive viral gene expression (Chen et al, 1997; Garber, Schaffer, and Knipe, 1997) could be involved. TSA induces neuronal apoptosis in vitro (Boutillier et al, 2002; Boutillier et al, 2003). Thus, LAT could be a viral response that attempts to protect the neuron during an event that induces apoptosis and/or reactivation (Gill and Windebank, 1998; Perng et al, 2000; Perng et al, 2001; Thompson and Sawtell, 2001). If LAT is serving a protective role during reactivation, a logical explanation would be the accumulation of LAT in cells that become protected and do not support reactivation (i.e., remain latently infected), whereas LAT would fail to accumulate to appreciable levels in cells supporting reactivation (Colgin et al, 2001; Halford et al, 1996). Because our analysis involved RT-PCR, it is not certain if the high levels of LAT are accumulating in the select few cells that reactivate virus, or lower levels are produced in the majority of cells that do not.
To our knowledge, this study provides the first evidence that treatment of neuronal cells harboring cryptic HSV-1 genomes with histone deacetylase inhibitors results in the production of infectious progeny. Interestingly, although LAT accumulates in response to TSA treatment and the LAT-ICP0 locus appears important in the histone regulatory state of latency, the absence of LAT did not affect the ability of the virus to reactivate in neuronal cells. Thus, LAT is dispensable for reactivation, but appears to enhance processes subsequent to reactivation that allow for the arrival of virus at, or close to, the site of original inoculation (i.e., recrudescence).
Material and methods
Viruses and cells
HSV-1 strain 17+ was obtained from N. Fraser of the Wistar Institute, Philadelphia, PA. The McKrae, dLAT2903, and dLAT2903-R strains were obtained from S. Wechsler, UCLA School of Medicine, Los Angeles, CA. Viral stocks were prepared in PRK cells for in vivo studies and in Vero cells for in vitro studies and maintained at −85°C. Rat pheochromocytoma (PC12) and African green monkey kidney (Vero cells) were grown as previously described (Danaher et al, 1999a). PRK cells were propagated and maintained at 37ºC in 2% CO2 as monolayers as previously described (Miller et al, 2003). All cells were obtained from American Type Culture Collection (Rockville, MD).
Neuronal differentiation
PC12 cells were dissociated by passage through a 22-gauge needle and plated in RPMI 1640 containing 0.1% fraction V bovine serum albumin (BSA) in 12-well tissue culture dishes coated with rat tail collagen type 1 (Becton Dickinson, Franklin Lakes, NJ) at 2.2 X 105 cells/well. Cells were differentiated and maintained in RPMI 1640 supplemented with 0.1% BSA and 50 ng/ml of 2.5S mouse nerve growth factor (NGF) (Becton Dickinson) (maintenance medium) beginning on the day of plating. Morphologic differentiation was confirmed by microscopic visualization of dendritic processes.
Establishment of a quiescent infection and reactivation of HSV-1
Quiescent HSV-1 infections in neuronally differentiated PC12 cells were established in 12-well plates using 100 μM ACV (Sigma; St. Louis, MO) as previously described (Danaher et al, 1999a; 2001; Miller et al, 2003). After ACV withdrawal, a quiescent state (i.e., free of detectable infectious virus in culture supernatants) was maintained for 7 days prior to induction. Quiescent cultures, that were free of detectable infectious virus, were induced to activate virus on day 17 postinfection, unless otherwise indicated, by subjecting the cells to heat stress (43°C for 3 h), as previously described (Danaher et al, 1999a), or by culturing the cells in maintenance medium supplemented with 50 μM forskolin (Sigma), 0.2 to 20 μM 5′-azacytidine (Sigma), 0.5 to 50 mM sodium butyrate (Sigma), or 0.4 ng/ml to 400 ng/ml TSA (Sigma). Sodium butyrate and TSA were prepared in DMSO. Treatment of QIF-PC12 cells with DMSO at the concentrations used for solubilization of these compounds does not induce detectable HSV-1 reactivation (Danaher et al, 1999b). Culture supernatants were monitored for virus production by plaque forming assays on monolayers of Vero cells as previously described (Miller et al, 2003).
Rabbit corneal inoculation
New Zealand white rabbits (2–3 kg) used in these studies were housed in American Association for Laboratory Animal Care-approved quarters. The corneas of the rabbits were topically anesthetized with proparacaine hydrochloride and inoculated with 2 x 105 PFU of virus following light scarification with a 27-gauge needle. To maximize viral adherence, the eyelids were gently rubbed. Corneal infection was monitored by SLE from day 3 to 14 post-inoculation and scored based on lesions characteristic of HSV-1 infection as previously described (Miller et al, 2003).
In vivo induced viral reactivation
Viral reactivation was induced at least 28 days post-inoculation, by which time viral latency was established. Only eyes of rabbits that had a primary infection confirmed by virus isolation and were clear of epithelial and stromal defects at the time of iontophoresis and negative for HSV-1 by eye swab on the day of harvest were used. Rabbits received transcorneal iontophoresis of 0.015% epinephrine (0.8 mA for 8 min) once a day for 3 days (Gebhardt et al, 1999; Hill et al, 1998; Kwon et al, 1981). The frequency of viral reactivation was determined by detecting viral shedding in the tear film collected on DacronTM swabs for 7 days following the first of three treatments of epinephrine iontophoresis. Swabs were placed onto PRK cells, and cells were monitored for cytopathic effects indicative of infectious virus.
Explant (cocultivation) assay
Following recovery from adrenergic induction, the trigeminal ganglia (TG) of randomly selected rabbits were removed, and the tissues were separated into four or five pieces and placed in Eagles’ modified essential medium containing 10% fetal bovine serum. Beginning 7 days after removal of the ganglia, the culture supernatants were assayed daily for infectious virus on PRK cells through day 20 post-removal. From day 20 to 30, the medium was assessed every other day.
Quantification of HSV DNA from QIF-PC12 cells
DNA was isolated from QIF-PC12 cells harvested on day 25 postinfection using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s directions. Real time PCR was performed on an ABI Prism 7700 Sequence Detection system (PE Applied Biosystems, Foster City, CA) in a 50 xl reaction volume consisting of final concentrations of 1X TaqMan Universal PCR master mix (PE Applied Biosystems), 900 nM primers, and 250 nM TaqMan probe. Analysis of host DNA was performed with rat GAPDH gene primers and probe (GAPDH sense 5′-GAACATCATCCCTGCATCCA-3′; GAPDH antisense 5′-CCAGTGAGCTTCCCGTTCA-3′; GAPDH probe 5′-CTTGCCCACAGCCTTGGCAGC-3′) as described elsewhere (Medhurst et al, 2000). Standards consisting of 10-fold serial dilutions of PC12 DNA ranging from 2 to 20,000 haploid genome equivalents per reaction were performed in triplicate. Primers and a probe specific to the HSV-1 gene encoding VP16 (forward 5′-TCGACGACTTGGGCTTTAGC-3′; reverse 5′-GAAAACAGATCCTCGTTCCAGGTA-3′; probe 5′-CCCCGCGCTATGTACCATGCTCG-3′) were designed with the use of the Primer Express 1.0 software (PE Applied Biosystems). Standards consisting of 10-fold serial dilutions of a plasmid containing the HSV-1 VP16 gene ranging from 2 X 102 to 2 X 106 genome equivalents per reaction were performed in triplicate. Probes were labeled at the 5′ end with the reporter fluorochrome, 6-carboxy-fluorescein (6-FAM), and at the 3′ end with quencher fluorochrome, 6-carboxy-tetramethyl-rhodamine (TAMRA) (PE Applied Biosystems).
Quantification of HSV DNA from TG
To quantitate the latent viral genome copies in rabbits, DNA was isolated from TG using a commercial extraction method (DNeasy Tissue Kit; Qiagen). All real-time PCR reactions were performed in a 50 xl mixture containing 100 ng of TG DNA (2 xl), 1X PCR buffer (Invitrogen, Carlsbad, CA), 4 mM MgCl2, 0.2 xM of each primer, 0.2 xM of probe, 0.2 mM dNTPs mix and 0.025 Unit of PlatinumTaq thermostable DNA polymerase (Invitrogen). Real-time quantitations were performed using the BIO-RAD iCycler iQ system (BioRad, Hercules, CA). The fluorescence threshold value was calculated using the iCycle iQ system software. Relative copy number was calculated using a standard curve generated from purified plasmid DNA (a generous gift from Dr. D.C. Bloom, University of Florida, Gainesville) carrying HSV-1 polymerase gene that had been serially diluted in water to contain from 1 copy to 1 million copies in 2 μl. The forward primer was 5′-CATCACCGACCCGAGGAGGAC-3′, the reverse primer was 5′-GGGCCAGGCGCTTGTTGGTGTA-3′, and the probe was 5′-CCGCCGAACTGAGCAGACACCCGCGC-3′. Primers were purchased from LSU Health Sciences Center Core Laboratories, New Orleans, LA. Probes were labeled at the 5′ end with the reporter fluorochrome, 6-FAM, and at the 3′ end with fluorochrome black hole quencher (BHQ) (Integrated DNA Technologies, Coralville, IA). Standard curves of the Ct values plotted against the logarithm of the copy number were found to be linear from 10 to 106 copies per well.
Quantification of RNA levels
PC12 cells were seeded at 8 X 105 cells/well in 6-well collagen-coated plates, infected with strain 17+ at an MOI of 5, and maintained in the presence of ACV for the duration of the experiment. Cultures were induced with TSA or mock-treated (maintenance medium), as described above. RNA was isolated at the indicated time points using the RNeasy Mini Kit (Qiagen) as recommended and quantified spectrophotometrically. Total RNA (2.5 μg) was treated with DNAse using DNA-freeTM (Ambion, Austin, TX) as recommended. DNase-treated RNA was reverse transcribed using random primers and SuperScript II (Invitrogen) as recommended. The quantity of cDNA synthesized for selected host and viral transcripts was determined by real-time PCR. Genes assessed included the 2.0-kb major LAT, 8.3- kb minor LAT, α0 (ICP0), α4 (ICP4), α27 (ICP27), UL39 (ribonucleotide reductase), and UL48 (VP16). Primers and probes are summarized in Table 1 and include those described by Cohrs et al 2000 for the major LAT and UL39, Medhurst et al 2000 for GAPDH, and others designed using Primer Express software (PE Applied Biosystems). There was comparable sensitivity for all of the gene sets and primers used. All PCR assays of negative control (−Rtase) and experimental (+RTase) samples were performed individually and in triplicate, respectively, from duplicate samples. Each real-time PCR assay included five standards examined in triplicate.
Statistical analysis
Data are presented as means. Results were analyzed with the two-tailed Student’s t test, using the statistical package in Microsoft Excel (Microsoft Corp., Redmond, WA).
Acknowledgments
We thank N. Fraser and S. Wechsler for the viral strains, D. Bloom for plasmid DNA, and M. Frost, J. Howard, S. Rice, A. Savells-Arb, and M. Simpson-Evans for providing technical assistance.
This research was supported by grants from the National Institute of Dental Craniofacial Research: DE014142 (CSM), and the National Eye Institute: EY006311 (JMH), EY002377 (LSU Eye Center Core), and Research to Prevent Blindness: unrestricted departmental grant (LSU Eye Center) and a Senior Scientific Investigator Award (JMH).
References
- Arthur JL, Scarpini CG, Connor V, Lachmann RH, Tolkovsky AM, Efstathiou S. Herpes simplex virus type 1 promoter activity during latency establishment, maintenance, and reactivation in primary dorsal root neurons in vitro. J Virol. 2001;75:3885–3895. doi: 10.1128/JVI.75.8.3885-3895.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birren BW, Taplitz SJ, Herschman HR. Butyrate-induced changes in nuclease sensitivity of chromatin cannot be correlated with transcriptional activation. Mol Cell Biol. 1987;7:3863–3870. doi: 10.1128/mcb.7.11.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom DC, Stevens JG, Hill JM, Tran RK. Mutagenesis of a cAMP response element within the latency-associated transcript promoter of HSV-1 reduces adrenergic reactivation. Virology. 1997;236:202–207. doi: 10.1006/viro.1997.8723. [DOI] [PubMed] [Google Scholar]
- Boutillier AL, Trinh E, Loeffler JP. Constitutive repression of E2F1 transcriptional activity through HDAC proteins is essential for neuronal survival. Ann NY Acad Sci. 2002;973:438–442. doi: 10.1111/j.1749-6632.2002.tb04679.x. [DOI] [PubMed] [Google Scholar]
- Boutillier AL, Trinh E, Loeffler JP. Selective E2F-dependent gene transcription is controlled by histone deacetylase activity during neuronal apoptosis. J Neurochem. 2003;84:814–828. doi: 10.1046/j.1471-4159.2003.01581.x. [DOI] [PubMed] [Google Scholar]
- Bryant H, Farrell PJ. Signal transduction and transcription factor modification during reactivation of Epstein-Barr virus from latency. J Virol. 2002;76:10290–10298. doi: 10.1128/JVI.76.20.10290-10298.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candido EP, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 1978;14:105–113. doi: 10.1016/0092-8674(78)90305-7. [DOI] [PubMed] [Google Scholar]
- Chen SH, Kramer MF, Schaffer PA, Coen DM. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J Virol. 1997;71:5878–5884. doi: 10.1128/jvi.71.8.5878-5884.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schmidt MC, Goins WF, Glorioso JC. Two herpes simplex virus type 1 latency-active promoters differ in their contributions to latency-associated transcript expression during lytic and latent infections. J Virol. 1995;69:7899–7908. doi: 10.1128/jvi.69.12.7899-7908.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohrs RJ, Randall J, Smith J, Gilden DH, Dabrowski C, van Der Keyl H, Tal-Singer R. Analysis of individual human trigeminal ganglia for latent herpes simplex virus type 1 and varicella-zoster virus nucleic acids using real-time PCR. J Virol. 2000;74:11464–11471. doi: 10.1128/jvi.74.24.11464-11471.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin MA, Smith RL, Wilcox CL. Inducible cyclic AMP early repressor produces reactivation of latent herpes simplex virus type 1 in neurons in vitro. J Virol. 2001;75:2912–2920. doi: 10.1128/JVI.75.6.2912-2920.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danaher RJ, Jacob RJ, Chorak M, Freeman CS, Miller CS. Heat stress activates production of herpes simplex virus type 1 from quiescently infected neurally differentiated PC12 cells. J Neurovirol. 1999a;5:374–383. doi: 10.3109/13550289909029478. [DOI] [PubMed] [Google Scholar]
- Danaher RJ, Jacob RJ, Miller CS. Establishment of a quiescent herpes simplex virus type 1 infection in neurally differentiated PC12 cells. J Neurovirol. 1999b;5:258–267. doi: 10.3109/13550289909015812. [DOI] [PubMed] [Google Scholar]
- Danaher RJ, Jacob RJ, Miller CS. Herpesvirus quiescence in neuronal cells. V: forskolin-responsiveness of the herpes simplex virus type 1 alpha0 promoter and contribution of the putative cAMP response element. J Neurovirol. 2003;9:489–497. doi: 10.1080/13550280390218797. [DOI] [PubMed] [Google Scholar]
- Danaher RJ, Savells-Arb AD, Black SA, Jr, Jacob RJ, Miller CS. Herpesvirus quiescence in neuronal cells IV: virus activation induced by pituitary adenylate cyclase-activating polypeptide (PACAP) involves the protein kinase A pathway. J Neurovirol. 2001;7:163–168. doi: 10.1080/13550280152058825. [DOI] [PubMed] [Google Scholar]
- Deshmane SL, Fraser NW. During latency, herpes simplex type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol. 1989;63:943–947. doi: 10.1128/jvi.63.2.943-947.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson AT, Sedarati F, Devi-Rao GB, Flanagan WM, Farrell MJ, Stevens JG, Wagner EK, Feldman LT. Identification of the latency-associated transcript promoter by expression of rabbit b-globin mRNA in mouse sensory nerve ganglia latently infected with a recombinant herpes simplex virus. J Virol. 1989;63:3844–3851. doi: 10.1128/jvi.63.9.3844-3851.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressler G, Rock DL, Fraser NW. Latent herpes simplex virus type 1 DNA is not extensively methylated in vivo. J Gen Virol. 1987;68:1761–1765. doi: 10.1099/0022-1317-68-6-1761. [DOI] [PubMed] [Google Scholar]
- Frazier DP, Cox D, Godshalk EM, Schaffer PA. The herpes simplex virus type 1 latency-associated transcript promoter is activated through Ras and Raf by nerve growth factor and sodium butyrate in PC12 cells. J Virol. 1996;70:7424–7432. doi: 10.1128/jvi.70.11.7424-7432.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galasinski SK, Lively TN, Grebe De Barron A, Goodrich JA. Acetyl coenzyme A stimulates RNA polymerase II transcription and promoter binding by transcription factor IID in the absence of histones. Mol Cell Biol. 2000;20:1923–1930. doi: 10.1128/mcb.20.6.1923-1930.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber DA, Schaffer PA, Knipe DM. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J Virol. 1997;71:5885–5893. doi: 10.1128/jvi.71.8.5885-5893.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt BM, Varnell ED, Hill JM, Kaufman HE (1999). Animal models of ocular herpes simplex virus infection (rabbits, primates, mice). In: Handbook of Animal Models of Infection: experimental models in antimicrobial chemotherapy. Zak O, Sande MA, (eds). Academic Press: San Diego, pp 919–926.
- Gill JS, Windebank AJ. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J Clin Invest. 1998;101:2842–2850. doi: 10.1172/JCI1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goins WF, Sternberg LR, Croen KD, Krause PR, Hendricks RL, Fink DJ, Straus SE, Levine M, Glorioso JC. A novel latency-active promoter is contained within the herpes simplex virus type 1 UL flanking repeats. J Virol. 1994;68:2239–2252. doi: 10.1128/jvi.68.4.2239-2252.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- Gwack Y, Byun H, Hwang S, Lim C, Choe J. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi’s sarcoma-associated herpesvirus open reading frame 50. J Virol. 2001;75:1909–1917. doi: 10.1128/JVI.75.4.1909-1917.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Gebhardt BM, Carr DJ. Mechanisms of herpes simplex virus type 1 reactivation. J Virol. 1996;70:5051–5060. doi: 10.1128/jvi.70.8.5051-5060.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Garza HH, Jr, Su YH, Meegalla R, Hanna LA, Loutsch JM, Thompson HW, Varnell ED, Bloom DC, Block TM. A 437-base-pair deletion at the beginning of the latency-associated transcript promoter significantly reduced adrenergically induced herpes simplex virus type 1 ocular reactivation in latently infected rabbits. J Virol. 1997;71:6555–6559. doi: 10.1128/jvi.71.9.6555-6559.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Sedarati F, Javier RT, Wagner EK, Stevens JG. Herpes simplex virus latent phase transcription facilitates in vivo reactivation. Virology. 1990;174:117–125. doi: 10.1016/0042-6822(90)90060-5. [DOI] [PubMed] [Google Scholar]
- Hill JM, Wen R, Halford WP (1998). Pathogenesis and molecular biology of ocular HSV in the rabbit. In: Herpes Simplex Virus Protocols Brown MS, MacLean AR, (eds). Humana Press/Wiley: Totowa, NJ, pp 291–315.
- Hill TJ, Field HJ, Roome AP. Intra-axonal location of herpes simplex virus particles. J Gen Virol. 1972;15:233–235. doi: 10.1099/0022-1317-15-3-253. [DOI] [PubMed] [Google Scholar]
- Hobbs WE, DeLuca NA. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J Virol. 1999;73:8245–8255. doi: 10.1128/jvi.73.10.8245-8255.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong L, Schroth GP, Matthews HR, Yau P, Bradbury EM. Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 "tail" to DNA. J Biol Chem. 1993;268:305–314. [PubMed] [Google Scholar]
- Hsia SC, Shi YB. Chromatin disruption and histone acetylation in regulation of the human immunodeficiency virus type 1 long terminal repeat by thyroid hormone receptor. Mol Cell Biol. 2002;22:4043–4052. doi: 10.1128/MCB.22.12.4043-4052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins PJ, Binne UK, Farrell PJ. Histone acetylation and reactivation of Epstein-Barr virus from latency. J Virol. 2000;74:710–720. doi: 10.1128/jvi.74.2.710-720.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. The pathogenesis of herpes virus encephalitis. I. Virus pathways to the nervous system of suckling mice demonstrated by fluorescent antibody staining. J Exp Med. 1964;119:343–356. doi: 10.1084/jem.119.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosz-Vnenchak M, Jacobson J, Coen DM, Knipe D. Evidence for a novel regulatory pathway for herpes simplex virus gene expression in trigeminal ganglion neurons. J Virol. 1993;67:5383–5393. doi: 10.1128/jvi.67.9.5383-5393.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruh J. Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Mol Cell Biochem. 1982;42:65–82. doi: 10.1007/BF00222695. [DOI] [PubMed] [Google Scholar]
- Kubat NJ, Tran RK, McAnany P, Bloom DC. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J Virol. 2004;78:1139–1149. doi: 10.1128/JVI.78.3.1139-1149.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon BS, Gangarosa LP, Burch KD, deBack J, Hill JM. Induction of ocular herpes simplex virus shedding by iontophoresis of epinephrine into rabbit cornea. Invest Ophthalmol Vis Sci. 1981;21:442–449. [PubMed] [Google Scholar]
- Low M, Hay J, Keir HM. DNA of herpes simplex virus is not a substrate for methylation in vivo. J Mol Biol. 1969;46:205–207. doi: 10.1016/0022-2836(69)90068-0. [DOI] [PubMed] [Google Scholar]
- Lundberg P, Welander P, Han X, Cantin E. Herpes simplex virus type 1 DNA is immunostimulatory in vitro and in vivo. J Virol. 2003;77:11158–11169. doi: 10.1128/JVI.77.20.11158-11169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medhurst AD, Harrison DC, Read SJ, Campbell CA, Robbins MJ, Pangalos MN. The use of TaqMan RT-PCR assays for semiquantitative analysis of gene expression in CNS tissues and disease models. J Neurosci Methods. 2000;98:9–20. doi: 10.1016/s0165-0270(00)00178-3. [DOI] [PubMed] [Google Scholar]
- Miller CS, Bhattacharjee PS, Higaki S, Jacob RJ, Danaher RJ, Thompson HW, Hill JM. Herpesvirus quiescence (QIF) in neuronal cells VI: correlative analysis demonstrates usefulness of QIF-PC12 Cells to examine HSV-1 latency and reactivation and deregulated LAT ORF Expression. Curr Eye Res. 2003;26:239–248. doi: 10.1076/ceyr.26.3.239.14901. [DOI] [PubMed] [Google Scholar]
- Millhouse S, Wigdahl B. Molecular circuitry regulating herpes simplex virus type 1 latency in neurons. J Neurovirol. 2000;6:6–24. doi: 10.3109/13550280009006378. [DOI] [PubMed] [Google Scholar]
- Nichol PF, Chang JY, Johnson EM, Jr, Olivo PD. Herpes simplex virus gene expression in neurons: viral DNA synthesis is a critical regulatory event in the branch point between the lytic and latent pathways. J Virol. 1996;70:5476–5486. doi: 10.1128/jvi.70.8.5476-5486.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicosia M, Deshmane SL, Zabolotny J, Valyi-Nagy T, Fraser NW. Herpes simplex virus type 1 latency-associated transcript (LAT) promoter deletion mutants can express a 2-kilobase transcript mapping to the LAT region. J Virol. 1993;67:7276–7283. doi: 10.1128/jvi.67.12.7276-7283.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol. 1994;68:8045–8055. doi: 10.1128/jvi.68.12.8045-8055.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, Wechsler SL. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science. 2000;287:1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- Perng GC, Slanina SM, Ghiasi H, Nesburn AB, Wechsler SL. The effect of latency-associated transcript on the herpes simplex virus type 1 latency-reactivation phenotype is mouse strain-dependent. J Gen Virol. 2001;82:1117–1122. doi: 10.1099/0022-1317-82-5-1117. [DOI] [PubMed] [Google Scholar]
- Poon AP, Liang Y, Roizman B. Herpes simplex virus 1 gene expression is accelerated by inhibitors of histone deacetylases in rabbit skin cells infected with a mutant carrying a cDNA copy of the infected-cell protein no. 0. J Virol. 2003;77:12671–12678. doi: 10.1128/JVI.77.23.12671-12678.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston CM. Repression of viral transcription during herpes simplex virus latency. J Gen Virol. 2000;81:1–19. doi: 10.1099/0022-1317-81-1-1. [DOI] [PubMed] [Google Scholar]
- Reeves R. Transcriptionally active chromatin. Biochim Biophys Acta. 1984;782:343–393. doi: 10.1016/0167-4781(84)90044-7. [DOI] [PubMed] [Google Scholar]
- Rock DL, Fraser NW. Detection of HSV-1 genome in central nervous system of latently infected mice. Nature. 1983;302:523–525. doi: 10.1038/302523a0. [DOI] [PubMed] [Google Scholar]
- Sawtell NM, Thompson RL. Rapid in vivo reactivation of herpes simplex virus in latently infected murine ganglionic neurons after transient hyperthermia. J Virol. 1992;66:2150–2156. doi: 10.1128/jvi.66.4.2150-2156.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JG, Cook ML. Latent herpes simplex virus in spinal ganglia of mice. Science. 1971;173:843–845. doi: 10.1126/science.173.3999.843. [DOI] [PubMed] [Google Scholar]
- Su YH, Meegalla RL, Chowhan R, Cubitt C, Oakes JE, Lausch RN, Fraser NW, Block TM. Human corneal cells and other fibroblasts can stimulate the appearance of herpes simplex virus from quiescently infected PC12 cells. J Virol. 1999;73:4171–4180. doi: 10.1128/jvi.73.5.4171-4180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM. The herpes simplex virus type 1 latency-associated transcript gene regulates the establishment of latency. J Virol. 1997;71:5432–5440. doi: 10.1128/jvi.71.7.5432-5440.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J Virol. 2001;75:6660–6675. doi: 10.1128/JVI.75.14.6660-6675.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trousdale MD, Steiner I, Spivack JG, Deshmane SL, Brown SM, MacLean AR, Subak-Sharpe JH, Fraser NW. In vivo and in vitro reactivation impairment of a herpes simplex virus type 1 latency-associated transcript variant in a rabbit eye model. J Virol. 1991;65:6989–6993. doi: 10.1128/jvi.65.12.6989-6993.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996;5:245–253. [PMC free article] [PubMed] [Google Scholar]
- Wechsler SL, Nesburn AB, Watson R, Slanina S, Ghiasi H. Fine mapping of the major latency-related RNA of herpes simplex virus type 1 in humans. J Gen Virol. 1988;69:3101–3106. doi: 10.1099/0022-1317-69-12-3101. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- Zwaagstra JC, Ghiasi H, Slanina SM, Nesburn AB, Wheatley SC, Lillycrop K, Wood J, Latchman DS, Patel K, Wechsle SL. Activity of herpes simplex virus type 1 latency-associated transcript (LAT) promoter in neuron-derived cells: evidence for neuron specificity and for a large LAT transcript. J Virol. 1990;64:5019–5028. doi: 10.1128/jvi.64.10.5019-5028.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]