

Abstract
Rationale: Acetylcholinesterase inhibitors are widely used for the treatment of patients with Alzheimer’s disease (AD). However, the relationship between the capacity of such drugs to ameliorate the symptoms of AD and their ability to alter the underlying disease process is not well understood. Transgenic mice that overexpress the human form of amyloid precursor protein and develop deposits of b-amyloid (Ab) and behavioral deficits during adulthood are useful for investigating this question. Objectives: The effects of administration of two acetylcholinesterase inhibitors, physostigmine and donepezil, on Ab plaque formation and memory-related behaviors were investigated in the Tg2576 transgenic mouse model of AD. At 9-10 months of age, Tg2576 transgenic (Tg(+)) mice develop Ab plaques and impairments on paradigms related to learning and memory as compared to transgene negative (Tg(-)) mice. Methods: Beginning at 9 months of age, increasing doses of physostigmine (0.03, 0.1, and 0.3mg/kg), donepezil (0.1, 0.3 and 1.0mg/kg) or saline were administered over six weeks to cohorts of Tg(+) and Tg(-) mice. Performance on tests of spatial reversal learning and fear conditioning was evaluated at each drug dose throughout the period of drug administration. After drug administration was completed, the animals were sacrificed and Ab plaque number was quantified. Results: Administration of physostigmine and donepezil improved deficits in contextual and cued memory in Tg(+) mice so that their behaviors became more similar to Tg(-) mice. However, administration of physostigmine and donepezil tended to improve cued memory and deficits in spatial learning in both Tg(+) and Tg(-) mice. Physostigmine administration demonstrated more prominent effects in improving contextual memory than donepezil, while donepezil was more effective than physostigmine in improving deficits in the acquisition of the spatial memory paradigm. Administration of neither drug altered the deposition of Aß plaques. Conclusions: These studies suggest that acetylcholinesterase inhibitors can ameliorate memory deficits in Tg(+) mice without necessarily altering the deposition of Aß plaques. Tg2576 mice may be useful as an animal model to further investigate the mechanisms by which aceytlcholinesterase inhibitors improve cognitive deficits in patients with AD.
Keywords: acetylcholinesterase inhibitor, Alzheimer’s disease, physostigmine, donepezil, ß-amyloid plaques, memory, Tg2576 mice
1. Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the most common cause of dementia in elderly individuals. The pathological hallmarks of AD are senile plaques and neurofibrillary tangles, which are distributed across many regions of the brain, including the structures of the medial temporal lobe involved in learning and memory (Price 1986, Jellinger 1990). Loss of cholinergic neurons and reduced activity of choline acetyltransferase in the cerebral cortex and hippocampus, have also been consistent findings in AD (Cummings et al. 1998, Francis et al. 1999, West and Gundersen 1990, Mullan 2000). In fact, reduced cholinergic activity in the brains of individuals with AD formed the rationale for the development of acetylcholinesterse inhibitors as drugs to ameliorate the dementia associated with AD (Davis et al. 1992, Rogers et al. 1998, Tariot et al. 2000, Csernansky, et al, 2002). Commonly prescribed cholinesterase inhibitors include donepezil, rivastigmine and galantamine (Moghul and Wilkinson, 2001, Shigeta and Homma, 2001).
The discovery that muscarinic receptor activation may regulate processing of the amyloid precursor protein (APP) (Nitsch et al. 1992) formed the basis for the hypothesis that the administration of cholinesterase inhibitors might slow the disease process of AD by decreasing the production of ß-amyloid (Aß) (Inestrosa et al. 1996, Nilsberth et al. 2002). However, it has been difficult to directly study the sequence of pathophysiological events in individuals with AD to test such hypotheses. Transgenic mice that overexpress human APP (hAPP) provide an alternative approach to investigate the pathophysiological processes underlying AD, and to test hypotheses related to the effects of drug therapies on the pathogenesis of AD. The Tg2576 mouse is one of the most well characterized strains of hAPP transgenic animals (Hsiao et al. 1996, Sturchler-Pierrat et al. 1997, Irizarry et al. 1997). At approximately 9 months of age, Aß deposits become visible in cortical and limbic brain regions of Tg2576 mice together with indications of cellular inflammation and behavioral deficits (Hsiao et al. 1996, Chapman et al. 1999). However, it should be noted that neuronal loss, including loss of cholinergic neurons is not a feature of Tg2576 mice (Jaffer, et al, 2001).
In this study, we administered the prototypical acetylcholinesterase inhibitor, physostigmine, as well as one of the acetylcholinesterase inhibitors used routinely for clinical purposes, donepezil, to Tg2576 mice in increasing doses over 6 weeks at the time Aß deposits and behavioral deficits usually begin to become apparent (i.e., 9 months of age). The principal aim of the study was to compare the effects of drug administration on the deposition of Aß deposits versus deficits in memory-related behavioral paradigms. The effect of physostigmine and donepezil on Aß deposition was assessed using immunohistochemistry and thioflavine S staining. Spatial working memory was assessed using a spatial reversal learning paradigm and contextual memory was assessed using a fear conditioning paradigm. In addition, spontaneous ambulation and sensitivity to the shock stimulus used in the fear conditioning paradigm were assessed to control for non-specific effects of drug treatment on behavior.
2. Methods
2.1. Animals
Heterozygous transgenic mice expressing the Swedish AD mutant gene, hAPP K670N, M671L (Tg2576; Hsiao et al. 1996), were used in this study. Tg2576 mice overexpress human APP 695, containing the double-mutation Lys670-Asn and Met671-Leu (K670N, M671L), which is driven by the hamster prion protein gene promoter in C57Bl6xSJL. Tg2576 males (Taconic Farms Inc. Germantown, NY) were bred with C57B6/SJL females (Jackson Laboratories, Bar Harbor, ME), and the offspring of both sexes were distributed equally among the experimental groups. Genotyping to confirm the presence of the hAPP DNA sequence in offspring (Tg(+)) was performed using DNA obtained from tail biopsies. Transgene-negative (Tg(-)) littermates were used as controls.
Animals were housed under standard conditions with a 12:12 light/dark cycle and food and water available ad libitum. Beginning at 9 months of age, 16 Tg(+) mice and 16 Tg(-) littermates were divided into three groups (each group containing five or six Tg(+) and five or six Tg(-) animals). The first two groups of animals received increasing doses of physostigmine or donepezil, over six weeks. The remaining control group received daily saline injections for six weeks.
2.2. Drug Administration
Physostigmine salicylate (Sigma, St. Louis, MO) and donepezil (Eisai, Teaneck, NJ) were dissolved in sterile saline so that all doses were administered in constant volume of 1.0 ml/kg body weight. Three doses of physostigmine (0.03, 0.1, and 0.3mg/kg), and donepezil (0.1, 0.3 and 1.0mg/kg) and physiological saline (1ml/kg) were given to Tg(+) and Tg(-) mice subcutaneously on a daily basis. On days when behavioral testing was performed, drugs were administered 30 minutes before testing.
2.3. Behavioral testing
Behavioral testing was performed at each drug dose using the same sequence over two weeks in all experimental groups: 1) spatial reversal learning, 2) locomotion, 3) fear conditioning and 4) shock sensitivity. This order was selected to minimize interference among testing paradigms. Separate experimental groups were used to test the effects of physostigmine, donepezil and saline; however, the three different doses of the two drugs were tested sequentially in the same experimental group to minimize the number of animals used in the study. All drug administration and testing was complete after six weeks.
Spatial reversal learning. Acquisition of the spatial learning paradigm and reversal learning were tested during the first five days of drug administration using a water T-maze as described in (Bardgett et al. 2003). The mice were habituated to the water T-maze during days 1-3, and then task acquisition began on day 4. On day 4, mice were trained to find the escape platform in one choice arm of the maze until 6 of 8 correct choices had been made on consecutive trials (Bardgett et al. 2003). The reversal learning phase was then conducted on day 5. During the reversal learning phase, mice were trained to find the escape platform in the choice arm opposite from the location of the escape platform on day 4. The same performance criterion and inter-trial interval were used as during task acquisition (Bardgett et al. 2003).
Ambulation. Because performance on a spatial reversal learning paradigm may be influenced by the capacity for ambulation, large ambulatory movements were assessed as previously described (Bardgett et al. 2003). After a rest period of two days, horizontal ambulatory movements (excluding vertical and fine motor movements) were assessed in a chamber equipped with a grid of motion-sensitive detectors on day 8. The numbers of movements accompanied by simultaneous blocking and unblocking of a detector in the horizontal dimension were measured during a one hour period.
Fear conditioning. The animals’ capacity for contextual and cued memory was tested using a fear conditioning paradigm beginning on day 9 (Bardgett et al. 2003). Testing took place in a chamber that contained a piece of absorbent cotton soaked in an odor-emitting solution (e.g., mint extract) placed below the grid floor. A 5 minute, 3 trial 80 dB, 2800 Hz tone-foot shock sequence was administered to train the animals on day 9. On day 10, memory for context was tested by returning each mouse to the chamber without exposure to the tone and foot shock, and recording the presence or absence of freezing behavior every 10 seconds for 8 minutes. Freezing was defined as no movement (ambulation, sniffing or stereotypy) other than respiration.
On day11, the animals’ response to an alternate context and to the auditory cue was tested. Coconut extract was placed in the cup and the 80 dB tone was presented, but no foot shock was delivered. The presence or absence of freezing in response to the alternate context was then determined during the first 2 minutes of the trail. Then, the tone was presented continuously for the remaining 8 minutes of the trial, and the presence or absence of freezing in response to the tone was determined.
Foot shock sensitivity. On day 12, the animals were tested to assess their sensitivity to the conditioning stimulus (i.e., foot shock), as previously described (Bardgett et al. 2003).
2.4. ß-amyloid plaque quantification.
Following the last day of behavioral testing, animals were deeply anesthetized with 150 mg/kg pentobarbital and transcardially perfused with 1% heparinized saline followed by 4% paraformaldehyde in 0.1 M PBS. Brains were removed and post-fixed overnight in 30% sucrose, 4% paraformaldehyde in 0.1 M PBS. Thirty-five mm sections were cut though the entire hippocampus on a freezing microtome and stored in PBS/sodium azide (0.06%).
The most anterior section containing the dorsal hippocampus was chosen as the first section, and thereafter every 6th section was selected until the entire hippocampus was covered (total 12-16 sections per brain). For staining of Aß plaques, selected sections were rinsed with 0.1 M PBS (pH 7.4) and treated with a blocking solution of 5% normal goat serum for 1 hour. Sections were then incubated in the primary antibody at 4° C overnight (1:1000, Biosource Camarillo, CA). The sections were then washed in PBS and incubated in biotinylated, goat anti-rabbit IgG for two hours. After washing again in PBS, the sections were treated with an avidin-biotin complex (Vector Laboratories, Burlingame, CA) for one hour. β-amyloid like immunoreactivity was visualized using a DAB kit (Vector Laboratories, Burlingame, CA). The total number of Ab-immunoreactive plaques was counted in each animal using a point grid superimposed over the predefined counting box (West and Gundersen 1990). The percentage of points overlying plaques was calculated to determine plaque number. Plaque burden was calculated by multiplying this percentage by the area covered by the grid. To confirm the presence of ß-amyloid plaques, thioflavine S was used to stain floating sections in a 1% thioflavine S aqueous solution for 5 minutes, and then differentiated in 70% alcohol 3-5 minutes (Guntern et al. 1992, Dong et al. 2004).
2.5. Data analysis.
The behavioral testing data were analyzed in stages to determine the effects of hAPP status (i.e., (Tg(+) versus Tg(-)) and drug administration. First, a two-way analysis of variance (ANOVA) was performed using the data from the highest dose of each of the two acetylcholinesterase inhibitors or saline to determine whether there were effects of hAPP status, drug condition or an interaction between hAPP status and drug administration for each of the behavioral paradigms. Then, the data collected in the groups of animals injected with ascending doses of either physostigmine or donepezil were analyzed separately using repeated measures ANOVA to determine whether there was a dose/time effect for each of the drug conditions using saline control adjusted scores. Saline control adjusted scores were obtained by taking the task score for each animal administered drug and dividing it by the mean for the respective saline control group (Tg(+) or Tg(-)). In each of these analyses, the effect of drug dose and the interaction between drug dose and hAPP status were tested.
The Aß plaque data were analyzed in Tg+ animals using a one-way ANOVA to determine whether there was an effect of drug condition on the number of plaques counted and on plaque burden.
For both behavioral testing and Aß plaque data, post-hoc between-group analyses were performed using a Fisher’s protected Least Squares Design (PLSD) test. P values of <.05 were used to indicate a significant group difference in all analyses.
3. Results
There was a significant effect of hAPP status (i.e., Tg(+) versus Tg(-)) on acquisition of the spatial learning paradigm (F=8.5, df =1,24, p<.01), but not on reversal learning. The effects of hAPP status on the capacity for contextual memory (F=6.57, df=1,24, p=.02) and cued memory (F=4.63, df=1,24, p=.04) in the fear conditioning paradigm were both significant. However, there was only a trend toward significance for the effect of hAPP status on the alternate context measure in the fear conditioning paradigm (F=3.91, df=1,24, p=.06), and the effects of hAPP status on shock sensitivity and ambulation were not significant.
There was a significant effect of drug condition (i.e., highest dose of each drug or saline) on acquisition of the spatial learning paradigm (F=5.75, df=2,24, p<.01), and a similar trend was observed for reversal learning (F=3.14, df=2,24, p=.06). The effects of drug condition on the capacity for contextual memory (F=4.13, df=2,24, p=.03) and cued memory (F=3.55, df=2,24, p<.05) in the fear conditioning paradigm were significant. There was also a significant effect of drug condition on memory of an alternate context in the fear conditioning paradigm (F=3.86, df=2,24, p=.04). Finally, the effects of drug condition on shock sensitivity and ambulation were not significant.
The interactions between hAPP status and drug condition on acquisition of the spatial learning paradigm and reversal learning were not significant. However, there was a significant interaction between hAPP status and drug condition on the capacity for contextual memory (F=4.57, df=2,4, p=.02). The interaction effects between hAPP status and drug condition for cued memory and for the alternate context were not significant. Finally, the interaction effects between hAPP status and drug condition on shock sensitivity and ambulation were not significant.
Drug treatment tended to reduce the number of trials required for acquisition of the spatial learning paradigm in both Tg(+) and Tg(-) animals (see Figures 1 and 2, panels a and b). In particular, the number of acquisition trials was significantly less in Tg(+) and Tg(-) animals injected with donepezil (p<.05); similar trends were observed in Tg(+) and Tg(-) animals injected with physostigmine (.05<p<.10). Similarly, the number of reversal learning trials was significantly less in Tg(-) animals injected with donepezil (p = 0.02), and tended to be significantly less in Tg(-) animals injected with physostigmine (p = 0.06).
Figure 1.
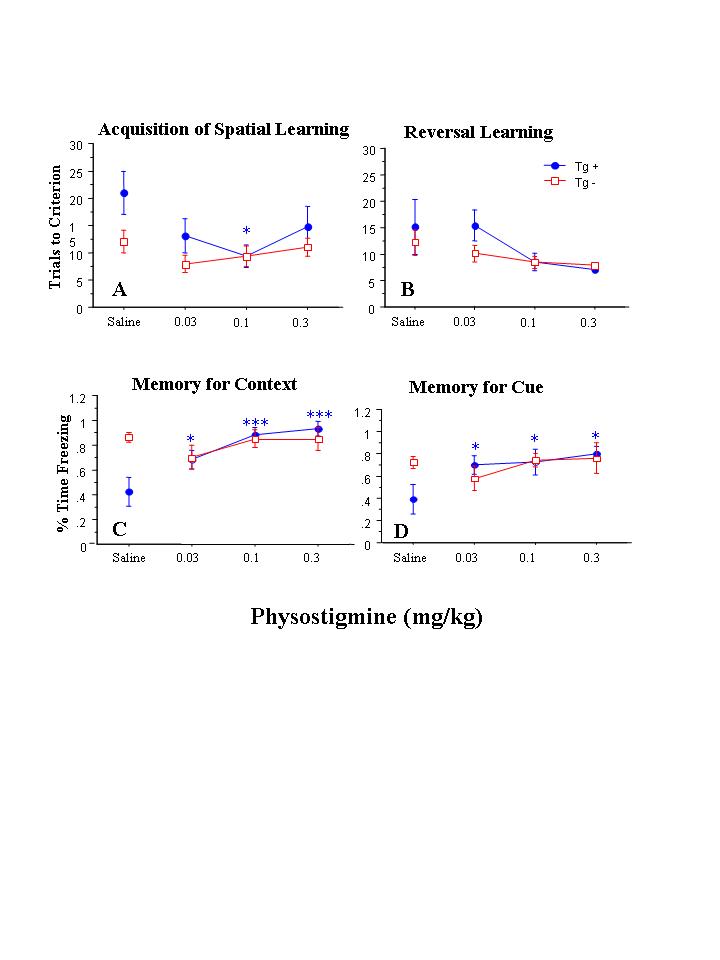
Behavioral performance in animals administered physostigmine (n = 5 Tg(+) and 6 Tg(-)) or saline (n = 5 Tg(+) and 5 Tg(-)). The effect of dose/time on acquisition of the spatial learning paradigm (panel a) and reversal learning (panel b) was not significant, nor were there any significant interactions between dose/time x APP status for these measures (see text for statistics). However, there was a significant effect of dose/time, and a significant interaction between dose/time and APP status, on the capacity for contextual memory (panel c). The effect of dose/time, and the interaction between dose/time and APP status, on the measurement of cued memory was not significant (panel d). Significant post-hoc between-group differences in groups of Tg(+) or Tg(-) mice administered a particular drug dose versus saline are indicated as follows: * - p<.05, **- p<.01, *** p<.001.
Figure 2.
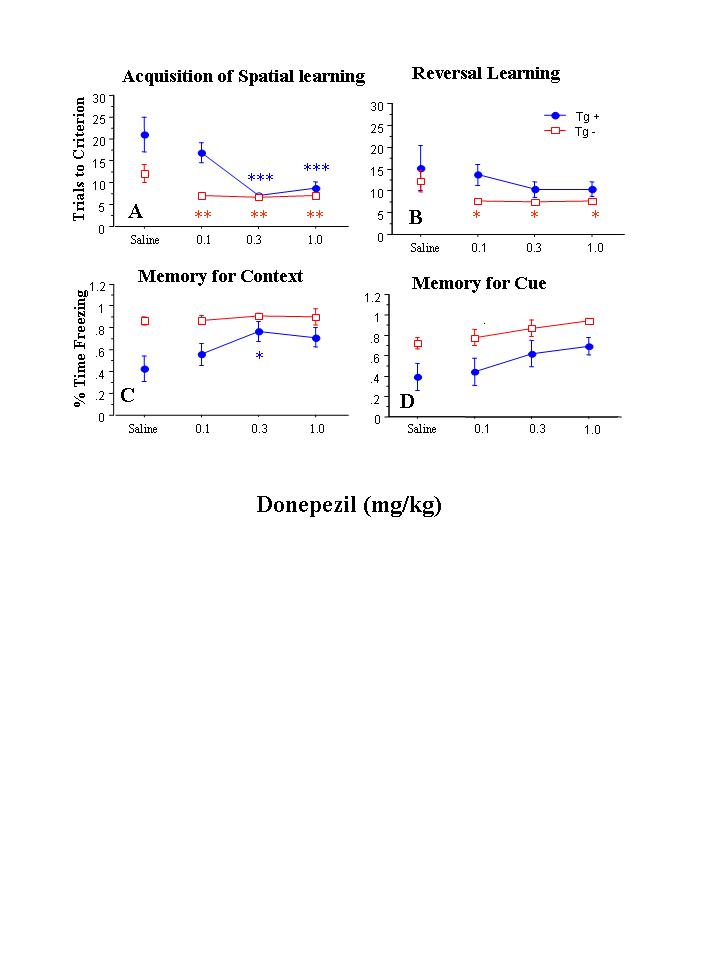
Behavioral performance in animals administered donepezil (n = 6 Tg(+) and 5 Tg(-)) or saline (n = 5 Tg(+) and 5 Tg(-)). The effect of dose/time, and the interaction between dose/time and APP status, was significant on acquisition of the spatial learning paradigm (panel A), but not on reversal learning (panel b) (see text for statistics). Similarly, the effect of dose/time on the measurement of contextual memory (panel c) and cued memory (panel d), as well as the interaction between dose/time and APP status on these measures was not significant. Significant post-hoc between-group differences in groups of Tg(+) or Tg(-) mice administered a particular drug dose versus saline are indicated as follows: * - p<.05, **- p<.01, *** p<.001.
Drug treatment tended to normalize the contextual memory deficit in Tg(+) animals so that they became more similar to Tg(-) animals (see Figures 1 and 2, panels c and d). Post-hoc testing showed that Tg(+) animals injected with physostigmine (p=.0007) or donepezil (p=.027) exhibited more freezing in response to context that saline-injected Tg(+) animals. Similarly, freezing was increased in response to cue in Tg(+) animals injected with physostigmine (p = 0.017), and a similar trend was observed in donepezil-injected Tg(+) animals (p=.064). Freezing was also increased in response to the altered context in Tg(+) animals injected with physostigmine (p = 0.049), but not donepezil.
Because all three doses of each drug were given consecutively to each drug group, the effects of dose and duration of drug administration could not be distinguished. The effects of dose/time for physostigmine and donepezil in the spatial reversal learning and fear conditioning paradigms are summarized below and in Figures 1 and 2, respectively.
3.1. Physostigmine
In physostigmine-injected animals, repeated measures ANOVA indicated that the effect of dose/time on acquisition of the spatial learning paradigm was not significant, nor was there a significant interaction between dose/time and hAPP status (see Figure 1, panel A). However, in post-hoc testing where each dose/time condition was compared separately to saline-injected animals that had the same APP status, acquisition was improved after the 0.1 mg/kg dose of physostigmine in Tg(+) animals. Also, in animals administered physostigmine, there was not a significant effect of dose/time on reversal learning (see Figure 1, panel B), nor was there a significant interaction between dose/time and hAPP status on reversal learning.
In physostigmine-injected animals, there was a significant effect of dose/time on the capacity for contextual memory (F=33.6, df=2,18, p<.0001) (see Figure 1, panel C), as well as a significant interaction between dose/time and hAPP status (F=9.90, df=2,18, p=.001). Post-hoc testing revealed that the three doses of physostigmine were associated with progressively larger improvements in contextual memory as compared to saline in Tg(+) animals (p<.05), but not in Tg(-) animals. The overall effect of physostigmine dose/time or the interaction effect between dose/time and hAPP status on the capacity for cued memory was not significant, although some improvement was noted in Tg(+) mice administered the three doses of physostigmine as compared to saline on post-hoc testing (see Figure 1, panel D). There was a significant effect of physostigmine dose/time on freezing during the alternate context condition (F=15.2, df=2,18, p=.0001), but the interaction between dose/time and hAPP status was not significant. Post-hoc between-group testing revealed that the 0.1 mg/kg dose of physostigmine resulted in an increase in freezing during the alternate context condition as compared to saline (p = 0.03) in Tg(-) animals only.
3.2. Donepezil
In donepezil-injected animals, repeated measures ANOVA indicated that there was a significant effect of dose/time on acquisition of the spatial learning paradigm (F=9.01, df=2,16, p=.002) (see Figure 2, panel A), as well as a significant interaction between dose/time and hAPP status on (F=5.44, df=2,16, p=.016). Post-hoc testing where each dose/time condition was compared separately to saline-injected animals that had the same APP status revealed that the two higher doses of donepezil were associated with improved acquisition of the spatial learning paradigm in Tg(+) mice (p<.001), while in Tg(-) mice, all three doses of donepezil were associated with improved acquisition of the spatial learning paradigm (p <.05). However, in animals administered donepezil, the effect of dose/time on reversal learning was not significant, nor was there a significant interaction between dose/time and hAPP status on reversal learning. Post-hoc testing, however, suggested that all three doses of donepezil as compared to saline improved reversal learning in Tg(-) animals (see Figure 2, panel B).
In animals administered donepezil, the overall effects of dose/time on the capacity for contextual memory or cued memory did not achieve significance (see Figure 2, panels C and D), nor was there a significant interaction between dose/time and hAPP status on the capacity for contextual memory or cued memory. However, post-hoc testing suggested that the 0.3 mg/kg dose of donepezil improved the capacity for contextual memory in Tg(+) animals. Also, the effect of dose/time on freezing during the alternate context condition was not significant, nor was there a significant interaction between dose and hAPP status on freezing during the alternate context condition.
3.7. ß - amyloid plaque analysis
ANOVA did not reveal a significant effect of drug condition on the total number of Aß-immunoreactive plaques counted in Tg (+) mice (F=0.53, df=2,13, p=.60) (see Figure 3). Also, there was no significant effect of drug condition on amyloid burden (i.e., the proportion of cortex or hippocampus occupied by Aß-immunoreactive plaques).
Figure 3.
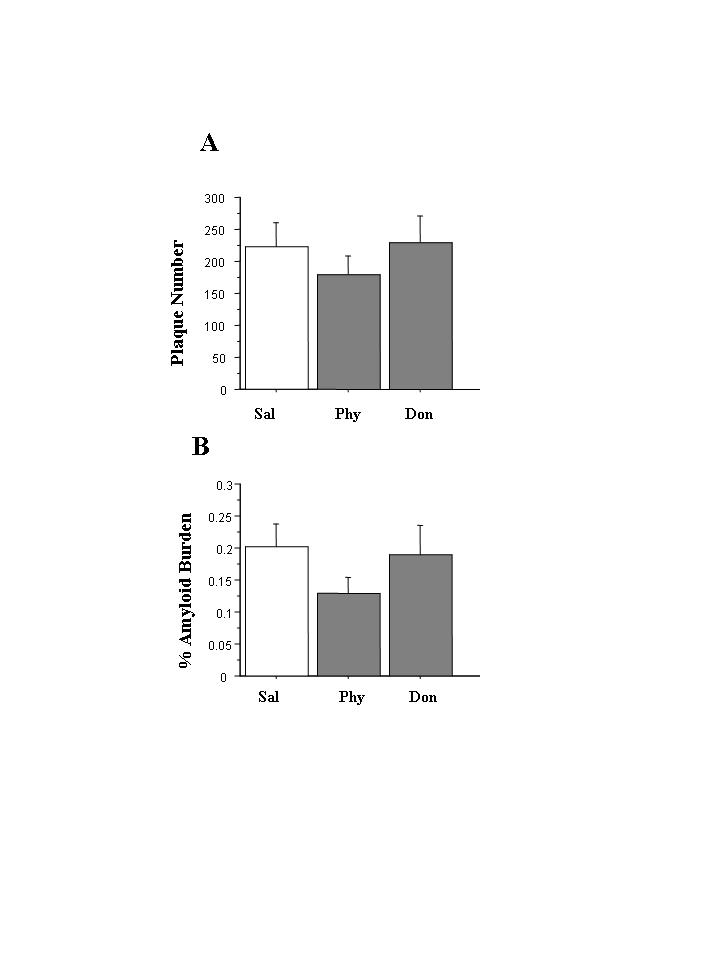
Aß plaque deposition in Tg(+) mice treated with physostigmine or donepezil (n= 5-7 per drug group). The effects of drug condition on the total number of Aß-amyloid plaques (panel a) and on the percentage of cortex and hippocampus occupied by Aß-immunoreactive plaques (panel b) were not significant.
4. Discussion
This study demonstrates that two acetylcholinesterase inhibitors, physostigmine and donepezil, had ameliorative effects on memory-related behavioral deficits in the Tg2576 mouse model of AD. As expected, deficits in spatial learning and in both contextual and cued memory associated with a fear conditioning paradigm were present in Tg(+) mice as compared to Tg(-) mice. In the first phase of the analysis of the data, we found that administration of the two acetylcholinesterase inhibitors improved deficits in contextual memory in Tg(+) mice so that their behaviors became more similar to Tg(-) mice. However, we unexpectedly found that donepezil administration improved acquisition of the spatial memory task in Tg(-) mice. Overall, our findings are consistent with the cognition-enhancing effects of acetylcholinesterase inhibitors in AD patients, but the findings with donepezil in Tg(-) mice suggest that the cognition-enhancing effects of such drugs may not be limited to the dementia associated with AD.
There were subtle but interesting differences in the effects of the two acetylcholinesterase inhibitors on memory-related behaviors in Tg(+) mice. In the first phase of the analysis, donepezil tended to be more effective than physostigmine in improving acquisition of the spatial learning paradigm, while physostigmine tended to be more effective than donepezil in improving reversal learning. In the fear conditioning paradigm, physostigmine also tended to be more effective than donepezil in ameliorating deficits in contextual, and to a lesser extent, cued memory. In the second phase of the analysis in which the effects of dose/time were investigated, dose/time effects were more evident in physostigmine-injected animals for contextual memory than other behavioral measures. However, in donepezil-injected animals, dose/time effects were more evident for acquisition of the spatial learning paradigm than other behavioral measures.
Little is known about the comparative efficacy of various acetylcholinesterase inhibitors to reverse specific groupings of dementia symptoms in AD patients. While physostigmine is not used clinically, donepezil has been shown to ameliorate impaired memory as well as a variety of other cognitive functions in patients with mild to moderate dementia of the Alzheimer type (Rogers, et al, 1998; Shigeta and Homma, 2001). Notably, the pharmacological features of physostigmine donepezil are slightly different, and could form a basis for differences in behavioral effects. For example, physostigmine inhibits both acetylcholinesterase and butylcholinesterase, while donepezil inhibits only acetylcholinesterase (Darvesh, et al, 2003). Moreover, physostigmine but not donepezil is a modulator at the nicotinic acetylcholine receptor (Storch, et al, 1995). These differences in the pharmacodynamic effects of the two drugs may underly the different behavioral effects of the two drugs in the two memory-related paradigms. However, the observed differences in the behavioral effects of the two drugs may also have been due to relative differences in the doses that were used, or may have been spurious because of the relatively small numbers of animals in each experimental group.
Despite the observed effects of the two acetylcholinesterase inhibitors on memory-related behaviors in Tg(+) mice, neither drug administered for a total of six weeks during the time that amyloid plaques usually appear had a significant effect on Aß deposition in the cortex or hippocampus. While six weeks of increasing doses of the two drugs may not have been adequate to test the capacity of such drugs to alter the deposition of amyloid plaques in Tg(+) mice, the data do suggest that the behavioral effects of physostigmine and donepezil in the Tg2576 mouse model of AD are not closely related to potential effects of the two drugs on APP processing of plaque deposition.
We had hoped to observe some effect of the two acetylcholinesterase inhibitors on the deposition of Ab plaques, and it is possible that higher doses and longer periods of drug administration at different ages may be needed to observe such effects. Acetycholinesterase inhibitors have been reported to affect APP processing (Pakaski and Kasa 2003, Mazzucchelli et al. 2003, Piazzi et al. 2003, Rees et al. 2003). Also, both physostigmine and donepezil have been reported to inhibit acetylcholinesterase-induced ß-amyloid aggregation (Bartolini et al. 2003), and physostigmine also may regulate APP metabolism (Beach et al. 2001).
The relationships between Aß plaque deposition, behavioral impairments and defects in cholinergic neurotransmission in animal models of AD remain poorly understood. Cholinergic dysfunction has been reported in the Tg2576 mouse (Boncristiano et al. 2002, Apelt et al. 2002, Fodero et al. 2002), and behavioral deficits appear to be better correlated with impairments in cholinergic neurotransmission than Aß deposition in these animals (Apelt et al. 2002, Fodero et al. 2002). However, it is also possible that soluble forms of Aß may be disrupting behavior in animal models of AD (Hsiao 2001, Westerman et al. 2002, Gong et al. 2003). Finally, fimbria-fornix lesions do not alter hippocampal Aß levels or the extent of Ab plaque accumulation in APP+PS1 mice (Liu et al. 2002). Our results are consistent with these prior findings in that the beneficial behavioral effects of physostigmine and donepezil were not correlated with measurable changes in Aß deposition.
Further research is necessary to investigate the long-term effects of acetylcholinesterase inhibitors and other putative treatments for AD on behavior and Aß deposition in mouse models of AD. In particular, studies of longer periods of drug administration, perhaps at different ages may help to elucidate the complex interaction between Aß deposition and cognition. Improving our understanding of this interaction may help to optimize the drug treatment of patients with AD.
5. Acknowledgements
This research was supported by PHS grant R-01 MH60883.
6. References
- Apelt J, Kumar A, Schliebs R. Impairment of cholinergic neurotransmission in adult and aged transgenic Tg2576 mouse brain expressing the Swedish mutation of human beta-amyloid precursor protein. Brain Res. 2002;953:17–30. doi: 10.1016/s0006-8993(02)03262-6. [DOI] [PubMed] [Google Scholar]
- Bardgett ME, Boeckman R, Krochmal D, Fernando H, Ahrens R, Csernansky JG. NMDA receptor blockade and hippocampal neuronal loss impair fear conditioning and position habit reversal in C57Bl/6 mice. Brain Res Bull. 2003;60:131–142. doi: 10.1016/s0361-9230(03)00023-6. [DOI] [PubMed] [Google Scholar]
- Bartolini M, Bertucci C, Vavrini V, Andrisano V. Beta-amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem Pharmacol. 2003;65:407–416. doi: 10.1016/s0006-2952(02)01514-9. [DOI] [PubMed] [Google Scholar]
- Beach TG, Kuo Y, Schwab C, Walker D, Roher A. Reduction of cortical amyloid levels in guinea pig brain after systemic administration of physostigmine. Neuroscience Letters. 2001;310:21–24. doi: 10.1016/s0304-3940(01)02076-6. [DOI] [PubMed] [Google Scholar]
- Boncristiano S, Calhoun ME, Kelly PH, Pfeifer M, Bondolfi L, Stalder M, Phinney AL, Abramowski D, Sturchler-Pierrat C, Enz A, Sommer B, Staufenbiel M, Jucker M. Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J Neurosci. 2002;22:3234–3243. doi: 10.1523/JNEUROSCI.22-08-03234.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nature Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- Csernansky J, Bargett M, Dong H, Humphrey W, Wang L. Hippocampal structure and the action of cholinomimetic drugs. Drug Dev Res. 2002;51:1–10. [Google Scholar]
- Cummings JL, Vinters HV, Cole GM, Khachaturian Alzheimer’s disease: Etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology. 1998;51:S2–S17. doi: 10.1212/wnl.51.1_suppl_1.s2. [DOI] [PubMed] [Google Scholar]
- Darvesh S, Walsh R, Kumar R, Caines A, Roberts S, Magee D, Rockwood K, Martin E. Inhibition of human cholinesterase by drugs used to treat Alzheimer disease. Alzheimer Disease & Associated Disorders. 2003;17:117–126. doi: 10.1097/00002093-200304000-00011. [DOI] [PubMed] [Google Scholar]
- Davis KL, Thal LJ, Gamzu ER, Davis CS, Woolson RF, Gracon SI, Drachman DA, Schneider LS, Whitehouse PJ, Hoover TM, The Tacrine Collaborative Study Group A double-blind placebo-controlled multicenter study of tacrine for Alzheimer’s disease. N Engl J Med. 1992;327:1253–1259. doi: 10.1056/NEJM199210293271801. [DOI] [PubMed] [Google Scholar]
- Dong H, Goico B, Martin M, Csernansky CA, Bertchume A, Csernansky JG. Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience. 2004;127:601–609. doi: 10.1016/j.neuroscience.2004.05.040. [DOI] [PubMed] [Google Scholar]
- Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J Neurol Neurosurg. Psychiatry. 1999;66:137–147. doi: 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodero LR, Saez-Valero J, McLean CA, Martins RN, Beyreuther K, Masters CL, Robertson TA, Small DH. Altered glycosylation of acetylcholinesterse in APP (SW) Tg2576 transgenic mice occurs prior to amyloid plaque deposition. J Neurochem. 2002;81:441–448. doi: 10.1046/j.1471-4159.2002.00902.x. [DOI] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: Presence of oligomeric Ab ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA. 2003;100:10417–22. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntern E, Bouras C, Hoff PR, Vallet PG. An improved thioflavine S method for staining neurofibrillary tangles ad senile plaques in Alzheimer’s disease. Experientia. 1992;48:8–10. doi: 10.1007/BF01923594. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aß elevation and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hsiao K. Learning and memory in transgenic mice modeling Alzheimer’s Disease. Learning & Memory. 2001;8:301–308. doi: 10.1101/lm.43701. [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Alvarez A, Perez CA, Moreno RD, Vicente M, Linker C, Casanueva OI, Soto C, Garrido J. Acetylcholinesterase accelerates assembly of amyloid-b-peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron. 1996;16:881–891. doi: 10.1016/s0896-6273(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Irizarry M, McNamara M, Fedorchak K, Hsiao K, Hyman B. APPsw transgenic mice develop age-related Ab deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropath Exper Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- Jellinger K. Morphology of Alzheimer’s disease and related disorders. Alzheimer’s disease, epidemiology, neurochemistry, and clinics. In: Maurer K, Riederer P, Beckman H, editors. Chapman and Hall. 1990. pp. 61–77. [Google Scholar]
- Liu L, Ikonen S, Tapiola T, Tanila H, Van Groen T. Fimbria-fornix lesion does not affect APP levels and amyloid deposition in the hippocampus of APP+PS1 double transgenic mice. Experimental Neurology. 2002;177:565–574. doi: 10.1006/exnr.2002.8015. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli M, Porrello E, Villetti G, Pietra C, Govoni S, Racchi M. Characterization of the effect of ganstigmine (CHF2819) on amyloid precursor protein metabolism in SH-SY5Y neuroblastoma cells. J Neural Transm. 2003;110:935–947. doi: 10.1007/s00702-003-0006-x. [DOI] [PubMed] [Google Scholar]
- Moghul S, Wilkinson D. Use of acetylcholinesterase inhibitors in Alzheimer’s disease. Expert Rev Neurotherapeutics. 2001;1:61–69. doi: 10.1586/14737175.1.1.61. [DOI] [PubMed] [Google Scholar]
- Mullan M. J Clin Psychiatry. 2000;61:307–315. doi: 10.4088/jcp.v61n0413. [DOI] [PubMed] [Google Scholar]
- Nilsberth C, Basun H, Eckman C, Lannfelt L, Younkin S. Plasma levels of Ab42 and Ab40 in Alzheimer patients during treatment with the acetylcholinesterase inhibitor tacrine. Dement Geriatr Cogn disord. 2002;14:156–160. doi: 10.1159/000063605. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992;258:304–307. doi: 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- Pakaski M, Kasa P. Role of acetycholinesterase inhibitors in the metabolism of amyloid precursor protein. Curr Drug Target CNS Neurol Disord. 2003;2:163–171. doi: 10.2174/1568007033482869. [DOI] [PubMed] [Google Scholar]
- Piazzi L, Rampa A, Bisi A, Gobbi S, Belluti F, Cavalli A, Bartolini M, Andrisano V, Valenti P, Recanatini M. 3-(4-[[Benzyl(methyl)amino]methyl]phenyl)-6,7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced beta-amyloid aggregation: A dual function lead for Alzheimer's disease therapy. J Med Chem. 2003;46(12):2279–2282. doi: 10.1021/jm0340602. [DOI] [PubMed] [Google Scholar]
- Price D. New perspectives on Alzheimer’s disease. Ann Rev Neurosci. 1986;9:489–512. doi: 10.1146/annurev.ne.09.030186.002421. [DOI] [PubMed] [Google Scholar]
- Rees T, Hammond PI, Soreq H, Younkin S, Brimijoin S. Acetylcholinesterase promotes beta-amyloid plaques in cerebral cortex. Neurobiology of Aging. 2003;24:777–787. doi: 10.1016/s0197-4580(02)00230-0. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Farlow MR, Doody RS, Mohs R, Friedhoff LT. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology. 1998;50:136–145. doi: 10.1212/wnl.50.1.136. [DOI] [PubMed] [Google Scholar]
- Shigeta M, Homma A. Donepezil for Alzheimer’s disease: pharmacodynamic, pharmacokinetic, and clinical profiles. CNS Drug Rev. 2001;7:353–368. doi: 10.1111/j.1527-3458.2001.tb00204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch A, Schrattenholz A, Cooper JC, Abdel Ghani EM, Gutbrod O, Weber KH, Reinhardt S, Lobron C, Hermsen B, Soskic V, et al. Physostigmine, galanthamine and codeine act as noncompetitive nicotinic agonists on clonal rat pheochromocytoma cells. Eur J Pharmacol. 1995;290:207–219. doi: 10.1016/0922-4106(95)00080-1. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94(24):13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariot PN, Solomon PR, Morris JC, Kershaw P, Lilienfeld S, Ding C, Galatamine USA-10 Study Group A 5-month, randomized placebo-controlled trial of galantamine in AD. Neurology. 2000;54:2269–2276. doi: 10.1212/wnl.54.12.2269. [DOI] [PubMed] [Google Scholar]
- West M, Gundersen H. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol. 1990;296:1–22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. The relationship between Ab and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]