

Abstract
Protein conformational disorders, which include certain types of retinitis pigmentosa, are a set of inherited human diseases in which mutant proteins are misfolded and often aggregated. Many opsin mutants associated with retinitis pigmentosa, the most common being P23H, are misfolded and retained within the cell. Here, we describe a pharmacological chaperone, 11-cis-7-ring retinal, that quantitatively induces the in vivo folding of P23H-opsin. The rescued protein forms pigment, acquires mature glycosylation, and is transported to the cell surface. Additionally, we determined the temperature stability of the rescued protein as well as the reactivity of the retinal-opsin Schiff base to hydroxylamine. Our study unveils novel properties of P23H-opsin and its interaction with the chromophore. These properties suggest that 11-cis-7-ring retinal may be a useful therapeutic agent for the rescue of P23H-opsin and the prevention of retinal degeneration.
Rhodopsin (Rh)1 is the visual pigment protein of the rod cell and belongs to a large family of transmembrane G protein-coupled receptor, which are involved in numerous physiological functions (1, 2). This superfamily of membrane glycoprotein receptors is characterized by seven transmembrane α-helices that are anchored within the lipid bilayer. In contrast to other G protein-coupled receptors, which respond to diffusible ligands, Rh (∼40,000 daltons) has a covalently bound reverse agonist, 11-cis-retinal, which yields a distinct UV-visible spectrum with a λmax of ∼500 nm (3). The crystal structure of Rh has been previously elucidated (4).
In recent years, more than 100 opsin mutants (apoprotein = opsin, opsin bound with chromophore = Rh) have been linked to various genetic forms of retinitis pigmentosa, the most common form of hereditary retinal degeneration. Retinitis pigmentosa leads to photoreceptor death and subsequent severe loss of peripheral and night vision (5, 6). These mutants account for nearly 50% of all the autosomal dominant retinitis pigmentosa cases with the most frequent mutation being P23H, which accounts for ∼10% of all cases (7). Although they display distinct biochemical features, the mutant phenotypes fall mainly into three basic classes (8-10). Class I mutants are expressed at nearly WT levels and form stable pigment with 11-cis-retinal in the dark. These mutations cluster at the C terminus of Rh and disrupt vectorial transport to the rod outer segment. Some mutations also inefficiently activate transducin (5, 11). Class II mutants do not bind 11-cis-retinal and are retained in the endoplasmic reticulum. Class III mutants, like P23H, form small amounts of pigment and mainly remain in the endoplasmic reticulum or form aggresomes (12, 13). These mutants are targeted for degradation by the ubiquitin proteasome system (13). The results obtained from the heterologous expression of the opsin mutants in mammalian cells correlate well with the findings using transgenic Rh animals (14). Other phenotypic properties acquired as a result of a mutation may include the destabilization of the structure formed within the rod outer segment by mutated Rh molecules (15) or the disruption of other biological processes unique to the rod outer segment.
Patients with the P23H mutation (Fig. 1A) usually have milder disease progression than those harboring other Rh mutations. Based on the crystal structure of Rh, Pro23 is located in the N-terminal tail within one of the β-strands that make up an integral part of the N-terminal plug (16). The plug keeps the chromophore in its proper position (3), and mutations within this region result in improper folding of opsin and poor binding of the chromophore (5).
Fig. 1.
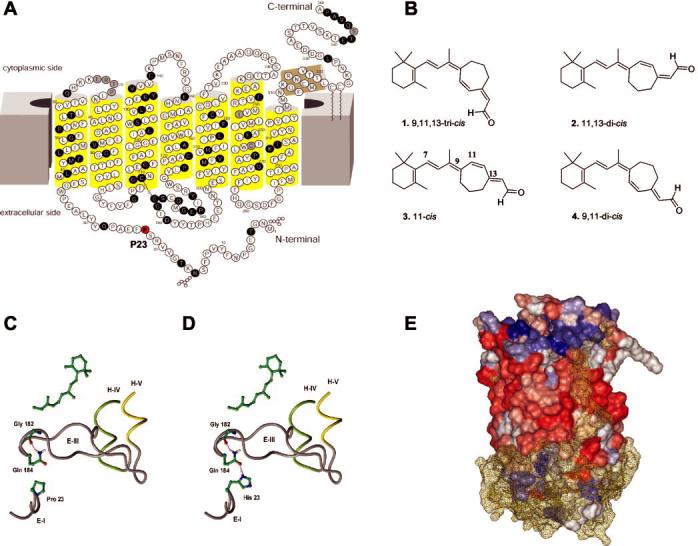
Models of Rh and structures of 7-locked-retinals. A, two-dimensional model of Rh (4). White letters on a black background indicate a mutation associated with retinitis pigmentosa. Pro23 is shown on a red background. B, chemical structures of the four 11-cis-7-ring retinal isomers. Carbon atom numbering is as accepted in retinoid nomenclature. C, WT Rh. Gln184 binds to Gly182 on the plug between helices IV and V. Pro23 is located on the first extracellular loop. D, P23H mutation of Rh. Gln184 bridges Gly182 and His23 and stabilizes this portion of the mutant protein. E, molecular dynamics simulation of Rh and nearby water cap. The same results were obtained for WT and the P23H mutant. The water cap was put on the extracellular part of Rh (together with that part buried in membrane in contact with polar heads of phospholipids). Only water and the extracellular part of Rh were allowed to move. During 100 ps of simulations, water found its way toward the cytoplasmic part of Rh going through the highly hydrophobic (red) surface part of the membrane of Rh. The hydrophilic surface is colored in blue, and neutral parts are white. In every simulation, water took the same route. During the whole course of simulation, the hydrogen bond involving His23 and Gln184 was stable. Water is shown as yellow mesh.
In previous studies, we demonstrated that P23H-opsin (opsin with proline at position 23 mutated to histidine) forms the pigment poorly, does not acquire the Golgi-related glycosylation, and is retained within the cell, collectively providing evidence that it is misfolded (9). In this paper, we show that P23H-opsin can be induced to properly fold by providing cells with a seven-membered ring variant of 11-cis-retinal during opsin biosynthesis. Furthermore, the remarkable affinity and selectivity of mutant opsin for 11-cis-7-ring retinal can be explained based on the crystal structure of Rh. These findings open the possibility for pharmacological intervention of this otherwise incurable retinal degenerative disease.
EXPERIMENTAL PROCEDURES
Synthesis of 11-cis-7-Ring Retinals—Synthesis of 11-cis-7-ring retinals was performed as described previously with some modifications (17-19). All of the reactions were performed in a dried nitrogen atmosphere unless otherwise specified. 2-Cycloheptenone was first converted into allyl acetate by N-bromosuccinimide bromination in CCl4 followed by treatment with KOAc in hexamethylphosphoramide. Purified 4-acetoxy-2-cycloheptenone (46% from 2-cycloheptenone) was subjected to a Horner-Emmons reaction with diethyl (2-cyanoethyl)phosphonate, which gave an isomeric mixture of two trans/cis (E/Z) cyanoacetates in a 2:1 ratio. The mixture was hydrolyzed with K2CO3 in MeOH:H2O (5:1), and then the hydroxy group of the resulting allylic alcohol was protected with tert-butyldimethylsilyl chloride in pyridine (80% from cycloheptenonyl acetate). The resulting cyano compound was reduced with diisobutylaluminium hydride in CH2Cl2 to an aldehyde and purified by flash chromatography on a silica gel (63%). β-Cyclocitral was reduced with NaBH4 to β-cyclogeraniol and then reacted with triphenylphosphine hydrobromide in MeOH over 3 days to afford β-cyclogeranyltriphenylphosphonium bromide after the removal of solvent and drying of the residue in vacuum. Wittig reaction of the silylated aldehyde with an excess of phosphonium salt in the presence of potassium tert-butoxide and a catalytic amount of 18-crown-6 in methylene chloride at ambient temperature afforded protected cyclic alcohol in 75% yield. The tert-butyldimethylsilyl protecting group was removed by treatment with tetrabutylammonium fluoride in dry THF, and the resulting alcohol was oxidized with MnO2 in CH2Cl2 to a mixture of two (E/Z) cyclic ketones (2:1 ratio) in 96% yield. This mixture was condensed with triethyl phosphonoacetate under Horner-Emmons conditions, followed by lithium aluminum hydride reduction of the resulting isomeric mixture of ethyl 7-ring retinoates and oxidation of retinols with MnO2 (86%) in CH2Cl2.
The following isomers were used: isomer 1 (Fig. 1B), 9,11,13-tri-cis-7-ring retinal: 1.04 (s, 6H, 2xCH3-1), 1.45–1.50 (m, 2H, CH2-2), 1.59–1.67 (m, 2H, CH2-3), 1.74 (s, 3H, CH3-5), 1.90 (m, 2H, J 6.74 Hz, CH2-22), 1.97 (s, 3H, CH3-9), 2.03 (t, 2H, CH2-4), 2.46 (t, 2H, J 7.26 Hz CH2-20), 2.51 (t, 2H, J 6.74 Hz CH2-21), 5.78 (d, 1H, J 7.78 Hz, H-14), 6.20 (d, 1H, J 11.42 Hz, H-7), 6.54 (d, 1H, J 11.42 Hz, H-8), 6.95 (d, 1H, J 16.08 Hz, H-12), 7.08 (d, 1H, J 16.08 Hz, H-11), 10.11 (d, 1H, J 7.78 Hz, H-15); isomer 2 (Fig. 1B), 11,13-di-cis-7-ring retinal: 1.04 (s, 6H, 2xCH3-1), 1.47–1.50 (m, 2H, CH2-2), 1.60–1.67 (m, 2H, CH2-3), 1.73 (s, 3H, CH3-5), 1.86 (m, 2H, J 6.74 Hz, CH2-22), 2.00 (s, 3H, CH3-9), 2.04 (t, 2H, CH2-4), 2.45 (t, 2H, J 7.27 Hz CH2-20), 2.54 (t, 2H, J 6.74 Hz CH2-21), 5.79 (d, 1H, J 7.78 Hz, H-14), 6.32 (d, 1H, J 16.08 Hz, H-7), 6.52 (d, 1H, J 16.08 Hz, H-8), 7.02 (m, 2H, H-11, H-12), 10.12 (d, 1H, J 7.78 Hz, H-15); isomer 3 (Fig. 1B), 11-cis-7-ring retinal: 1.04 (s, 6H, 2xCH3-1), 1.44–1.52 (m, 2H, CH2-2), 1.57–1.67 (m, 2H, CH2-3), 1.74 (s, 3H, CH3-5), 1.86 (m, 2H, J 6.74 Hz, CH2-22), 2.00 (s, 3H, CH3-9), 2.04 (t, 2H, CH2-4), 2.58 (t, 2H, J 6.75 Hz CH2-20), 2.87 (t, 2H, J 6.75 Hz CH2-21), 5.93 (d, 1H, J 8.3 Hz, H-14), 6.22 (d, 1H, J 11.42 Hz, H-12), 6.32 (d, 1H, J 16.08 Hz, H-7), 6.52 (d, 1H, J 16.08 Hz, H-8), 6.91 (s, 1H, J 11.42 Hz, H-11), 10.03 (d, 1H, J 7.79 Hz, H-15); and isomer 4 (Fig. 1B), 9,11-di-cis-7-ring retinal: 1.04 (s, 6H, 2xCH3-1), 1.44 –1.52 (m, 2H, CH2-2), 1.57–1.67 (m, 2H, CH2-3), 1.74 (s, 3H, CH3-5), 1.86 (m, 2H, J 6.74 Hz, CH2-22), 2.00 (s, 3H, CH3-9), 2.04 (t, 2H, CH2-4), 2.58 (t, 2H, J 6.75 Hz CH2-20), 2.87 (t, 2H, J 6.75 Hz CH2-21), 5.93 (d, 1H, J 8.3 Hz, H-14), 6.17 (d, 1H, J 11.41 Hz, H-12), 6.21 (d, 1H, J 16.08 Hz, H-7), 6.53 (d, 1H, J 16.08 Hz, H-8), 6.98 (s, 1H, J 11.41 Hz, H-11), 10.03 (d, 1H, J 7.78 Hz, H-15).
Cell Lines and Growth Conditions—For the construction and selection of tetracycline-inducible opsin cell lines, we used the Invitrogen T-REx™ system. Briefly, wild type opsin and the P23H mutant were excised as EcoRI-NotI fragments from pMT4 and then cloned into the EcoRI-NotI site within the polylinker of pcDNA4. Using opsin-specific forward and reverse primers, the entire gene was sequenced for verification. These plasmids were then separately transfected by calcium-phosphate precipitation into T-REx™-293 cells that already stably expressed the tetracycline repressor; these cells were routinely grown under blasticidin selection. After transfection, zeocin was added to the culture medium, and surviving colonies of cells were isolated and subsequently expanded into larger 6-well plates. Each of these clones was exposed to tetracycline, and 48 h after induction, the cells were harvested and solubilized with 1% DM. Separately, uninduced cells were also solubilized. The samples were run on SDS-PAGE and immunoblotted with the monoclonal antibody 1D4. Cell lines were chosen on the basis of production of the least amount of opsin (i.e. nondetectable opsin levels on immunoblots) in the absence of tetracycline and moderate amounts of opsin in the presence of the drug. HEK293 cells were grown in Dulbecco's modified Eagle's medium containing high glucose (Invitrogen) at 37 °C in the presence of 5.0% CO2. In all of the experiments, the cells were harvested after 48 h of induction with tetracycline (1 μg/ml).
Cell Culture and Regeneration—The cells were washed three times with PBS, harvested, and incubated with different analogs of 11-cis-retinal (50 μM) for 45 min at 4 °C. The cells were then lysed with 1.0% n-dodecyl-β-maltoside (DM) (Anatrace) in the presence of protease inhibitors (complete protease inhibitor mixture tablets; Roche Molecular Biochemicals) and centrifuged at 36,000 rpm in a Beckman ultracentrifuge for 30 min at 4 °C. The supernatant was incubated with 1D4-coupled CNBr-activated Sepharose 4B (Pharmacia Corp.) beads overnight. The beads were then washed three times with PBS containing 0.1% DM. Bound pigments were eluted off the beads using 0.1 mM synthetic peptide (last 9 amino acid residues of the C terminus of Rh) in 0.1% DM. In experiments when retinoids (50 μM) were added during biosynthesis, a Me2SO solution of retinoids (10 μl of 100 mM) was added directly to the cell culture medium after 2 and 24 h of induction. The cells were harvested at 48 h and lysed with 1.0% DM. Pigment was purified by immunoaffinity chromatography. The UV-visible spectra of the eluted pigment samples were then recorded on a Perkin Elmer Lambda 800 UV-visible spectrophotometer in the range of 250–650 nm.
SDS Gel Electrophoresis and Immunoblotting—The samples were loaded on 10% SDS-polyacrylamide gels (20) and transferred to Immobilon-NC membrane (Millipore) according to established protocols. The membranes were blocked with blocking buffer (Licor) for 1 h and incubated at room temperature with 1D4 antibody (1:1000) for 1 h. The membranes were then washed with PBS containing 0.1% Tween 20 and then incubated for 1 h at room temperature with IRDye800TM-conjugated affinity purified goat anti-mouse IgG (Licor), diluted 1:5000. The membranes were then washed with PBS containing Tween 20 and scanned using an Odyssey Infrared imager (Licor).
Glycosylation Status—Purified 7-Rh and 7-P23H-Rh (Rh and P23H-Rh regenerated with 11-cis-7-ring retinal, respectively) were treated with N-glycanase according to the manufacturer's recommendations (Glyko). The samples were incubated at 37 °C for 16 h and then loaded onto a 10% polyacrylamide gel. The immunoblots were developed as described above.
Photosensitivity Experiments—Whole cells expressing WT and P23Hopsin treated with 11-cis-7-ring retinals were harvested and exposed for 20 min to light emitted from a Fiber-Lite, MI 150, High Intensity Illuminator (Dolan-Jenner). The cells were then washed and then lysed with 1.0% DM as described above. In a separate experiment, immunoaffinity purified 7-Rh and 7-P23H-Rh samples were exposed to light from Fiber-Lite, and UV-visible spectra were recorded at different times.
Hydroxylamine Sensitivity—Whole cells expressing WT and P23Hopsin treated with 11-cis-7-ring retinals were harvested and treated with 500 mM neutral solution of hydroxylamine for 45 min. The cells were thoroughly washed with PBS to remove all traces of hydroxylamine. Rh was then purified, and spectra were taken. In a separate experiment, purified 7-Rh and 7-P23H-Rh were treated with 20 mM neutral hydroxylamine, and UV-visible spectra were recorded at different times.
Thermostability of Pigments and Acid Hydrolysis—Purified 7-Rh and 7-P23H-Rh were incubated at 37 °C, and the spectra were recorded at indicated times. In acid treatment experiments, purified 7-Rh and 7-P23H-Rh were treated with 100 mM sulfuric acid, and spectra were recorded.
HPLC Analysis of Retinoids—A 450-μl aliquot of either purified 7-Rh, purified 7-P23H-Rh, or cell medium were treated with 50 μl of 10% SDS and 100 μl of 1 M NH2OH (freshly prepared, pH 7.5). The mixture was kept at room temperature for 30 min, and then 400 μl of MeOH and 400 μl of hexane were added. The mixture was shaken on a vortex for 5 min and centrifuged at 14,000 rpm to separate layers. The hexane layer was collected. Another 400 μl of hexane was added to the aqueous layer, and extraction was repeated. Combined hexane layers were dried down and redissolved in 120 μl of hexane, and 100-μl fractions were analyzed by normal phase HPLC as previously described (21).
Immunocytochemistry—HEK293 cells expressing WT Rh or P23Hopsin under a tetracycline-inducible promoter were cultured in Dulbecco's modified Eagle's medium (Invitrogen). The cells were attached to glass bottom microwell dishes (MatTek Corp.) with Cell-Tak (Becton Dickinson Labware). Expression of opsin or P23H-opsin was induced by the addition of 1 μg/ml tetracycline as recommended by the manufacturer's protocol (Invitrogen). The cells were treated with 50 μM 11-cis-7-ring retinal 2 h after induction. The cells were harvested after 24 h and fixed with 4% paraformaldehyde (Fisher) in PBS (136 mM NaCl, 11.4 mM sodium phosphate, pH 7.4) for 10 min and washed by PBS. To block nonspecific labeling, the cells were incubated in 1.5% normal goat serum (Vector Lab., Inc.) in PBST (136 mM NaCl, 11.4 mM sodium phosphate, 0.1% Triton X-100, pH 7.4) for 15 min at room temperature. The cells were incubated overnight at 4 °C in purified 1D4 antibody diluted with PBST. The sections were rinsed in PBST and incubated with indocarbocyanine (Cy3)-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Lab., Inc.) and Hoechst 33342 dye (Molecular Probes). The cells were rinsed in PBST and mounted in 50 μl of 2% 1,4-diazabicyclo-[2.2.2]octane (Sigma) in 90% glycerol to retard photobleaching. For confocal imaging, the cells were analyzed on a Zeiss LSM510 laser scanning microscope (Carl Zeiss, Inc.).
Protein Simulation and Modeling—Coordinates for Rh were taken from the Protein Data Bank (1HZX) (22). The addition of hydrogen atoms and all of the optimizations were done in Insight II (InsightII release 2000, Accelrys, Inc., San Diego, CA). Crystallographic water was removed, and water molecules were introduced based on the accessible space in the extracellular region. No minimization was performed before water was added.
A water layer (5 Å thick) was used to coat the extracellular part of Rh as well as residues in contact with polar phospholipids heads. All of the water molecules were allowed to move freely, as was the extracellular half of Rh, which contains P23H and retinal. Because no water cap was put on the cytoplasmic part of Rh, this part of the molecule was frozen to prevent degradation of the model.
RESULTS
Rationale—The chromophore of Rh, 11-cis-retinal, plays a central role in the photoactivation process (28) and is also important for the stabilization of the receptor. For example, Rh is stable for months in mild detergents, although opsin precipitates in a few hours (29). The chromophore may also induce a proper folding of Rh mutants (12, 30). The P23H mutation may destabilize the native chromophore-accepting conformation of opsin, leading to aberrant folding and subsequent aggregation (13). However, an analog of 11-cis-retinal may facilitate native-like folding of P23H-opsin and therefore play the role of pharmacological chaperone. Recently, we demonstrated that WT opsin regenerated with 11-cis-7-ring retinal (7-Rh), containing a chromophore with a seven-membered ring to prevent isomerization around the C11=C12 double bond (Fig. 1B), is very stable in vivo and in vitro (31). In contrast, bleaching of the WT opsin regenerated with 11-cis-6-ring retinal (6-Rh), which contains a more rigid chromophore, produces multiple isomers with modest changes in protein conformations (28). These results reveal that 11-cis-7-ring retinals, particularly isomer 3 (Fig. 1B), are easily accepted into the binding site of opsin and provide additional contact sites by virtue of the seven-membered ring and the 9-methyl group with the protein moiety (31). Because of its intrinsic flexibility, this analog could potentially influence/affect protein folding during biosynthesis, which is particularly important for those mutant proteins that fold less efficiently. Clearly, the binding pocket of WT opsin, and possibly that of the P23H mutant as well, adopt specific conformations that allow it to bind preferentially certain retinoids (32). This observation provided the impetus to pursue the experiments described below.
11-cis-7-Ring Retinal Chaperones Folding of P23H-opsin—Rh is regenerated with 11-cis-7-ring retinals in the harvested membranes (Fig. 2B), whereas only a small amount of pigment was formed when membranes containing P23H-opsin were treated with 11-cis-retinal (9), 11-cis-6-ring retinal, or 11-cis-7-ring retinal after the cells were harvested (Fig. 2B). If the mutant proteins are structurally unstable, we reasoned that addition of retinal during the course of protein biosynthesis might induce a more native-like folding of the protein and its subsequent stabilization. Our first attempts utilized 11-cis-locked versions of 11-cis-retinal in stable cell lines expressing WT and P23H-opsin. The UV-visible spectra of the immunoaffinity purified samples from 11-cis-6- or 11-cis-7-ring retinals provided to the cells during biosynthesis showed that both retinals recombined with WT opsin (Fig. 2C). Supporting our prediction, P23H-opsin formed significant amounts of pigment when 11-cis-7-ring retinal was added during biosynthesis (Fig. 2D). The λmax of the pigment was ∼490 ± 3 nm, unlike the WT protein (λmax = ∼500 ± 3 nm) (Fig. 2, C and D). The UV-visible spectra with a λmax of ∼440 nm of acid-denatured purified 7-P23H-Rh (Fig. 2D, inset) provided evidence that the chromophore forms a Schiff base with rescued P23H-opsin. Importantly, no significant pigment was seen when 11-cis-6-ring retinal, which is photoisomerable along C9- and C13-C=C double bonds (28), was added to P23H-opsin-expressing cells (Fig. 2D). Furthermore, 11-cis-9-demethyl-7-ring retinal was also ineffective in pigment formation with P23H-opsin (data not shown). These experiments illustrate unique specificity of the binding interaction between 11-cis-7-ring retinal and P23H-opsin.
Fig. 2.
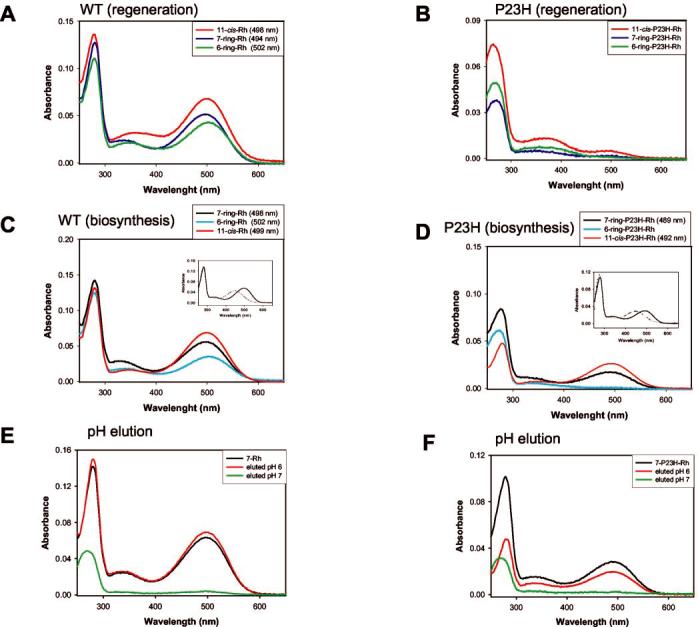
Pigment formation when the chromophores were added to harvested cell membranes or provided during biosynthesis. A, regeneration of WT Rh in cell membranes with 11-cis-retinal, 11-cis-6-ring retinal, and 11-cis-7-ring retinal. B, lack of regeneration of P23H-Rh in cell membranes with 11-cis-retinal, 11-cis-6-ring retinal, and 11-cis-7-ring retinal. C, regeneration of WT Rh with 11-cis-6-ring retinal, 11-cis-7-ring retinal, and 11-cis-retinal when retinals were added during biosynthesis. D, regeneration of P23H-Rh with 11-cis-7-ring retinal and 11-cis-retinal, but not with 11-cis-6-ring retinal, when retinals were added during biosynthesis. Insets in C and D, acid denaturation of purified 7-Rh and 11-cis-7-ring-P23H-Rh, respectively. E, elution of 7-Rh at pH 6.0 and pH 7.0. F, elution of 7-P23H-Rh at pH 6.0 and 7.0.
Immunoblots showed that the rescued P23H-opsin had a high molecular weight and a Golgi-associated glycosylation pattern (Fig. 3B, lane 5) similar to the WT protein (Fig. 3A), suggesting that the protein is sufficiently folded to proceed along the secretory pathway. This is in striking contrast to P23H-opsin that was not treated with 11-cis-7-ring retinal during P23H-opsin biosynthesis (Fig. 3B, lanes 1–4). Purified 7-Rh and 7-P23H-Rh were efficiently deglycosylated by N-glycanase, and the mutant protein spontaneously aggregated, forming high molecular weight aggregates (Fig. 3, C and D).
Fig. 3.
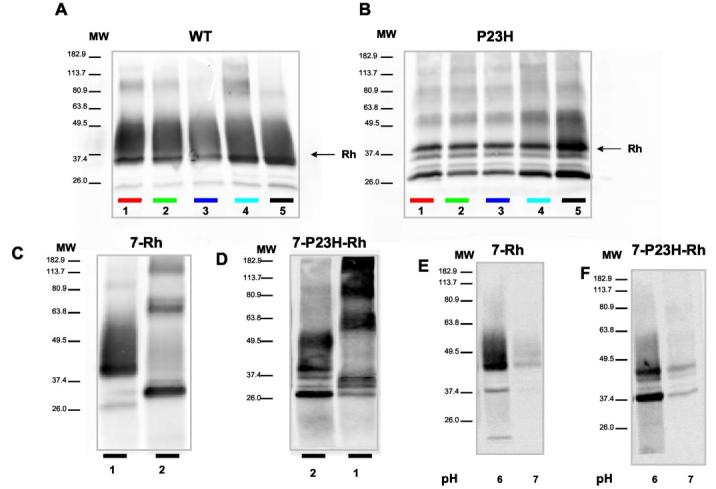
Rescued 7-P23H-Rh acquires mature glycosylation. A, WT Rh acquires mature glycosylation with all of the analogs, both in the case of regeneration in cell membranes and when retinals were added during biosynthesis.Lanes 1–3, regeneration in membranes with 11-cis-retinal, 11-cis-6-ring retinal, and 11-cis-7-ring retinal, respectively; lanes 4 and 5, 11-cis-6-ring retinal or 11-cis-7-ring retinal added during biosynthesis. B, 7-P23H-Rh acquires Golgi-associated glycosylation only with 7-locked analogs and only when retinals were added during biosynthesis. Lanes 1–3, regeneration in membranes with 11-cis-retinal, 11-cis-6-ring retinal, and 11-cis-7-ring retinal, respectively; lanes 4 and 5, 11-cis-6-ring retinal or 11-cis-7-ring retinal added during biosynthesis. The usage of different retinoids is coded using bars, the colors of which correspond to those in Fig. 2. C and D, purified 7-Rh and 7-P23H-Rh treated with N-glycanase. E and F, immunoblotting of 7-Rh and 7-P23H-Rh eluted at pH 6.0 and 7.0. MW, molecular weight.
In control experiments, we tested 9-cis-retinal and 11-cis-retinal for proper folding of P23H-opsin. It should also be noted that 9-cis-retinal promoted transport of P23H-opsin to the cell surface as previously shown by another group of investigators (12). We observed that 11-cis-retinal also induced the in vivo folding and stabilization of P23H-opsin forming visual pigments (λ499 nm and λ492 nm , for Rh and P23H-Rh, respectively) (Fig. 2, C and D). Moreover, these pigments also acquired a mature state of glycosylation.2 11-cis-retinoids are unstable in vitro andin vivo and rapidly undergo “reverse isomerization” (33). Therefore, locked analogs were used throughout the rest of this study.
Properties and Stability of 7-P23H-Rh—When the mixture of the four 7-ring retinal isomers (Fig. 1B) was added to cells expressing opsin and P23H-opsin during biosynthesis, both WT opsin and P23H-opsin selectively bound only isomer 3 (Fig. 4, A and D). These observations further support the extraordinary specificity of WT and P23H-opsin for binding this 11-cis-retinal isomer (spectrum in Fig. 4D). Addition of 11-cis-retinal to cell membranes containing either WT or P23H-opsin that were already treated with the 11-cis-7-ring retinal during their biosynthesis did not lead to its substitution after photobleaching, as measured spectrophotometrically and by retinoid analysis of the bound chromophores (data not shown). 7-Rh, purified in the detergent, is stable to light (Fig. 5A), whereas purified 7-P23H-Rh undergoes bleaching under the same conditions (Fig. 5B). In contrast, both Rhs are stable in the membranes of HEK293 cells (Fig. 5, C and D). This result suggests that, in detergent, the chromophore in 7-P23H-Rh is more flexible, thus allowing photoisomerization. The mechanism by which 7-P23H-Rh undergoes bleaching in detergent most likely involves isomerization of isomer 3 to three other isomers and its subsequent hydrolysis (Fig. 4F). The membranes apparently stabilized 7-P23H-Rh, thus preventing isomerization of its bound chromophore (Figs. 4E and 5D). In tissue culture samples, small amounts of non-Rh-bound retinals eluted between 30 and 40 min and remained in equilibrium with the vast majority of retinols, governed by the redox potential of the cells (Fig. 4A, inset).
Fig. 4.

HPLC analysis of 11-cis-7-ring Rh and 11-cis-7-ring-P23H-Rh. Trace A, analysis of an isomeric composition of seven-membered locked retinals bound to Rh when added during biosynthesis. Only syn- (3′) and anti- (3″) oximes of 11-cis-7-ring retinal (isomer 3) were observed. Inset, HPLC analysis of retinoids in the cell culture after the experiment. Mainly an isomeric mixture of 7-ring retinols is present. Trace B, analysis of isomeric composition of 7-ring retinals when added during Rh biosynthesis. Rh was purified and bleached for 6 min in detergent. Trace C, analysis of the isomeric composition of seven-membered locked retinals when added during Rh biosynthesis. Rh was bleached for 20 min, and then 11-cis-retinal was added. Only 11-cis-7-ring isomer 3 is found. Trace D, same as trace A but for P23H. Inset, UV-visible spectrum of peak 3′, showing syn-oxime of 11-cis-7-ring retinal. Trace E, bleaching of 7-P23H-Rh in membranes. Trace F, isomeric chromophore composition after 7-P23H-Rh was purified and bleached for 20 min in detergent. Note that the bleaching of P23H-Rh in detergent, but not in membranes, produces all four isomers of the 7-ring- retinals. The absorbance was recorded at 325 nm.
FIG. 5.

Photosensitivity of 7-Rh and 7-P23H-Rh in detergent and in cell membrane. A, purified 7-Rh is stable to light in detergent. B, purified 7-P23H-Rh was bleached after 5 min of exposure to light in detergent. C and D, 7-Rh and 7-P23H-Rh in cell membranes were not bleached by light. Insets, changes in absorption at 494 nm as a function of time.
To further characterize the rescued protein, we eluted the affinity bound 7-Rh and 7-P23H-Rh under conditions that selectively released only the folded form of the protein (10 mm phosphate, pH 6.0) (34). The spectra of the eluates (Fig. 2, E and F) and the corresponding immunoblots (Fig. 3, E and F) indicated that ∼80% of 7-P23H-Rh folded to form pigment. Additionally, immunoblots of these eluates demonstrated that both contained a mature glycosylated band and unglycosylated bands; however, only the material eluted at pH 6.0 had the Golgi-specific glycosylation pattern.
We also probed the structure of the rescued protein by determining its sensitivity to neutral hydroxylamine. 7-P23H-Rh is resistant to hydroxylamine treatment when embedded in the lipid bilayer of HEK293 cells, suggesting that the rescued protein adopts a conformation that sequesters the Schiff base linkage within the protein and protects it from chemical modification (Fig. 6B). Unlike the purified WT protein (Fig. 6A), the mutant chromophore is accessible to hydroxylamine in detergent (0.1% DM), as evidenced by the formation of the retinaloximes (λmax = ∼360 nm). These data imply that the structure of 7-P23H-Rh is less tightly packed than that of the 7-Rh. Finally, Fig. 6 (C and D) shows the temperature stability of the purified 7-Rh and 7-P23H-Rh. There was no change in the amount of chromophore after incubation of 7-Rh at either 4 °C (not shown) or 37 °C (Fig. 6C). However, for the rescued 7-P23H-Rh, there is time-dependent thermal bleaching that is accelerated by increased temperature, with a half-life of ∼4 min at 37 °C (Fig. 6D) and 10 days at 4 °C in the detergent solution (data not shown).
Fig.6.

Stability of 7-Rh and 7-P23H-Rh. A, stability of membrane-bound and detergent-solubilized 11-cis-7-ring Rh to hydroxylamine. B, sensitivity of 7-P23H-Rh to hydroxylamine. The pigment is stable in the presence of cell membranes (up to 500 mm hydroxylamine) but loses the chromophore when exposed to 20 mm hydroxylamine in detergent. C, stability of 7-Rh to thermal bleaching. 7-Rh remains unchanged up to 2 h at 37 °C. D, thermal stability of 7-P23H-Rh. 7-P23H-Rh loses its chromophore after 10 min at 37 °C. Insets, changes in absorption at 494 nm as a function of time.
Localization of the Rescued 7-Locked-P23H-Rh—Immunofluorescence microscopy demonstrated that P23H-opsin is retained intracellularly, predominantly in a perinuclear distribution with punctuated fluorescence consistent with aggresomes (Fig. 7C) (9, 12, 13). In contrast, the rescued 7-P23H-Rh is predominantly found in a diffuse pattern with significantly greater staining at the cell surface (Fig. 7D) similar to the 7-Rh and WT opsin, which are found predominantly at the plasma membrane (Fig. 7, A and B). In summary, this result suggests that the rescued P23H-Rh was not only correctly glycosylated and formed a pigment but was also correctly routed to the cell membranes.
Fig. 7.
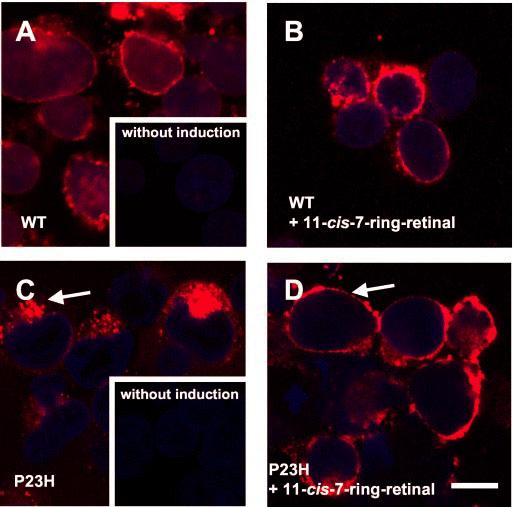
Subcellular localization of Rh and mutant P23H-opsin in HEK293 cells. Rh (red) was immunolabeled by anti-Rh monoclonal antibody 1D4. DNA (blue) was stained by Hoechst 33342. A, cells were fixed 24 h after inducing the expression of Rh. The most intense labeling (red) was observed on the surface of cells. Inset, the cells were cultured without inducing the expression of Rh. No staining (red) is observed. B, cells were treated with 11-cis-7-ring retinals and cultured for 24 h. Intense labeling (red) was observed on the surface of cells. C, cells were fixed 24 h after inducing the expression of P23H-opsin. Intense labeling (red) was observed in intracellular structure. Inset, the cells were cultured without inducing the expression of P23H-opsin. No staining (red) is observed. D, cells were treated with 11-cis-7-ring retinals and cultured for 24 h. Intense labeling (red) was observed on the surface of cells. The bar indicates 10 μm.
DISCUSSION
Protein Conformational Disorders—Dominantly inherited diseases can result from (a) haploinsufficiency of a gene, e.g. as for the melanocortin-4 receptor (35, 36); (b) constitutive activity (gain of function) in some opsin mutants (37) or lack of activity because of somatic mutation (38); and (c) loss of function because of mutant protein misfolding and aggregation (39). The diseases caused by misfolded proteins are known as protein conformational disorders and include Alzheimer's disease, Huntington's disease, Parkinson's disease, diabetes insipidus, cystic fibrosis, prion disease, emphysema, dominant cataracts, and oculopharyngeal muscular dystrophy (reviewed in Refs. 40 and 41). Gene delivery is one method of correcting haploinsufficiency. Constitutive activity has been successfully blocked by reverse agonist-like compounds (42, 43), and conformation and function can be rescued by small chemical agents (38). The medically most prevalent forms of dominant disorders are the protein conformational disorders. Although they are the most challenging to treat, some successes have been reported. Examples include the rescue of misfolded cystic fibrosis trans-membrane regulator (CFTR) variant F508Δ (44) and the Z-variant of α1-antitrypsin (45). In the instance of CFTR, the osmolytes glycerol and trimethylamino-oxide have been used (46 - 48). The protein acquires mature glycosylation and is transported to the cell surface where it pumps Cl− ions. The chemical chaperone 4-phenylbutyric acid mediated a marked increase in secretion of α1-antitrypsin (49). Recent studies, however, suggest that the rescued protein is less stable than the WT (47). Other approaches to inducing the folding of proteins have been developed. For example, mini-chaperones that prevent β-sheet formation prevent toxic aggregation of amyloid-β found in Alzheimer's disease and the PrP protein associated with prion disease (50).
P23H Rescue—We demonstrate that P23H-opsin can be induced to properly fold and be stabilized with a seven-membered locked ring version of 11-cis-retinal, the inverse agonist of opsin. Furthermore, our model system provides a quantitative means to study the folding of this membrane protein. The rescued 7-P23H-Rh contains a Schiff base linkage with Lys296, producing pigment with a λmax of 490 nm (Fig. 2D) that can be used as a quantitative measure of the amount of correctly folded protein. Using chromatographic conditions that selectively elute the folded form of the protein, 11-cis-7-ring retinal leads to the rescue of ∼80% of the P23H-opsin. We have also demonstrated that the rescued protein is not only folded but also proceeds along the secretory pathway. Like the WT, the rescued protein acquires the heterogeneous glycosylation associated with oligosaccharide processing within the Golgi apparatus. Clearly, the protein is of a sufficiently native-like conformation and is not sequestered by the cellular quality control apparatus, at least that which exists at the level of endoplasmic reticulum.
Furthermore, we show that the rescued protein is structurally different from the WT protein. The detergent-purified protein is light-sensitive, displays thermal instability, and is sensitive to neutral hydroxylamine. The bleaching of detergent-solubilized 7-P23H-Rh occurs because of isomerization of double bonds along the polyene chain of the chromophore producing a mixture of isomers. However, in the membranes of the HEK cells, 7-P23H-Rh is stable, and therefore, it is expected this pigment will also be stable in vivo.
Chemical and Structural Considerations of the Rescue—The mutant P23H protein is less stable because it is misfolded and is thus retained intracellularly. The rescue effect with 11-cis-7-ring retinal leads to increased amounts of the correctly folded mutant and is extraordinarily specific as shown by the lack of activity observed with several structural homologs (11-cis-9-demethyl-ring retinal and 11-cis-6-ring retinal). However, unlike the WT protein, purified 7-P23H-Rh appears to be in a less compact conformation, as evidenced by its sensitivity to neutral hydroxylamine in detergent. We have also observed that the rescued protein spontaneously releases the locked retinoid over time (Fig. 6D). Previous studies have demonstrated that 11-cis-7-ring retinal is inherently more flexible than 11-cis-6-ring retinal (31). Perhaps the rigidity of 11-cis-6-ring retinal does not allow the P23H-opsin binding pocket to accept this chromophore. Alternatively, it is possible that P23H-opsin binds 11-cis-6-ring retinal but that the resulting protein is less stable and maybe more susceptible to water-assisted hydrolysis of the Schiff base linkage, as it is in the case of 9-cis-retinal.
Our molecular modeling studies provide a possible explanation for the defect caused by P23H mutation. Two alterations appear to be present in P23H mutant: (a) The mutation disrupts ionic interactions of Pro23, Gln182, and Glu184 on the tip of the plug between helices 4 and 5 (Fig. 1, C and D). Mutation of Pro to His leads to formation of conjugated hydrogen bonding that exposes the chromophore-binding site. This interaction stiffens the plug and possibly opens the binding site of opsin, allowing water to hydrolyze the Schiff base between the retinal chromophore and γ-amino group of Lys296. This prediction is consistent with a hipsochromic shift in the absorption maximum of the chromophore and accessibility to hydrolysis in the presence or absence of hydroxylamine. (b) There is a hydro-philic channel in the interior of the protein that could accommodate water molecules (Fig. 1E). In the model, Rh was soaked with water on the external side, only including the part of the membranes that is charged and without constraints imposed on water molecules. During simulation, a route was established where water molecules drifted from one side to another (cytoplasmic). This route was along helix II and partly along helices I and III. The key residue facilitating this transport was Thr93 on helix II (close to Glu113). Collectively, these two effects may alter the electronic/dipole environment around the retinal, explaining the nearly 10-nm blue shift.
In summary, we have shown using a cell culture model system that the retinitis pigmentosa mutant P23H-opsin can be induced to properly fold by providing cells with a seven-membered ring variant of 11-cis-retinal during opsin biosyn-thesis. Furthermore, we explain this remarkable affinity and selectivity of mutant opsin for 11-cis-7-ring retinal on the basis of the crystal structure of Rh. These findings open the possibility for pharmacological rescue of the P23H mutant associated with this incurable retinal degenerative disease.
Acknowledgments
We thank Dr. Robert Molday for providing the 1D4 monoclonal antibody, Dr. M. Gelb for help during retinoid syntheses, Dr. Karen Smith for generating the cell lines, and Yunie Kim for help with the manuscript preparation. Dr. Kaushal especially thanks Sri Sathya Sai Baba for inspiration for this work.
Footnotes
This work was supported by a Career Development Award from the Foundation Fighting Blindness (to S. K.), funds from Research to Prevent Blindness (to S. K.) and the British Retinitis Pigmentosa Society (to S. K.), NEI, National Institutes of Health Grant EY01730 (to S. K. and K. P.), and funds from the Department of Ophthalmology Vision Foundation (to S. K.) and the Institute of Human Genetics (to S. K.). Computational tasks were partly done in the ICM computer center (University of Warsaw, Warsaw, Poland). This work was also supported by National Institutes of Health Grants EY09339 and EY13385 (to K. P.), a grant from Research to Prevent Blindness, Inc. (to the Department of Ophthalmology at the University of Washington), and grants from Foundation Fighting Blindness, Inc. and the E. K. Bishop Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: Rh, rhodopsin; DM, n-dodecyl-β-malto-side; GPCR, G protein-coupled receptor; WT, wild type; PBS, phosphate-buffered saline; HPLC, high pressure liquid chromatography; ICM, Interdisciplinary Center of Modeling.
S. M. Noorwez, R. Malhotra, and S. Kaushal, manuscript in preparation.
REFERENCES
- 1.Okada T, Palczewski K. Curr. Opin. Struct. Biol. 2001;11:420–426. doi: 10.1016/s0959-440x(00)00227-x. [DOI] [PubMed] [Google Scholar]
- 2.Ballesteros J, Palczewski K. CurR. Opin. Drug Discovery Dev. 2001;4:561–574. [PMC free article] [PubMed] [Google Scholar]
- 3.Filipek S, Stenkamp RE, Teller DC, Palczewski K. Annu. Rev. Physiol. 2003;65:851–879. doi: 10.1146/annurev.physiol.65.092101.142611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 5.Rattner A, Sun H, Nathans J. Annu. Rev. Genet. 1999;33:89–131. doi: 10.1146/annurev.genet.33.1.89. [DOI] [PubMed] [Google Scholar]
- 6.Dryja TP. Am. J. Ophthalmol. 2000;130:547–563. doi: 10.1016/s0002-9394(00)00737-6. [DOI] [PubMed] [Google Scholar]
- 7.Chapple JP, Grayson C, Hardcastle AJ, Saliba RS, van der Spuy J, Cheetham ME. Trends Mol. Med. 2001;7:414–421. doi: 10.1016/s1471-4914(01)02103-7. [DOI] [PubMed] [Google Scholar]
- 8.Sung CH, Davenport CM, Nathans J. J. Biol. Chem. 1993;268:26645–26649. [PubMed] [Google Scholar]
- 9.Kaushal S, Khorana HG. Biochemistry. 1994;33:6121–6128. doi: 10.1021/bi00186a011. [DOI] [PubMed] [Google Scholar]
- 10.Sung CH, Schneider BG, Agarwal N, Papermaster DS, Nathans J. Proc. Natl. Acad. Sci. U. S. A. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papermaster DS. Invest. Ophthalmol. Vis. Sci. 2002;43:1300–1309. [PubMed] [Google Scholar]
- 12.Saliba RS, Munro PM, Luthert PJ, Cheetham ME. J. Cell Sci. 2002;115:2907–2918. doi: 10.1242/jcs.115.14.2907. [DOI] [PubMed] [Google Scholar]
- 13.Illing ME, Rajan RS, Bence NF, Kopito RR. J. Biol. Chem. 2002;277:34150–34160. doi: 10.1074/jbc.M204955200. [DOI] [PubMed] [Google Scholar]
- 14.Frederick J, Bronson JD, Baehr W. Methods Enzymol. 2000;316:515–526. doi: 10.1016/s0076-6879(00)16746-1. [DOI] [PubMed] [Google Scholar]
- 15.Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. Nature. 2003;421:127–128. doi: 10.1038/421127a. [DOI] [PubMed] [Google Scholar]
- 16.Okada T, Ernst OP, Palczewski K, Hofmann KP. Trends Biochem. Sci. 2001;26:318–324. doi: 10.1016/s0968-0004(01)01799-6. [DOI] [PubMed] [Google Scholar]
- 17.Fujimoto Y, Xie R, Tully SE, Berova N, Nakanishi K. Chirality. 2002;14:340–346. doi: 10.1002/chir.10076. [DOI] [PubMed] [Google Scholar]
- 18.Akito H, Tanis SP, Adams M, Balogh-Nair V, Nakajima K. J. Am. Chem. Soc. 1980;102:6370–6372. [Google Scholar]
- 19.Caldwell CG, Derguini F, Bigge CF, Chen AH, Hu S, Sastry L, Nakanishi K. J. Org. Chem. 1993;58:3533–3537. [Google Scholar]
- 20.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Van Hooser JP, Liang Y, Maeda T, Kuksa V, Jang GF, He YG, Rieke F, Fong HK, Detwiler PB, Palczewski K. J. Biol. Chem. 2002;277:19173–19182. doi: 10.1074/jbc.M112384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teller DC, Okada T, Behnke CA, Palczewski K, Stenkamp RE. Biochemistry. 2001;40:7761–7772. doi: 10.1021/bi0155091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deleted in Proof.
- 24. Deleted in Proof.
- 25. Deleted in Proof.
- 26. Deleted in Proof.
- 27. Deleted in Proof.
- 28.Jang GF, Kuksa V, Filipek S, Bartl F, Ritter E, Gelb MH, Hofmann KP, Palczewski K. J. Biol. Chem. 2001;276:26148–26153. doi: 10.1074/jbc.M102212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okada T, Le Trong I, Fox BA, Behnke CA, Stenkamp RE, Palczewski K. J. Struct. Biol. 2000;130:73–80. doi: 10.1006/jsbi.1999.4209. [DOI] [PubMed] [Google Scholar]
- 30.Li T, Sandberg MA, Pawlyk BS, Rosner B, Hayes KC, Dryja TP, Berson EL. Proc. Natl. Acad. Sci. U. S. A. 1998;95:11933–11938. doi: 10.1073/pnas.95.20.11933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuksa V, Bartl F, Maeda T, Jang GF, Ritter E, Heck M, Van Hooser JP, Liang Y, Filipek S, Gelb MH, Hofmann KP, Palczewski K. J. Biol. Chem. 2002;277:42315–42324. doi: 10.1074/jbc.M206014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lou J, Tan Q, Karnaukhova E, Berova N, Nakanishi K, Crouch RK. Methods Enzymol. 2000;315:219–237. doi: 10.1016/s0076-6879(00)15846-x. [DOI] [PubMed] [Google Scholar]
- 33.McBee JK, Van Hooser JP, Jang GF, Palczewski K. J. Biol. Chem. 2001;276:48483–48493. doi: 10.1074/jbc.M105840200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ridge KD, Lu Z, Liu X, Khorana HG. Biochemistry. 1995;34:3261–3267. doi: 10.1021/bi00010a016. [DOI] [PubMed] [Google Scholar]
- 35.Vaisse C, Clement K, Guy-Grand B, Froguel P. Nat. Genet. 1998;20:113–114. doi: 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- 36.Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O'Rahilly S. Nat. Genet. 1998;20:111–112. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- 37.Rao VR, Cohen GB, Oprian DD. Nature. 1994;367:639–642. doi: 10.1038/367639a0. [DOI] [PubMed] [Google Scholar]
- 38.Foster BA, Coffey HA, Morin MJ, Rastinejad F. Science. 1999;286:2507–2510. doi: 10.1126/science.286.5449.2507. [DOI] [PubMed] [Google Scholar]
- 39.Muchowski PJ, Ning K, D'Souza-Schorey C, Fields S. Proc. Natl. Acad. Sci. U. S. A. 2002;99:727–732. doi: 10.1073/pnas.022628699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soto C. FEBS Lett. 2001;498:204–207. doi: 10.1016/s0014-5793(01)02486-3. [DOI] [PubMed] [Google Scholar]
- 41.Kopito RR, Ron D. Nat. Cell Biol. 2000;2:207–209. doi: 10.1038/35041139. [DOI] [PubMed] [Google Scholar]
- 42.Govardhan CP, Oprian DD. J. Biol. Chem. 1994;269:6524–6527. [PubMed] [Google Scholar]
- 43.Yang T, Snider BB, Oprian DD. Proc. Natl. Acad. Sci. U. S. A. 1997;94:13559–13564. doi: 10.1073/pnas.94.25.13559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seibert FS, Loo TW, Clarke DM, Riordan JR. J. Bioenerg. Biomembr. 1997;29:429–442. doi: 10.1023/a:1022478822214. [DOI] [PubMed] [Google Scholar]
- 45.Bathurst IC, Travis J, George PM, Carrell RW. FEBS Lett. 1984;177:179–183. doi: 10.1016/0014-5793(84)81279-x. [DOI] [PubMed] [Google Scholar]
- 46.Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR. J. Biol. Chem. 1996;271:635–638. doi: 10.1074/jbc.271.2.635. [DOI] [PubMed] [Google Scholar]
- 47.Sharma M, Benharouga M, Hu W, Lukacs GL. J. Biol. Chem. 2001;276:8942–8950. doi: 10.1074/jbc.M009172200. [DOI] [PubMed] [Google Scholar]
- 48.Bai C, Biwersi J, Verkman AS, Matthay MA. J. Pharmacol. Toxicol. Methods. 1998;40:39–45. doi: 10.1016/s1056-8719(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 49.Burrows JA, Willis LK, Perlmutter DH. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1796–1801. doi: 10.1073/pnas.97.4.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soto C, Kascsak RJ, Saborio GP, Aucouturier P, Wisniewski T, Prelli F, Kascsak R, Mendez E, Harris DA, Ironside J, Tagliavini F, Carp RI, Frangione B. Lancet. 2000;355:192–197. doi: 10.1016/s0140-6736(99)11419-3. [DOI] [PubMed] [Google Scholar]