

Abstract
The impact of muscarinic receptor stimulation was examined on apoptotic signaling induced by DNA damage, oxidative stress, and mitochondrial impairment. Exposure of human neuroblastoma SH-SY5Y cells to the DNA-damaging agent camptothecin increased p53 levels, activated caspase-3, and caused cell death. Pretreatment with oxotremorine-M, a selective agonist of muscarinic receptors that are expressed endogenously in these cells, did not affect the accumulation of p53 but greatly attenuated caspase-3 activation and protected from cell death to nearly the same extent as treatment with a general caspase inhibitor. Treatment with 50–200 μm H2O2 caused the activation of caspase-3 beginning after 2–3 h, followed by eventual cell death. Oxotremorine-M pretreatment protected cells from H2O2-inducedcaspase-3 activation and death, and this was equivalent to protection afforded by a caspase inhibitor. Muscarinic receptor stimulation also protected cells from caspase-3 activation induced by exposure to rotenone, a mitochondrial complex 1 inhibitor, but no protection was evident from staurosporine-induced caspase-3 activation. The mechanism of protection afforded by muscarinic receptor activation from camptothecin-induced apoptotic signaling involved blockade of mitochondrial cytochrome c release associated with a bolstering of mitochondrial bcl-2 levels and blockade of the translocation of Bax to mitochondria. Likely the most proximal of these events to muscarinic receptor activation, mitochondrial Bax accumulation, also was attenuated by oxotremorine-M treatment after treatment with H2O2 or rotenone. These results demonstrate that stimulation of muscarinic receptors provides substantial protection from DNA damage, oxidative stress, and mitochondrial impairment, insults that may be encountered by neurons in development, aging, or neurodegenerative diseases. These findings suggest that neurotransmitter-induced signaling bolsters survival mechanisms, and inadequate neurotransmission may exacerbate neuronal loss.
No decision is more important for an individual cell than that which determines whether it will survive or die. It is not surprising that recent research has found this biochemical decision-making process encompasses a regulatory web with a complexity matching the importance of the decision (1, 2). Thus, throughout life cells mobilize an active effort to repress programs that could lead to cell death. In many instances, continued survival is dependent upon the environment of the cell, because cell-to-cell contact and signals received from neighboring cells may provide the necessary impetus to bolster defenses against death programs. Eventually, through senescence, insults, and/or loss of adequate inputs, cells are eliminated, often to make room for replacements. In the central nervous system, however, replacement neurons are only available marginally if at all (3, 4), putting a greater burden on the survival mechanisms of mature neurons. Determining how neurons are able to survive manyfold longer than the average mammalian cell poses one of the great challenges of research in neurobiology (5, 6). This question is more than of academic interest because failure to survive, either during the developmental process, during aging, or upon exposure to potentially lethal insults, is the basis for innumerable neurodegenerative conditions.
Neurodegenerative diseases often arise as the result of increased burdens placed on the survival-promoting mechanisms of neurons (7). The accumulation of mutated proteins or toxic insults can challenge survival mechanisms, causing neurons to prematurely succumb, which in some instances of adult-onset neurodegenerative disorders follows decades of heightened challenge (8). For example, in Alzheimer’s disease (AD),1 it is apparent that neurons survive many years of heightened stress caused by mutations and/or elevated stressors before the catastrophic effects are evident (9). Because the establishment of synaptic contacts is a critical factor for neuronal survival during development (10, 11), synaptic activity also may be critical for bolstering cellular defenses from toxic insults that contribute to neurodegenerative diseases. In this regard, it is well known that activation of receptors by neurotrophins, such as nerve growth factor, has a profound influence on cell survival (12). However, much less is known about the survival-promoting potential of receptors for classical neurotransmitters, such as cholinergic muscarinic receptors.
The present investigation was undertaken to determine the extent to which activation of plasma membrane neurotransmitter receptors are capable of providing protection from widely encountered toxic conditions. Considering the substantial but still controversial evidence that loss of cholinergic signaling activity may be an early event in AD (13), activation of cholinergic muscarinic receptors was used as the model for neuro-transmitter input. Three toxic conditions were examined: DNA damage, oxidative stress, and impaired mitochondrial function. DNA damage is an important initiator of neuronal apoptosis, which is evident in conditions including ischemia, oxidative stress, several neurodegenerative diseases, and after exposure to chemotherapeutic agents, as recently reviewed (14). Experimentally, camptothecin, a topoisomerase 1 inhibitor, has been used to generate DNA damage in neuronal model systems. Camptothecin treatment induces p53-dependent neuronal death, which follows activation of the pro-apoptotic protein Bax (15–17). Oxidative stress appears to be one of the most prevalent causes of neuronal dysfunction and demise in neurodegenerative disorders and can cause DNA damage. This overproduction of reactive oxygen species or diminished anti-oxidant capacity, is evident in Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, aging, stroke, and many other conditions associated with impaired function and loss of neurons (18–22). In addition, oxidative stress is an early event in AD pathology, and the extent and distribution of neurodegeneration in AD parallels indices of oxidative damage (9, 23–25). Mitochondrial dysfunction also has been implicated in numerous neurodegenerative conditions (26, 27). Therefore, the mitochondrial complex I inhibitor rotenone (28) was used in this study because rotenone administration has been used to model the neuronal dysfunction of Parkinson’s disease (29).
The results show that muscarinic receptor stimulation provides remarkably effective protection from each of these three disparate insults. These findings support the notion that synaptic activity, through activation of neurotransmitter receptors, can provide substantial support of cellular survival mechanisms, suggesting that loss of such synaptic input increases vulnerability to insult-induced programmed cell death.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatments
Human neuroblastoma SH-SY5Y cells were grown in RPMI medium (Cellgro, Herndon, VA) supplemented with 10% horse serum (Invitrogen), 5% fetal clone II (Hyclone, Logan, UT), 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells were maintained at 37 °C in 95% air, 5% CO2. Experimental agents used include H2O2, 3-aminobenzamide (3AB), oxotremorine-M, camptothecin, nicotine, carbachol, mecamylamine, atropine, and rotenone from Sigma and boc-aspartyl(OMe)-fluoromethyl ketone (BAF) from Alexis Biochemicals (San Diego, CA).
Enzyme Assays
Caspase-3 activity was assessed by measuring cleavage of a fluorogenic substrate, as described previously (30). Lactate dehydrogenase release was detected using the cytotoxicity detection kit (Roche Molecular Biochemicals) according to the manufacturer’s protocol.
Immunoblotting
Cells were harvested, washed twice with phosphate-buffered saline, and lysed in lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 2 mm EDTA, 2 mm EGTA, 1 mm sodium orthovanadate, 100 μm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin, 1 nm okadaic acid, and 0.5% Nonidet P-40). The lysates were sonicated for 10 s on ice and centrifuged at 16,000 ×g for 15 min, and supernatants were collected. Protein concentrations were determined using the Bradford method (31) or the bicinchoninic (BCA) method (Pierce). Where indicated, nuclear fractions were prepared as described previously (32). For preparation of cytosolic and mitochondrial fractions, harvested cells were disrupted by nitrogen cavitation (33) followed by sucrose gradient ultracentrifugation (34), and immunoblots of tubulin and cytochrome oxidase were used as markers of cytosolic and mitochondrial fractions, respectively. Proteins were resolved in SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and incubated with primary antibodies followed by incubation with horseradish peroxidase-conjugated secondary antibodies. The primary antibodies used were: poly (ADP-ribose) polymerase (PARP), p21, cytochrome c (BD Pharmingen), p53, bcl-2, Bax, tubulin (Upstate Biotechnology, Lake Placid, NY), cytochrome oxidase (Molecular Probes, Eugene, OR). Immunoblots were developed with enhanced chemiluminescence (Amersham Biosciences) and were quantified by densitometry.
RESULTS
Oxotremorine-M Protects against Camptothecin-Induced Caspase-3 Activation and Cell Death
We first examined if muscarinic receptor activation is capable of providing protection from apoptosis after DNA damage. To do this, we used camptothecin, a topoisomerase 1 inhibitor, to generate DNA damage-induced apoptosis, which we recently characterized in SH-SY5Y cells (35). As reported, treatment with 1 μm camptothecin caused a large and relatively rapid increase in the activity of caspase-3 (Fig. 1A). Activation of muscarinic receptors with the selective agonist oxotremorine-M (300 μm; 30 min of pretreatment) attenuated by ~75% caspase-3 activation induced by camptothecin (above the basal level of activity). This protective effect of oxotremorine-M was completely blocked by the specific muscarinic receptor antagonist 1 μm atropine. Equivalent results were obtained when the proteolysis of PARP was used as an indicator of the degree of apoptotic signaling activation. PARP proteolysis increased time-dependently after camptothecin treatment, and oxotremorine-M pretreatment greatly attenuated this response (Fig. 1B). Muscarinic receptor stimulation did not block the initial response to camptothecin, as evidenced by the lack of effect of oxotremorine-M treatment on the accumulation of p53 that followed camptothecin treatment (Fig. 1C). Additionally, the transcriptional activity of p53 was not blocked by oxotremorine-M pretreatment, as increases in the level of p21waf1/cip1, which is regulated by p53, followed the accumulation of p53 equivalently in the absence or presence of oxotremorine-M (Fig. 1C). This indicates that muscarinic receptor stimulation provided protection at a site downstream from the initial toxic insult, in this case DNA damage and the subsequent accumulation and activation of p53. Examination of lactate dehydrogenase release into the medium as a measure of cell death revealed that oxotremorine-M greatly attenuated cell death (Fig. 1D). Oxotremorine-M pretreatment provided a degree of protection that was nearly as great as that afforded by the general caspase inhibitor BAF (Fig. 1D), an inhibitor previously reported to block the early, caspase-3-dependent phase of cell death after camptothecin treatment of primary cortical neurons (16). Furthermore, protection by oxotremorine-M was blocked by atropine. Thus, caspase-3 activation and cell death induced by the DNA-damaging agent camptothecin was substantially attenuated by stimulation of muscarinic receptors.
Fig. 1. Oxotremorine-M protects cells from camptothecin-induced caspase-3 activation and cell death.
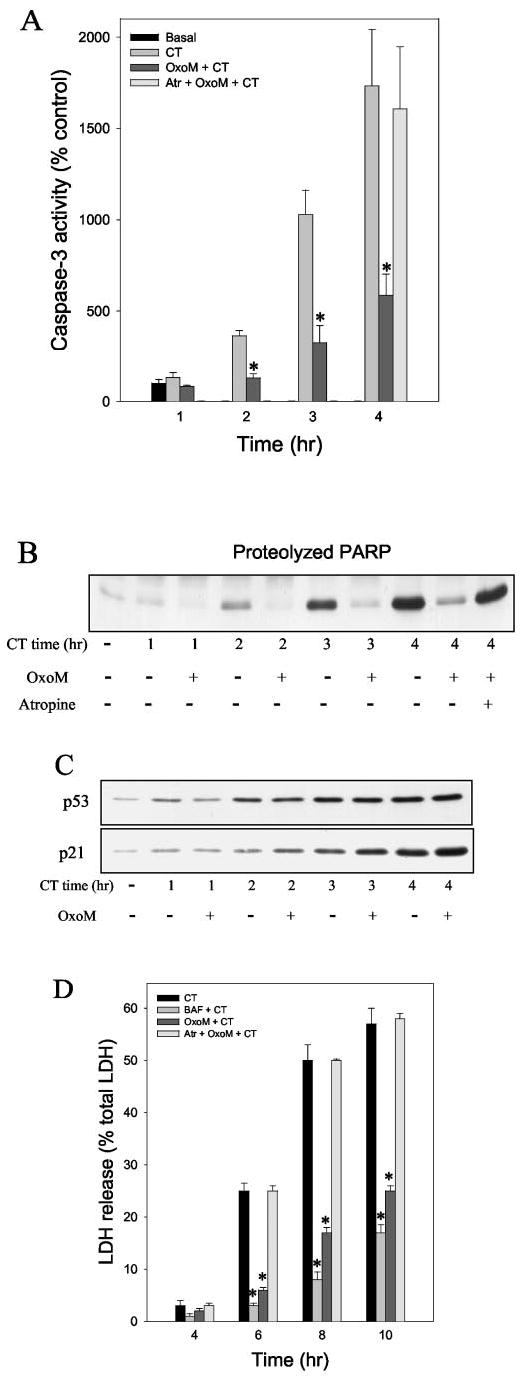
Caspase-3 activity (A) and PARP proteolysis (B) were measured in cells treated with 1 μm camptothecin (CT), 300 μm oxotremorine-M (OxoM) 30 min before camptothecin, and 1 μm atropine (Atr) 15 min before oxotremorine-M followed by camptothecin. C, p53 and p21 levels were measured in cells treated with camptothecin alone or with pretreatment with 300 μm oxotremorine-M for 30 min. D, lactate dehydrogenase (LDH) release into the media was measured 4 –10 h after treatment with camptothecin with or without pretreatments with 100 μm BAF or 300 μm oxotremorine-M and in cells treated with 1 μm atropine before oxotremorine-M treatment. Quantitative values are means ±S.E.; n =4 –5; *, p < 0.05.
Muscarinic Receptor Activation Protects against H2O2-induced Caspase-3 Activation and Cell Death
To test if muscarinic receptor stimulation provided protection from oxidative stress, it was first necessary to characterize the H2O2 treatment conditions needed to activate caspase-3 and induce cell death in SH-SY5Y cells. The primary mechanism of apoptotic signaling after oxidative stress ultimately involves activation of the effector enzyme, caspase-3 (1). Subsequently, activated caspase-3 cleaves susceptible substrates, such as PARP (36). Caspase-3 activity increased after 2.5 h of treatment with 100 μm H2O2 and began to plateau between 4.5 and 5.5 h aftertreatment (Fig. 2A). The activation of PARP (37), which may follow oxidative stress and DNA damage, can lead to depletion of cellular NAD+and ATP, an effect that can be avoided by the use of PARP inhibitors, such as 3AB (38, 39). Therefore, in addition to samples treated with 100 μm H2O2 alone, in parallel H2O2-treated samples PARP was inhibited by pretreatment with 100 μm 3AB. Similar rates and magnitudes of caspase-3 activation were obtained after treatment with 100 μm H2O2 with or without inhibition of PARP (Fig. 2A). The effects of H2O2-induced caspase-3 activation were reflected in measurements of the proteolysis of PARP, which is mediated by caspase-3 and results in the production of a stable 85-kDa product cleaved from the 116-kDa intact PARP. Treatment with 50–200 μm H2O2 caused a concentration-dependent increase in PARP proteolysis (Fig. 2B) and in caspase-3 activity (see below) that was similar in the presence and absence of 3AB. Cell death, assessed by measuring the release of lactate dehydrogenase into the media, was prominent between 8 and 24 h after treatment with 100 μm H2O2 in the absence or presence of 3AB (Fig. 2C). After 24 h, 60% of cells treated with H2O2 had died, and only slightly less (50%) had died after treatment with H2O2 plus 3AB.
Fig. 2. H2O2 treatment induces apoptosis.
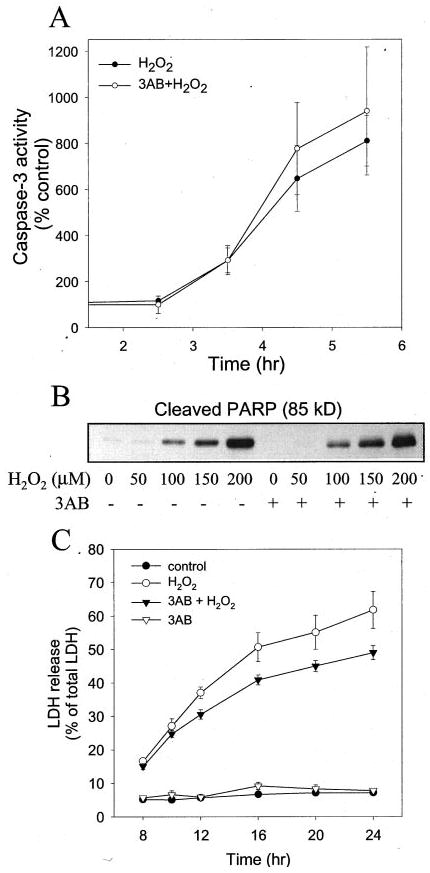
A, the time course of caspase-3 activation was measured in cells treated with 100 μm H2O2 with or without a 30-min pretreatment with 100 μm 3AB. (B) PARP proteolysis was measured in cells treated with 50 to 200 μm H2O2 for 5 h, with or without a 30 min pretreatment with 100 μm 3AB, by immunoblot analysis. The immunoblot shows the 85-kDa product of PARP cleaved by caspase-3. C, the time course of lactate dehydrogenase release was measured in cells treated with 100 μmH2O2 with or withouta 30-min pretreatment with 100 μm 3AB, in cells treated with only 3AB, and in control untreated cells. Quantitative data are means ±S.E.; n =3–4.
Using these established conditions, we examined if muscarinic receptor stimulation with oxotremorine-M modulated cellular responses to H2O2. Pretreatment with 300 μm oxotremorine-M (30 min) greatly reduced caspase-3 activity induced by 100, 150, and 200 μm H2O2 in the presence or absence of 3AB (Fig. 3A). Remarkably, nearly complete blockade of caspase-3 activation was attained with oxotremorine-M in cells incubated with 100 μm H2O2, and even with the highest concentration of H2O2 tested, 200 μm, caspase-3 activity was reduced more than 50% by treatment with oxotremorine-M. Measurements of PARP proteolysis showed that it mirrored changes in caspase-3 activity, as oxotremorine-M treatment also greatly attenuated the H2O2 concentration-dependent induction of PARP proteolysis (Fig. 3B). Thus, stimulation of muscarinic receptors provided substantial protection from the activation of caspase-3 caused by oxidative stress induced by H2O2.
Fig. 3. Oxotremorine-M treatment protects cells from H2O2-induced apoptosis.
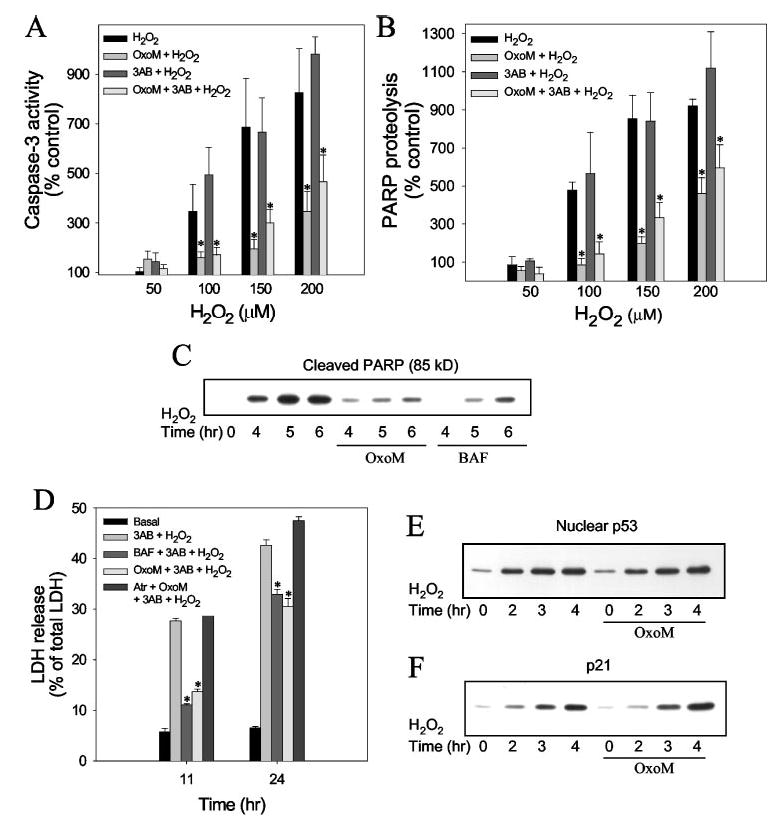
SH-SY5Y cells were pretreated with 100 μm 3-AB for 30min, 100 μm BAF for 60 min, or 300 μm oxotremorine-M (OxoM) (with or without a 15-min pretreatment with 1 μm atropine (Atr)) for 30 min wherem indicated followed by treatment with H2O2. Caspase-3 activity (A) and PARP cleavage (B) were measured 6 h after treatment with 50 –200 μM H2O2. C, proteolyzed PARP was measured 4, 5, and 6 h after treatment with 100 μmH2O2 in cells pretreated with 3AB and, where indicated, with oxotremorine-M or BAF. D, lactate dehydrogenase (LDH) release into the media was measured 11 and 24 h after treatment with 100 μm H2O2 and the indicated agents. Nuclear p53 levels (E) and p21 levels (F) were measured 2, 3, and 4 h after treatment with 100 μm H2O2 without or with a 30-min pretreatment with 300 μM oxotremorine-M. Quantitative values are the means ±S.E.; n = 3–4; *, p < 0.05.
To evaluate the significance of the protective effect of muscarinic receptor activation, the protection provided by oxotremorine-M was compared with that of a general caspase inhibitor, BAF. These experiments demonstrated that the substantial protection provided by treatment with oxotremorine-M against 100 μm H2O2-induced PARP proteolysis was similar to that afforded by 100 μm BAF (Fig. 3C). Furthermore, the effects of oxotremorine-M and BAF were compared on H2O2-inducedcell death, as assessed by measurements of lactate dehydrogenase release into the media at early (11 h) and late (24 h) times of cell death that were determined previously (Fig. 2C). Treatment with the general caspase inhibitor, 100 μm BAF, blocked cell death by 80% 11 h after treatment, indicating that early cell death induced by 100 μm H2O2 plus 3AB was predominantly caspase-dependent (Fig. 3D). However, after 24 h, pretreatment with BAF only reduced cell death by 30%, indicating that later cell death is mediated by a caspase-independent mechanism. A similar transition, with protection by BAF at shorter times after oxidative stress followed by loss of protection at 24 h, has been reported previously (40). In the same experiments in which BAF was tested, parallel samples were used to determine whether cell death was modulated by activation of muscarinic receptors. Once again, 300 μm oxotremorine-M (30 min preincubation) afforded the same extent of protection as did inhibition of caspases with BAF (Fig. 3D),reducing cell death by 80% 11 h after treatment with H2O2. The protective effect of oxotremorine-M was completely blocked by pretreatment with the muscarinic antagonist atropine (1 μm), confirming that oxotremorine-M action was mediated by a muscarinic receptor-dependent mechanism. There was a lower protective effect of oxotremorine-M 24 h after treatment with H2O2, which matched the diminished effect of BAF. Thus, treatment with H2O2 caused activation of caspase-3 and an early caspase-dependent cell death, and these effects were greatly attenuated by the caspase inhibitor BAF and to a similar extent by muscarinic receptor activation.
The complete signaling pathway mediating H2O2-induced caspase-3 activation is not known, but an early event in the response to H2O2 is stabilization, accumulation, and transcriptional activation of the tumor suppressor p53. Treatment with 100 μm H2O2 caused a large increase in the level of p53 that was evident 2 h after treatment, before activation of caspase-3 (Fig. 3E). There was a corresponding increase in the level of the p53-regulated protein p21, indicating the p53 was transcriptionally active (Fig. 3F). Pretreatment with 300 μm oxotremorine-M (30 min) did not reduce the H2O2-induced increases in p53 or p21 (Fig. 3, E and F), indicating that oxotremorine-M did not exert an antioxidant effect blocking initial responses to H2O2 but, rather, attenuated signaling leading to caspase-3 activation and cell death.
Stimulation of Muscarinic Receptors Protects against Rotenone-induced Caspase Activation but Not from Staurosporine
To examine if the protection afforded by muscarinic receptor stimulation also was evident in another condition that causes apoptosis, experiments were carried out using rotenone to inhibit the mitochondrial complex 1, which causes oxidative stress (41). In accordance with our recent report that characterized rotenone-induced caspase-3-mediated apoptosis in SH-SY5Y cells (42), there was a nearly 6-fold increase in caspase-3 activity after treatment with 5 μm rotenone (Fig. 4A). Pretreatment with 300 μm oxotremorine-M almost completely prevented rotenone-induced activation of caspase-3, whereas activation of nicotinic cholinergic receptors with nicotine provided no protection from rotenone. Treatment with carbachol, a non-specific cholinergic receptor agonist, prevented caspase-3 activation by rotenone to a similar extent as oxotremorine-M, and the protective effect of carbachol was prevented by the muscarinic receptor antagonist atropine but was unaffected by the nicotinic receptor antagonist mecamylamine. Thus, activation of muscarinic receptors greatly attenuated caspase-3 activation induced by rotenone as well as by camptothecin and H2O2.
Fig. 4. Muscarinic receptor stimulation attenuates caspase-3 activation induced by rotenone but not staurosporine.
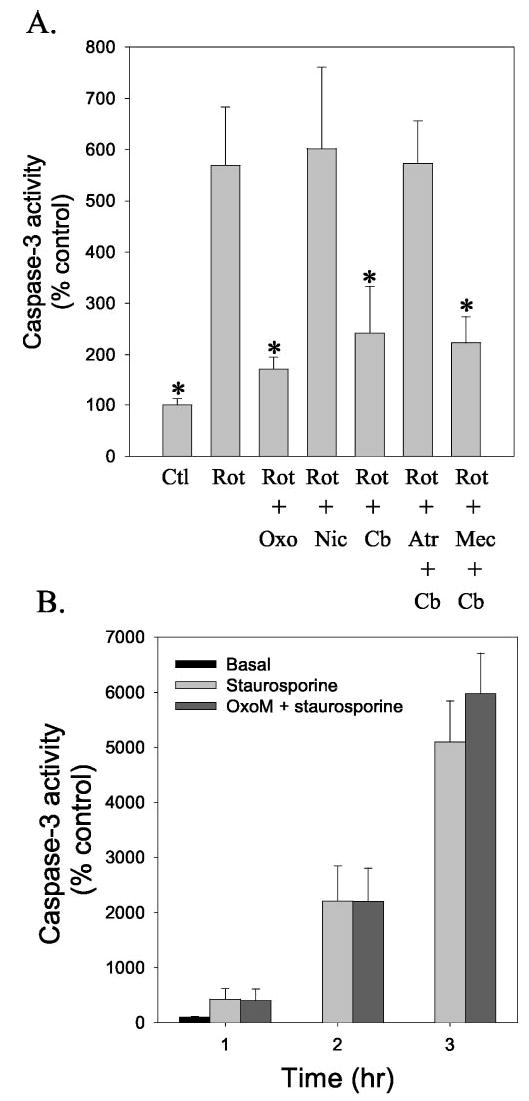
A, caspase-3 activity was measured in cells 16 h after treatment with 5 μm rotenone (Rot). Where indicated, cells were pretreated for 30 min with 300 μm oxotremorine-M (Oxo), 300 μm carbachol (Cb), or 10 μm nicotine (Nic). Cells were treated with the antagonists 10 μm atropine (Atr) or 10 μm mecamylamine (Mec) 15 min before treatment with agonists. Values are presented as the percent of caspase-3 activity in untreated control (Ctl) cells and are the means ±S.E.; n = 3–4; *, p < 0.05 compared with values from samples treated with rotenone alone. B, caspase-3 activity was measured in cells treated with 0.5 μm staurosporine alone or after a 30-min pretreatment with 300 μm oxotremorine-M. Values are presented as the percent of caspase-3 activity in untreated control cells (means ±S.E.; n = 3).
It is important to note that not all apoptotic-inducing insults are attenuated by stimulation of muscarinic receptors. Staurosporine is one of the most widely used agents to activate caspase-3-mediated apoptosis, and we previously characterized the staurosporine concentration- and time-dependent activation of apoptosis in SH-SY5Y cells (30). Treatment with staurosporine caused a time-dependent increase in caspase-3 activity, and there was no attenuation in cells pretreated with oxotremorine-M (Fig. 4B). Thus, muscarinic receptor activation does not cause a global inhibition of the activation of caspase-3 caused by apoptotic stimuli, but it provides protection from a discreet group of insults.
Oxotremorine-M Treatment Prevents Camptothecin-induced Mitochondrial Cytochrome c Release, bcl-2 Depletion, and Bax Accumulation
We focused on the actions of oxotremorine-M against the deleterious effects of camptothecin to examine the mechanism of protection. To examine if the protective effect of stimulated muscarinic receptors was upstream or downstream of the mitochondrial release of cytochrome c, we monitored the mitochondrial and cytosolic levels of cytochrome c. Treatment with camptothecin (1 μm; 4 h) caused a massive release of cytochrome c into the cytosol, resulting in a nearly 4-fold increase in the cytosolic cytochrome c level and a 60% decline in the mitochondrial cytochrome c content (Fig. 5). Pretreatment with oxotremorine-M (300 μm; 30 min) completely blocked this displacement of cytochrome c from the mitochondria into the cytosol. Thus, the protective effect of muscarinic receptor stimulation targeted a step in the apoptotic signaling pathway preceding cytochrome c release from the mitochondria.
Fig. 5. Muscarinic receptor stimulation blocked cytochrome c release from the mitochondria caused by camptothecin treatment.
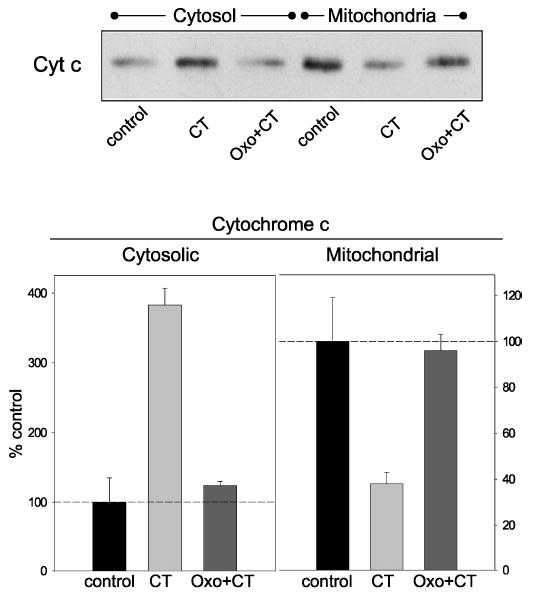
Cells were treated with 1 μm camptothecin (CT) or with 300 μm oxotremorine-M (Oxo) 30 min before camptothecin, and cytosolic and mitochondrial fractions were immunoblotted for cytochrome c (Cyt c). Quantitative values are the means ±S.E.; n = 3.
Members of the bcl-2 family of proteins are key regulators of the mitochondrial-dependent apoptotic pathway. For example, the proapoptotic Bax can translocate to the mitochondria to transmit apoptotic signals, an event that can reduce the mitochondrial level of anti-apoptotic bcl-2. Therefore, we tested if treatment with oxotremorine-M influenced changes in the mitochondrial levels of Bax and bcl-2 after exposure to camptothecin. Treatment with camptothecin increased the level of Bax in the mitochondria by ~4-fold (Fig. 6A). This translocation of Bax to the mitochondria induced by camptothecin was nearly completely blocked by pretreatment with oxotremorine-M. The mitochondrial bcl-2 levels were regulated opposite to those of Bax. As shown in Fig. 6B, camptothecin treatment caused a reduction in the mitochondrial level of bcl-2, and pretreatment with oxotremorine-M prevented this loss of mitochondrial bcl-2. Thus, the protective action of stimulated muscarinic receptors after camptothecin treatment is associated with blockade of the activation of proapoptotic Bax, which was associated with retention of mitochondrial bcl-2 and cytochrome c.
Fig. 6. Mitochondrial Bax and bcl-2 levels.
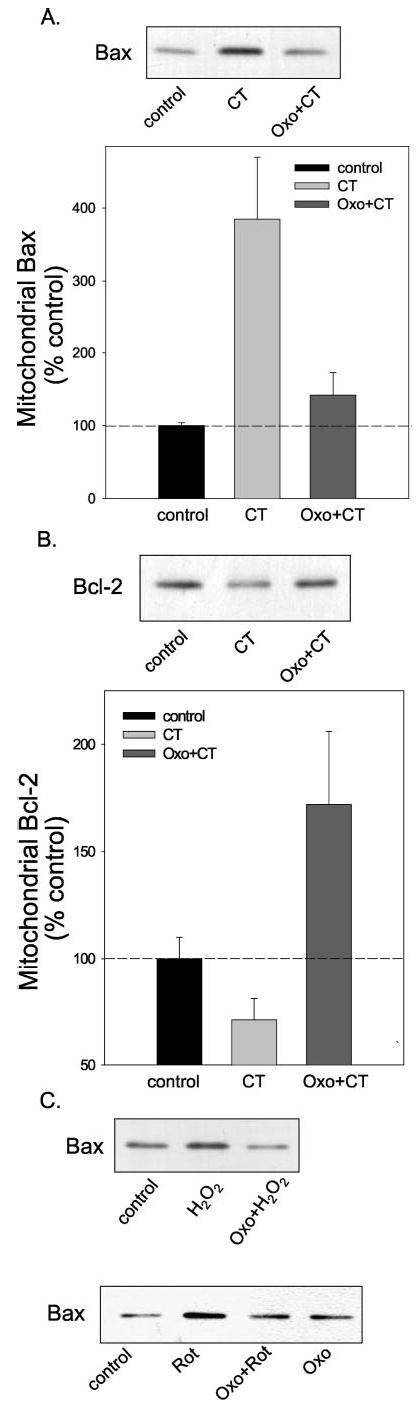
Cells were treated with 1 μm camptothecin (CT) or with 300 μm oxotremorine-M (Oxo) 30 min before camptothecin, and mitochondrial fractions were immunoblotted for Bax (A) or bcl-2 (B). Quantitative values are the means ±S.E.; n = 3. C, mitochondrial Bax levels were measured after treatment with 100 μm H2O2 (5 h) or 5μm rotenone (16 h) with or without a 30-min pretreatment with 300 μm oxotremorine-M.
Because the mitochondrial accumulation of Bax is likely most proximal to muscarinic receptors of these events, we examined mitochondrial Bax levels after treatment with H2O2 and rotenone. A similar oxotremorine-M-induced diminution of Bax translocation to the mitochondria was detected after treatment with H2O2 or with rotenone (Fig. 6C), suggesting that acommon survival-promoting mechanism, blockade of Bax activation, is induced by stimulation of muscarinic receptors after each of these insults.
DISCUSSION
This investigation found that activation of muscarinic receptors provided substantial protection from the induction of apoptotic signaling caused by three common insults, DNA damage, oxidative stress, and impaired mitochondrial function. Although cells eventually succumbed because of the extremity of the toxic treatments employed, remarkable protection from the activation of caspase-3 induced by each of these conditions was evident upon stimulation of muscarinic receptors, a protection that was associated with reduced translocation of the proapoptotic protein Bax to the mitochondria. To study this effect in cells with a relatively uniform level of expression of muscarinic receptors, human neuroblastoma SH-SY5Y cells were used because they endogenously express muscarinic receptors predominantly of the M3 subtype (43), as opposed to mixed populations of cells and receptors present in most other neuronal preparations. These properties of physiologically relevant levels of receptors and the uniformity of receptor expression were deemed sufficiently advantageous to balance the limitations imposed by the utilization of proliferating cells. In fact, many studies show that exposure to differentiating agents enhances defenses against many insults, such as oxidative stress (44–46), suggesting that this model system imposed a greater challenge for rescuing cells from these insults. However, with this identification of the protective capacity of activated muscarinic receptors, further efforts should be directed toward examining these interactions in mature neurons and determining whether or not stimulation of other G-protein-coupled receptors also provides protection.
Stimulation of muscarinic receptors provided protection from three relatively common insults. Oxidative stress contributes to neuronal dysfunction and loss in many neurodegenerative conditions (21, 22). For example, much evidence links oxidative stress to the neuropathology of AD (9, 18, 19, 20, 25), where there is a concomitant loss of cholinergic function (13), including loss of muscarinic receptor-coupled signaling activities (47). Based on this co-existence of oxidative stress and loss of muscarinic receptor function, it was of interest to investigate if loss of muscarinic receptor-coupled signaling could contribute to the deleterious effects of oxidative stress by testing if active muscarinic receptors provide protection from caspase activation. The results demonstrated that activation of muscarinic receptors with the selective agonist oxotremorine-M provided significant protection from the apoptosis-signaling cascade induced by H2O2. Muscarinic receptor activation also provided substantial protection from caspase-3 activation ensuing from inhibition of mitochondrial function or DNA damage. These findings suggest that loss of muscarinic receptor activity in neurodegenerative conditions, such as AD, likely increases the vulnerability of cells to the detrimental effects of these insults.
The protection provided by muscarinic receptor activation was of a similar magnitude to that of a general caspase inhibitor. For both of these, protection was greatest at lower concentrations of H2O2 and at short times of treatment with H2O2 or camptothecin. The former suggests that muscarinic receptor activation may provide considerable protection from physiologically relevant levels of oxidative stress. The delayed death observed even in the presence of either oxotremorine-M or the caspase inhibitor BAF could be due to necrotic mechanisms contributing to the later cell death, against which these agents were ineffective, or the agents themselves may have lost the ability to provide protection after long incubation periods. For example, the agents may have been degraded or sequestered, or prolonged exposure to oxotremorine-M may have down-regulated relevant signaling activities, which lessened the protection provided from cell death. Regardless of the cause of the temporally limited protection, it is notable that muscarinic receptor activation was essentially equivalent to inhibition of caspases in providing protection, indicating that at physiologically relevant levels of toxic insults, considerable protection is provided by intracellular signals emanating from muscarinic receptors.
The signaling mechanisms responsible for the protective effect of muscarinic receptor activation on the detrimental effects of these insults appeared to be directed at Bax. The results clearly showed that oxotremorine-M did not prevent the initial detrimental effects of H2O2 or camptothecin, as exemplified by the equal activation of p53 by each of these agents in the presence and absence of oxotremorine-M. However, oxotremorine-M treatment blocked the translocation of Bax to mitochondria, blocked loss of mitochondrial bcl-2, and blocked cytochrome c release to the cytosol. The latter action leads to activation of caspase-3, indicating that the muscarinic receptor-induced protection from caspase-3 activation likely results from these actions, originating at the point of Bax activation, which is known to mediate apoptosis after a variety of insults, such as DNA damage (48). The mechanism by which stimulated muscarinic receptors block apoptotic activation of Bax remains to be determined. In the absence of apoptotic conditions, Bax is predominantly cytosolic, at least partially sequestered by the scaffold protein 14-3-3 (49, 50). Conformational changes in Bax appear to lead to its translocation to the mitochondria and oligomerization (51–53) followed by release of cytochrome c (54), although precise details of the molecular mechanisms underlying this transition during apoptotic signaling remain unknown. Our findings suggest that signals from muscarinic receptors attenuate apoptotic activation of Bax, an action that may be mediated by signals (e.g. kinase activities, calcium transients) impinging on Bax, 14-3-3, or other regulatory molecules known to participate in Bax activation (e.g. Refs. 55 and 56), retarding its activation and translocation to the mitochondria. However, the precise intermediate steps accounting for Bax activation as well as muscarinic receptor-induced neuroprotection await identification.
AD is associated with both increased neuronal oxidative stress and loss of cholinergic function. The results of this study demonstrate that these two conditions likely exacerbate neuronal dysfunction since activation of muscarinic receptor-coupled signaling provides protection from oxidative stress-induced apoptosis. Thus, as muscarinic receptor-coupled signaling activity becomes impaired during the progression of AD, neurons lose this important mechanism of counteracting the detrimental effects of oxidative stress. Interpretation of the results presented here must take into consideration the current controversy about the extent to which apoptosis contributes to Alzheimer’s disease as well as other neurodegenerative conditions (57). Therefore, we suggest that the protection provided by muscarinic receptors against oxidative stress and the other insults examined might be of greater importance for maintaining neural function and plasticity, which likely are impaired temporally before apoptosis after exposure to these insults, although indicators of apoptotic signaling were used as outcome measures in this study. The findings reported here also may have relevance for developmental regulation of the central nervous system. There is a wealth of information that cholinergic afferents to the cortex and hippocampus serve a growth regulatory function in ontogeny (reviewed in Refs. 58 and 59). The present results suggest that one action of cholinergic activity may be to promote survival of innervated neurons during development by inhibiting apoptotic mechanisms. In this regard, it is of interest that cell loss induced by alcohol exposure has been hypothesized to stem at least in part from inhibitory actions of alcohol on muscarinic receptor signaling (60). We would speculate that stimulation of other G-protein-coupled receptors activating similar signaling pathways also would bolster neuronal survival, providing a means for innervated cells to survive under conditions that are lethal to cells not connected to sufficiently active networks.
In conclusion, these results indicate that in many instances continued neuronal survival is dependent upon the environment of the cell, as cell-to-cell contact and signals received from neighboring cells may provide the necessary impetus to bolster defenses against death programs.
Footnotes
This research was supported by grants from the National Institutes of Health and the Alzheimer’s Association.
The abbreviations used are: AD, Alzheimer’s disease; 3AB, 3-aminobenzamide; BAF, boc-aspartyl(OMe)-fluoromethyl ketone; PARP, poly (ADP-ribose) polymerase.
References
- 1.Zornig M, Hueber A, Baum W, Evan G. Biochim Biophys Acta. 2001;1551:1–37. doi: 10.1016/s0304-419x(01)00031-2. [DOI] [PubMed] [Google Scholar]
- 2.Bortner CD, Cidlowski JA. Annu Rev Pharmacol Toxicol. 2002;42:259–281. doi: 10.1146/annurev.pharmtox.42.083101.143836. [DOI] [PubMed] [Google Scholar]
- 3.Jacobs BL, van Praag H, Gage FH. Mol Psychiatry. 2000;5:262–269. doi: 10.1038/sj.mp.4000712. [DOI] [PubMed] [Google Scholar]
- 4.Kempermann G. Bipolar Disord. 2002;4:17–33. doi: 10.1034/j.1399-5618.2002.40101.x. [DOI] [PubMed] [Google Scholar]
- 5.Miller FD, Pozniak CD, Walsh GS. Cell Death Differ. 2000;7:880–888. doi: 10.1038/sj.cdd.4400736. [DOI] [PubMed] [Google Scholar]
- 6.Morrison RS, Kinoshita Y. Cell Death Differ. 2000;7:868–879. doi: 10.1038/sj.cdd.4400741. [DOI] [PubMed] [Google Scholar]
- 7.Yuan J, Yankner BA. Science. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 8.Price DL, Sisodia SS, Borchelt DR. Science. 1998;282:1079–1083. doi: 10.1126/science.282.5391.1079. [DOI] [PubMed] [Google Scholar]
- 9.Pratico D, Clark CM, Liun F, Lee VY, Trojanowski JQ. Arch Neurol. 2002;59:972–976. doi: 10.1001/archneur.59.6.972. [DOI] [PubMed] [Google Scholar]
- 10.Castagne V, Gautschi M, Lefevre K, Posada A, Clarke PG. Prog Neurobiol (NY) 1999;59:397–423. doi: 10.1016/s0301-0082(99)00012-x. [DOI] [PubMed] [Google Scholar]
- 11.Ikonomidou C, Bittigau P, Koch C, Genz K, Hoerster F, Felderhoff-Mueser U, Tenkova T, Dikranian K, Olney JW. Biochem Pharmacol. 2001;62:401–405. doi: 10.1016/s0006-2952(01)00696-7. [DOI] [PubMed] [Google Scholar]
- 12.Patapoutian A, Reichardt LF. Curr Opin Neurobiol. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- 13.Bartus RT. Exp Neurol. 2000;163:495–529. doi: 10.1006/exnr.2000.7397. [DOI] [PubMed] [Google Scholar]
- 14.Ghahremani MH, Keramaris E, Shree T, Xia Z, Davis RJ, Flavell R, Slack RS, Park DS. J Biol Chem. 2002;277:35586–35596. doi: 10.1074/jbc.M204362200. [DOI] [PubMed] [Google Scholar]
- 15.Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keramaris E, Stefanis L, MacLaurin J, Harada N, Takaku K, Ishikawa T, Taketo MM, Robertson GS, Nicholson DW, Slack RS, Park DS. Mol Cell Neurosci. 2000;15:368–379. doi: 10.1006/mcne.2000.0838. [DOI] [PubMed] [Google Scholar]
- 17.Morris EJ, Keramaris E, Rideout HJ, Slack RS, Dyson NJ, Stefanis L, Park DS. J Neurosci. 2001;21:5017–5026. doi: 10.1523/JNEUROSCI.21-14-05017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markesbery WR. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 19.Mattson MP. Alzheimer’s Dis Rev. 1997;2:1–14. [Google Scholar]
- 20.Behl C. Prog Neurobiol. 1999;57:301–323. doi: 10.1016/s0301-0082(98)00055-0. [DOI] [PubMed] [Google Scholar]
- 21.Halliwell B. Drugs Aging. 2001;18:685–716. doi: 10.2165/00002512-200118090-00004. [DOI] [PubMed] [Google Scholar]
- 22.Sayre LM, Smith MA, Perry G. Curr Med Chem. 2001;8:721–738. doi: 10.2174/0929867013372922. [DOI] [PubMed] [Google Scholar]
- 23.Lovell M, Ehmann W, Butler S, Markesbery W. Neurology. 1995;45:1594 –1601. doi: 10.1212/wnl.45.8.1594. [DOI] [PubMed] [Google Scholar]
- 24.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 25.Gibson GE, Huang HM. Front Biosci. 2002;7:1007–1015. doi: 10.2741/A827. [DOI] [PubMed] [Google Scholar]
- 26.Tatton WG, Olanow CW. Biochim Biophys Acta. 1999;1410:195–213. doi: 10.1016/s0005-2728(98)00167-4. [DOI] [PubMed] [Google Scholar]
- 27.Manfredi G, Beal MF. Brain Pathol. 2000;10:462–472. doi: 10.1111/j.1750-3639.2000.tb00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolvetang EJ, Johnson KL, Krauer K, Ralph SJ, Linnane AW. FEBS Lett. 1994;339:40–44. doi: 10.1016/0014-5793(94)80380-3. [DOI] [PubMed] [Google Scholar]
- 29.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 30.Bijur GN, De Sarno P, Jope RS. J Biol Chem. 2000;275:7583–7590. doi: 10.1074/jbc.275.11.7583. [DOI] [PubMed] [Google Scholar]
- 31.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 32.Bijur GN, Jope RS. J Biol Chem. 2001;276:37436–37442. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gottlieb RA, Adachi S. Methods Enzymol. 2000;322:213–221. doi: 10.1016/s0076-6879(00)22022-3. [DOI] [PubMed] [Google Scholar]
- 34.Pallotti F, Lenaz G. Methods Cell Biol. 2001;65:1–33. doi: 10.1016/s0091-679x(01)65002-7. [DOI] [PubMed] [Google Scholar]
- 35.Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, Johnson GVW, Jope RS. Proc Natl Acad Sci U S A. 2002;99:7951–7955. doi: 10.1073/pnas.122062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 37.Ha HC, Snyder SH. Neurobiol Dis. 2000;7:225–239. doi: 10.1006/nbdi.2000.0324. [DOI] [PubMed] [Google Scholar]
- 38.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Biochem J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- 39.Pieper AA, Verma A, Zhang J, Snyder SH. Trends Pharmacol Sci. 1999;20:171–181. doi: 10.1016/s0165-6147(99)01292-4. [DOI] [PubMed] [Google Scholar]
- 40.Krishnamurthy PK, Mays JL, Bijur GN, Johnson GVW. J Neurosci Res. 2000;61:515–523. doi: 10.1002/1097-4547(20000901)61:5<515::AID-JNR6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Dawson VL, Dawson TM. Neurobiol Dis. 2000;7:240–250. doi: 10.1006/nbdi.2000.0319. [DOI] [PubMed] [Google Scholar]
- 42.King TD, Bijur GN, Jope RS. Brain Res. 2001;919:106–114. doi: 10.1016/s0006-8993(01)03005-0. [DOI] [PubMed] [Google Scholar]
- 43.Lambert DG, Ghataorre AS, Nahorski SR. Eur J Pharmacol. 1989;165:71–77. doi: 10.1016/0014-2999(89)90771-1. [DOI] [PubMed] [Google Scholar]
- 44.Jackson GR, Werrbach-Perez K, Pan Z, Sampath D, Perez-Polo JR. Dev Neurosci. 1994;16:285–290. doi: 10.1159/000112121. [DOI] [PubMed] [Google Scholar]
- 45.Mattson MP, Lovell MA, Furukawa K, Markesbery WR. J Neurochem. 1995;65:1740–1751. doi: 10.1046/j.1471-4159.1995.65041740.x. [DOI] [PubMed] [Google Scholar]
- 46.Kamata H, Tanaka C, Yagisawa H, Hirata H. Neurosci Lett. 1996;212:179–182. doi: 10.1016/0304-3940(96)12806-8. [DOI] [PubMed] [Google Scholar]
- 47.Jope RS. J Alz Dis. 1999;1:287–295. [Google Scholar]
- 48.Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Science. 2000;290:989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- 49.Samuel T, Weber HO, Rauch P, Verdoodt B, Eppel JT, McShea A, Hermeking H, Funk JO. J Biol Chem. 2001;276:45201–45206. doi: 10.1074/jbc.M106427200. [DOI] [PubMed] [Google Scholar]
- 50.Nomura M, Shimizu S, Sugiyama T, Narita M, Ito T, Matsuda H, Tsujimoto Y. J Biol Chem. 2003;278:2058–2065. doi: 10.1074/jbc.M207880200. [DOI] [PubMed] [Google Scholar]
- 51.Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gross A, Jockel J, Wei MC, Korsmeyer SJ. EMBO J. 1998;17:3878–3885. doi: 10.1093/emboj/17.14.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphy KM, Streips UN, Lock RB. J Biol Chem. 2000;275:17225–17228. doi: 10.1074/jbc.C900590199. [DOI] [PubMed] [Google Scholar]
- 54.Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Proc Natl Acad Sci U S A. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nomura M, Shimizu S, Ito T, Narita M, Matsuda H, Tsujimoto Y. Cancer Res. 1999;59:5542–5548. [PubMed] [Google Scholar]
- 56.Soucie EL, Annis MG, Sedicy J, Filmus J, Leber B, Andrews DW, Penn LZ. Mol Cell Biol. 2001;21:4725–4736. doi: 10.1128/MCB.21.14.4725-4736.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roth KA. J Neuropathol Exp Neurol. 2001;60:829–838. doi: 10.1093/jnen/60.9.829. [DOI] [PubMed] [Google Scholar]
- 58.Hohmann CF, Berger-Sweeney J. Perspect Dev Neurobiol. 1998;5:401–425. [PubMed] [Google Scholar]
- 59.Nguyen L, Rigo JM, Rocher V, Belachew S, Malgrange B, Rogister B, Leprince P, Moonen G. Cell Tissue Res. 2001;305:187–202. doi: 10.1007/s004410000343. [DOI] [PubMed] [Google Scholar]
- 60.Costa LG, Guizzetti M. Biochem Pharmacol. 1999;57:721–726. doi: 10.1016/s0006-2952(98)00278-0. [DOI] [PubMed] [Google Scholar]