

Abstract
AP-1 (Activating Protein 1) transcription factor activity is tightly regulated at multiple levels, including dimer formation (i.e., Fos/Jun). Here we show that the intermediate filament protein lamin A/C suppresses AP-1 function through direct interaction with c-Fos, and that both proteins can interact and colocalize at the nuclear envelope (NE) in mammalian cells. Perinuclear localization of c-Fos is absent in Lmna-null cells but can be restored by lamin A overexpression. In vitro, preincubation of c-Fos with lamin A prior to the addition of c-Jun inhibits AP-1 DNA-binding activity. In vivo, overexpression of lamin A reduces the formation of c-Fos/c-Jun heterodimers, and suppresses AP-1 DNA-binding and transcriptional activity. Notably, c-Fos colocalizes with lamin A/C at the NE in starvation-synchronized quiescent cells lacking detectable AP-1 DNA binding. In contrast, serum-induced AP-1 DNA-binding activity coincides with abundant nucleoplasmic c-Fos expression without changes in lamin A/C localization. We also found that Lmna-null cells display enhanced proliferation. In contrast, lamin A overexpression causes growth arrest, and ectopic c-Fos partially overcomes lamin A/C-induced cell cycle alterations. We propose lamin A/C-mediated c-Fos sequestration at the NE as a novel mechanism of transcriptional and cell cycle control.
Keywords: c-Fos, AP-1, leucine zipper, lamin A/C, transcriptional regulation, nuclear envelope
The members of the Fos, Jun, ATF (Activating Transcription Factor), and MAF (Musculo Aponeurotic Fibrosarcoma) protein families are components of the dimeric AP-1 (Activating Protein 1) transcription factor complex. AP-1 participates in the regulation of a variety of cellular processes, such as cell proliferation, cell differentiation, neoplastic transformation, and apoptosis (Angel and Karin 1991; Karin et al. 1997; Eferl and Wagner 2003). AP-1 transcription factor activity is regulated at multiple levels, including transcriptional control, post-translational modifications, dimer composition, and interactions with many structurally divergent regulatory proteins. AP-1 proteins are prototype transcription factors that harbor several functional domains: (1) several transactivation regions, (2) a basic domain that interacts with sequence elements in the promoters and enhancers of target genes, and (3) the adjacent leucine-zipper domain (ZIP) required for dimerization, a prerequisite for AP-1 DNA-binding activity and for transcriptional regulation of target genes (Angel and Karin 1991; Karin et al. 1997; Eferl and Wagner 2003).
The importance of protein–protein interactions in the control of AP-1 function is suitably illustrated by the participation of Fos and Jun in multiple dimeric transcription complexes including: (1) Jun/Jun homodimers, (2) Fos/Jun heterodimers, (3) heterodimers between Fos or Jun and other “basic-ZIP” (bZIP) family proteins (e.g., ATF, MAF, Nrf-1, Nrf-2), and (4) heterodimers between Fos or Jun and structurally unrelated DNA-binding proteins (e.g., NFAT, Ets, Smad, Stat, and basic helix–loop–helix [bHLH]) (Chinenov and Kerppola 2001). Additional complexity in the control of AP-1 function arises from the participation of AP-1 proteins in higher-order transcriptional complexes and in heterodimers with non-DNA-binding proteins (e.g., Fos/Sug1, Jun/Jab1, Jun/menin) (Wang et al. 1996; Chinenov and Kerppola 2001).
Dimer composition determines the repertoire of genes that are regulated by AP-1 and thus contributes to the differential functions of specific AP-1 transcription complexes. For example, Jun–ATF and ATF–ATF dimers bind preferentially to the cAMP-responsive element (CRE: 5′-TGACGTCA-3′), whereas Jun–Jun and Jun–Fos dimers prefer to bind to the 12-O-tetradecanoyl-phorbol-13-acetate (TPA)-responsive element (TRE: 5′-TGACTCA-3′) in the promoter of target genes (Angel and Karin 1991; Karin et al. 1997; Eferl and Wagner 2003).
In this study, we sought to identify and characterize novel protein–protein interactions potentially involved in the regulation of AP-1 function. To this end, we used the yeast two-hybrid assay (Ma and Ptashne 1988; Fields and Song 1989) with the bZIP region of c-Fos as a bait. We show that lamin A and lamin C, two splice variants of a single Lmna gene that are often referred to as A-type lamins (Zastrow et al. 2004; Gruenbaum et al. 2005), are novel heterodimerization partners of c-Fos. Together with B-type lamins, lamin A/C are the major components of the nuclear lamina. Besides having a ubiquitous role in the maintenance of nuclear architecture, A-type lamins are also thought to provide cell-specific regulatory functions (Zastrow et al. 2004; Gruenbaum et al. 2005). Here we provide evidence suggesting that AP-1 suppression by lamin A/C-dependent c-Fos sequestration at the nuclear envelope (NE) contributes to transcriptional and cell cycle control in mammalian cells.
Results
c-Fos and lamin A/C interact in the yeast two-hybrid system and in vitro
Since the two-hybrid system is based on reconstitution of a functional transcription factor, we carried out preliminary experiments to identify appropriate c-Fos “baits” that did not autoactivate the system. We constructed various fusion proteins containing the DNA-binding domain of Lex A attached in frame to the N terminus of either full-length or different N- and C-terminal deletion mutants of rat c-Fos. Among these, only the bZIP (amino acids 123–230) failed to induce reporter gene activity on its own (data not shown). Therefore, pBTM-c-Fos-bZIP (amino acids 123–230) was used as the “bait” protein. To screen for potential heterodimerization partners, we used a thymocyte cDNA library subcloned in pAD-Gal4-2.1 (encoding the activation domain of Gal4). In addition to previously known c-Fos interaction partners, such as Jun proteins (JunD and JunB) (Angel and Karin 1991; Karin et al. 1997; Eferl and Wagner 2003) and Sug1 (Wang et al. 1996), we identified A-type lamins as novel potential heterodimerization partners of c-Fos.
Like other intermediate filament proteins, A-type lamins have a short N-terminal “head” domain, a long α-helical “coiled-coil” structure (rod domain), and a globular C-terminal tail that includes an immunoglobulin (Ig)-fold domain (Fig. 1B; Supplementary Fig. S1; Zastrow et al. 2004; Gruenbaum et al. 2005). In vitro pull-down assays using hexahistidine (His6)-tagged full-length c-Fos and GST fusion proteins containing different regions of rat lamin A/C demonstrated direct binding of c-Fos to lamin A/C (Supplementary Fig. S2). Furthermore, we found that His6-bZIP-c-Fos interacts with GST fusions of either the rod domain (coiled-coil region: coil 1b + coil 2) or the Ig-fold domain of rat lamin A/C (Fig. 1A), both of which harbor several leucine residues. However, His6-bZIP-c-Fos did not interact with either GST or GST-lamin A/C (amino acids 390–452) (Fig. 1A), a truncated mutant that only contains one leucine residue. Figure 1B summarizes the results of these studies. It has been well established that the ZIP domain of AP-1 factors is formed by an α-helix in which every seventh amino acid is a leucine that protrudes from one side of the α-helix to form a hydrophobic surface (LHR: “leucine heptad repeat”) (Supplementary Fig. S1A), and that AP-1 dimerization occurs in a manner very similar to classical “coiled-coil” interactions (Angel and Karin 1991; Lupas 1996). We noted the presence of LHRs in rat and mouse lamin A/C (see examples in Supplementary Figs. S1B, S3A). Remarkably, disruption of one such LHR in the murine lamin A/C (amino acids 117–227) L158G-E159T-L172S-E173T mutant (Supplementary Fig. S1C) caused reduced binding to His6-bZIP-c-Fos (Fig. 1C). Together, these in vitro studies suggest that binding of c-Fos to lamin A/C involves hydrophobic interactions between leucine residues, similar to the ZIP-mediated dimerization among AP-1 family members (see Discussion).
Figure 1.

The interaction between lamin A/C and c-Fos is mediated by leucine heptad repeats. (A) GST fusion proteins spanning different regions of rat lamin A/C (LMN A/C) were tested for their interaction with hexahistidine-tagged c-Fos basic-leucine-zipper (His6-bZIPc-Fos, rat amino acids 116–211). Protein complexes were precipitated using glutathione-sepharose 4B and washed extensively. The presence of His6-bZIPc-Fos and GST proteins in the precipitated material was monitored using anti-His antibody and Coomassie blue staining, respectively. (B) Summary of pull-down experiments showing the domains of rat lamin A/C. (NLS) Nuclear localization signal; (+) positive interaction; (-) negative interaction. (C) Pull-down assay using His6-bZIPc-Fos and GST fused to the murine lamin A/C region containing a portion of coil 1 (amino acids 117–227), either wild-type (WT) or the mutant sequence L158G-E159T-L172S-E173T (Mutant) in which one “leucine heptad repeat” has been disrupted (see Supplementary Fig. S1C). (Left) Representative example showing the amount of GST-lamin A/C 117–227 (wild type [WT] and Mutant) and His6-bZIPc-Fos in the precipitate. (Right) Quantification of the interaction as determined by densitometric analysis of four independent experiments (wild type [WT] set as 100%).
c-Fos localization at the NE in mammalian cells depends on the expression of A-type lamins
We next investigated the localization of lamin A/C and c-Fos in HeLa cells by confocal indirect immunofluorescence microscopy. In agreement with previous studies (Gruenbaum et al. 2005), we found prominent lamin A/C expression at the nuclear periphery and a low level of nucleoplasmic expression (Fig. 2A,B). Cytoplasmic and nuclear c-Fos expression was detected by both confocal immunofluorescence microscopy (Fig. 2A,B) and Western blot analysis (data not shown). Three-dimensional reconstruction of confocal images demonstrated the colocalization of lamin A/C and c-Fos at specific points along the perinuclear rim (Fig. 2A), and fluorescence line profile analysis showed extensive overlap of c-Fos and lamin A/C signals at the nuclear periphery (Fig. 2B), suggesting that these proteins colocalize predominantly at this location. We also observed perinuclear localization of c-Fos in asynchronously growing wild-type mouse embryonic fibroblasts (MEFs), both in cells expressing low and high levels of nucleoplasmic c-Fos (Fig. 2C, asterisk and arrowhead, respectively). In contrast, perinuclear c-Fos was scarcely seen in asynchronously growing Lmna-deficient MEFs (Lmna-/-) with either low or high levels of c-Fos expression (Fig. 2D, asterisk and arrowhead, respectively). Quantitative analysis of these cultures revealed perinuclear c-Fos localization in 33% of wild-type MEFs versus 7% of Lmna-/- MEFs, and these differences were more prominent in starvation-synchronized cells (89% vs. 7%, respectively) (Fig. 2E).
Figure 2.

(A) Localization of endogenous lamin A/C (LMN A/C) (red) and c-Fos (green) in HeLa cells examined by confocal indirect double immunofluorescence microscopy. Colocalization (yellow) was noted at the nuclear periphery. Arrows in three-dimensional reconstructions show examples of coexpression at specific perinuclear locations. (B) Fluorescence line profile analysis of c-Fos (red) and LMN A/C (green) expression across the nuclear section depicted by the discontinuous white line. Arrows in the graph indicate points of prominent colocalization at the perinuclear rim. (C,D, top) Indirect immunofluorescence analysis of c-Fos expression in asynchronously growing wild-type (Lmna+/+) and Lmna-null (Lmna-/-) MEFs. (Middle) Nuclear Hoechst counterstain of the same microscopic field. (Bottom) Merged image. Asterisks and arrowheads mark cells displaying low and high levels of nucleoplasmic c-Fos expression, respectively. (E) Quantification of the percentage of cells with c-Fos perinuclear localization in starvation-synchronized (0.5% FBS) and in asynchronously growing (10% FBS) MEFs (n = 300 of each Lmna+/+ and Lmna-/- cells were scored). (F) Representative photomicrographs of Lmna-/- MEFs transfected with YFP-Fos (control) or YFP-Fos + CFP-LMN A and grown in 10% FBS showing the distribution of both proteins. Bar, 8 μm. The graph shows the percentage of cells that displayed c-Fos perinuclear localization.
To examine whether ectopic lamin A can restore NE localization of c-Fos in Lmna-/- MEFs, we ectopically expressed the fusion proteins enhanced cyan fluorescent protein-lamin A (CFP-LMN A) and enhanced yellow fluorescent protein-c-Fos (YFP-Fos) in these cells. As shown in Figure 2F, perinuclear expression of YFP-Fos was scant in the absence of ectopic lamin A, but markedly increased upon CFP-LMN A cotransfection (10% vs. 66%, respectively; p < 0.001). To further examine whether lamin A/C was sufficient to recruit c-Fos to the perinuclear rim, we performed overexpression studies in transiently transfected COS-7 cells. Ectopic HA-lamin A accumulated at the NE and recruited YFP-Fos to this location (Fig. 3A). In contrast, perinuclear YFP-Fos was not apparent in control cells transfected with empty pcDNA (Fig. 3B). Consistent with these findings, Western blot analysis of HA-lamin A-transfected cells revealed high levels of ectopic HA-lamin A and endogenous c-Fos in membrane-enriched extraction-resistant nuclear fractions (Fig. 3C, lane 4). In contrast, endogenous c-Fos expression was scarce in the nuclear extraction-resistant fraction obtained from control (mock-transfected) cells (Fig. 3C, lane 3). Expression of the nucleoporin Nup50 in the extraction-resistant nuclear fraction and cyclin A and p27 in the soluble fraction remained unchanged upon HA-lamin A overexpression (Fig. 3C). Together, these findings indicate that lamin A/C expression is necessary and sufficient for the localization of c-Fos within the NE of mammalian cells.
Figure 3.

(A,B) Representative photomicrographs of COS-7 cells cotransfected with either YFP-c-Fos (YFP-Fos) + pcDNA-HA-lamin A (HA-LMN A) or YFP-Fos + pcDNA. YFP-Fos is shown in green and HA-LMN A in red (visualized by indirect immunofluorescence using anti-HA monoclonal antibody). (C) Western blot analysis of the soluble and extraction-resistant nuclear fractions of COS-7 cells transfected with HA-LMN A (+) or with empty pcDNA (-) using the indicated antibodies. (Nup50) Nucleoporin 50-kDa/nuclear pore-associated protein 60L. The short exposure of the lamin A/C blot precludes the visualization of the endogenous proteins, which requires longer exposures (cf. Fig. 6A). (D) Immunoprecipitation (IP) in HeLa cell lysates was performed using mouse monoclonal anti-lamin A/C or control (anti-HA) antibody. Both the input (5% of starting material) and the whole immunoprecipitated material were subjected to Western blot (WB) analysis using anti-c-Fos and anti-lamin A/C antibodies. (E) Immunoprecipitation (IP) in HeLa cells was performed with rabbit polyclonal anti-c-Fos antibodies or normal rabbit IgG (control), and the presence of lamin A/C in the precipitate was examined by Western blot (WB).
c-Fos and lamin A/C interact in mammalian cells
Having demonstrated that c-Fos and lamin A/C interact in vitro and colocalize in mammalian cells, we next sought to examine whether c-Fos and lamin A/C can interact in vivo. We found that endogenous lamin A/C and c-Fos can be specifically coimmunoprecipitated from HeLa cell lysates using either anti-lamin A/C (Fig. 3D) or anti-c-Fos (Fig. 3E) antibodies. Further evidence that c-Fos and lamin A/C can interact in mammalian cells was obtained from experiments of fluorescence resonance energy transfer (FRET) confocal microscopy, which allows the measurement of protein–protein interactions at a molecular resolution of 1–10 nm (Kenworthy 2001). COS-7 cells were cotransfected with CFP-LMN A + YFP-Fos or CFP-LMN A + YFP as negative control, and FRET at the sites of CFP and YFP colocalization was measured using the acceptor photobleaching method described by Kenworthy (2001). As shown in Figure 4A, CFP-LMN A and YFP-Fos colocalized at the nuclear periphery, in agreement with the findings of Figure 2F. Moreover, the area of CFP fluorescence that increased after photobleaching in the perinuclear rim of cells cotransfected with YFP-Fos and CFP-LMN A was 4.5-fold higher than that observed in the negative control (YFP + CFP-LMN A; p < 0.01) (Fig. 4A), indicating that c-Fos and lamin A associate at the NE.
Figure 4.

FRET analysis demonstrates the interaction between A-type lamins and c-Fos and inhibition of c-Fos/c-Jun heterodimerization by ectopic lamin A. FRET was determined using the acceptor photobleaching method in COS-7 cells cotransfected with various combinations of expression vectors (see details in Materials and Methods). The graphs show the quantification of protein–protein interactions calculated as the percentage of pixels in the donor channel that increased their intensity value after photobleaching in 20–30 cells from two to five independent experiments. Images show representative examples. Bar, 8 μm. (A) Cells were cotransfected with CFP-LMN A and either YFP (negative control) or YFP-Fos. The low-magnification images show CFP-LMN A and YFP-Fos in the same cell before and after photobleaching. CFP-LMN A fluorescence in the perinuclear rim regions in the three squares is shown at higher magnification before and after photobleaching. (B) To examine the effect of lamin A on c-Fos/c-Jun heterodimerization, cells were cotransfected with YFP-Fos + CFP-LMN A + CFP-Jun (red and dark-blue bars). The photomicrographs show representative examples of cells negative only for CFP-LMN A (left), positive for all three fluorescent proteins (middle), and negative only for CFP-Jun (right). The arrowhead and arrow indicate the localization of CFP-LMN A (perinuclear rim) and CFP-Jun (nucleoplasm), respectively (cf. Supplementary Fig. S5). The red bars represent the nucleoplasmic CFP-Jun/YFP-Fos interaction, and the dark blue bar represents the CFP-LMN A/YFP-Fos interaction at the NE. The purple and light-blue bars represent the amount of nonspecific FRET in cells cotransfected with YFP + CFP-Jun or YFP + CFP-LMN A, respectively; (purple) measurements in nucleoplasm; (light blue) measurements in NE. Results were analyzed using one-way ANOVA and Bonferroni's post hoc test. For simplicity, only comparisons with appropriate controls are shown.
Lamin A/C suppresses AP-1 function
Because the bZIP mediates c-Fos dimerization with other AP-1 family members to form active complexes (Angel and Karin 1991; Karin et al. 1997; Eferl and Wagner 2003) and with lamin A/C (this study), we hypothesized that the interaction of lamin A/C with c-Fos may cause suppression of AP-1 DNA-binding and transcriptional activity. The studies of Figure 5 using primary rat vascular smooth muscle cells (VSMCs) gave support to this possibility. As revealed by indirect immunofluorescence microscopy, both c-Fos and lamin A/C accumulated predominantly at the NE in quiescent serum-deprived VSMCs (Fig. 5A,D,G). Consistently, Western blot analysis of serum-deprived VSMCs revealed c-Fos and lamin A/C expression in the extraction-resistant lamin A/C-containing nuclear fraction (Fig. 5J, left), whereas both proteins were undetectable in the soluble nuclear fraction (Fig. 5I,J, right, first lane). Moreover, electrophoretic mobility shift assays (EMSAs) using the soluble nuclear fraction from these cultures disclosed undetectable AP-1 DNA-binding activity (Fig. 5K, lane 2). When serum-deprived VSMCs were shortly re-exposed to serum, the expression and nuclear localization of lamin A/C remained essentially unchanged (Fig. 5B,C). In contrast, short re-exposure to serum caused a rapid and transient induction of c-Fos mRNA steady-state level between 15 and 60 min (Fig. 5H), which was followed by the accumulation of nucleoplasmic c-Fos protein (Fig. 5E,I,J) and the induction of AP-1 DNA-binding activity (Fig. 5K, lane 3) between 1 and 2 h. AP-1 DNA-binding activity became negligible at later time points of serum restimulation (Fig. 5K, lanes 4–8), coinciding with the disappearance of nucleoplasmic c-Fos and its perinuclear localization (Fig. 5F,J). [3H]-thymidine incorporation assays demonstrated that all these regulatory events induced by serum decidedly preceded S-phase entry (Fig. 5L).
Figure 5.

Dynamics of serum-dependent regulation of c-Fos expression and AP-1 DNA-binding activity in primary cultures of rat VSMCs. Cells were serum starved and restimulated with 10% FBS for the indicated time. (A–F) Subcellular localization of endogenous A-type lamins (A–C) and c-Fos (D–F) assessed by indirect immunofluorescence. Both lamin A/C and c-Fos disclosed perinuclear staining at all time points examined. Note strong c-Fos staining throughout the nucleus at 2 h of serum restimulation. (G) Confocal immunofluorescence microscopy confirmed the accumulation of c-Fos at the perinuclear rim in serum-starved cultures. (H,I) Expression levels of c-Fos mRNA and protein as assessed by qRT–PCR (H) and Western blot (I), respectively. Results of qRT–PCR are represented as the c-Fos/18S rRNA ratio relative to serum-deprived cells (set as 1; n = 3 independent measurements). (J) Western blot analysis of the extraction-resistant and soluble nuclear fractions. (K) The same soluble nuclear fractions were analyzed by EMSA using a consensus AP-1 radiolabeled probe. (Lane 1) A binding reaction without nuclear extract. AP-1 DNA binding was undetectable under conditions of colocalization of lamin A/C and c-Fos at the NE (cf. D,F) and lack of nucleoplasmic c-Fos expression. Maximum AP-1 DNA-binding activity correlated with highest amount of soluble c-Fos protein (cf. E,I,J). (Lane 9) Same as lane 3, but a 50-fold molar excess of unlabeled AP-1 oligonucleotide was added to the reaction mixture as specific competitor. (L) DNA synthesis in serum-deprived and serum-restimulated VSMCs was investigated by [3H]-thymidine incorporation assay (n = 3 independent assays).
Consistent with the findings in primary VSMCs, we found that increased association of c-Fos with the extraction-resistant nuclear fraction as a consequence of HA-lamin A overexpression (cf. Fig. 3C) inhibited AP-1 DNA-binding activity in 293 and COS-7 cells (Fig. 6A). In contrast, ectopic HA-lamin A did not reduce Sp1 DNA-binding activity (Fig. 6B). Because the bZIP mediates the dimerization of c-Fos with lamin A/C, we hypothesized that this inhibitory effect of ectopic lamin A might result from reduced ZIP-mediated c-Fos heterodimerization with additional AP-1 factors. To test this possibility, we performed EMSAs with recombinant c-Fos and c-Jun fragments spanning the bZIP region of these proteins. It has been previously shown that Fos proteins cannot associate with each other and therefore do not bind DNA on their own; however, Fos and Jun proteins can form stable heterodimers that have higher DNA-binding activity than Jun/Jun homodimers (Angel and Karin 1991; Karin et al. 1997; Eferl and Wagner 2003). As expected, DNA-binding activity was only detected in reaction mixtures containing both c-Fos(bZIP) and c-Jun(bZIP) (Fig. 6C, lanes 1–3). However, preincubation of c-Fos(bZIP) with GST-lamin A/C (amino acids 4–361; rod domain) before the addition of c-Jun(bZIP) specifically abrogated AP-1 DNA-binding activity (Fig. 6C, lanes 4,5), suggesting that lamin A/C inhibits c-Fos/c-Jun heterodimerization in vitro.
Figure 6.

Lamin A/C suppresses AP-1 DNA-binding and transcriptional activity. (A) Soluble nuclear extracts were prepared from control and HA-lamin A-transfected 293 and COS-7 cells and analyzed by Western blot (WB) with the indicated antibodies and EMSA using an AP-1 consensus probe (only retarded bands are shown). Addition of cold AP-1 competitor demonstrated binding specificity (first lane in each EMSA). The graph shows the relative DNA-binding activity in COS-7 cells averaged from three independent EMSAs. (B) COS-7 cells were analyzed as in A, but a consensus Sp1 probe was used. (C) EMSA with recombinant Fos(bZIP) (His6-c-Fos, rat amino acids 116–211), Jun(bZIP) (His6-c-Jun, rat amino acids 257–318), and AP-1 probe. (Lane 1) Fos(bZIP) alone. (Lane 2) Jun(bZIP) alone. (Lane 3) Fos(bZIP) + Jun(bZIP). (Lanes 4,5) Fos(bZIP) was preincubated with GST or GST-lamin A (amino acids 4–361, rod domain), respectively, before the addition of Jun(bZIP) and AP-1 probe. The amount of each recombinant protein was 200 ng. (D) Luciferase activity in extracts from HeLa cells cotransfected with the AP-1-responsive coll73-luciferase or the E2F-responsive 4xE2F1bTATA-luciferase reporters, and either empty pMEX (control, white bars) or pMEX-lamin A (LMN A, black bars). Cells were serum-starved and restimulated with 10% FBS for the indicated time. Luciferase is expressed relative to serum-starved cells (=1). Bars represent the mean ± SE of six independent transfections.
We next used FRET confocal microscopy to test whether A-type lamins can inhibit c-Fos/c-Jun heterodimerization in vivo. COS-7 cells were cotransfected with enhanced cyan fluorescent protein-c-Jun (CFP-Jun), CFP-LMN A, and YFP-Fos. Figure 4B shows the three populations of transfected cells that were analyzed: (1) cells that coexpressed all three recombinant fluorescent proteins (CFP-LMN A and CFP-Jun within nuclear periphery and nucleoplasm, respectively) (cf. Supplementary Fig. S5) (Fig. 4B, middle); (2) cells that only coexpressed YFP-Fos and CFP-Jun (Fig. 4B, left); and (3) cells that only coexpressed YFP-Fos and CFP-LMN A (Fig. 4B, right). The latter yielded a significant increase in perinuclear CFP-LMN A fluorescence after photobleaching when compared with the nonspecific values obtained in the negative control (cotransfection of YFP + CFP-LMN A) (about fourfold; p < 0.001; Fig. 4B, cf. light- and dark-blue bars). Thus, in agreement with the results of Figure 4A, these findings support the notion that YFP-Fos and CFP-LMN A associate at the NE. In the absence of CFP-LMN A (Fig. 4B, left), perinuclear localization of either CFP-Jun or YFP-Fos was negligible, therefore the interaction between these proteins was quantified in nucleoplasmic regions displaying CFP-Jun/YFP-Fos colocalization. Compared with the “after photobleaching” values obtained in the negative control (cotransfection of YFP + CFP-Jun), we observed a significant increase in nucleoplasmic CFP-Jun fluorescence in cells coexpressing YFP-Fos and CFP-Jun (fivefold increase, p < 0.001; Fig. 4B, cf. purple bar and first red bar). These findings confirm the well-known capacity of c-Fos and c-Jun to heterodimerize in vivo. Notably, the level of nucleoplasmic CFP-Jun/YFP-Fos interaction was markedly diminished in cells coexpressing CFP-LMN A when compared with the level seen in cells lacking CFP-LMN A (Fig. 4B, cf. red bars, p < 0.001). A similar inhibition of nucleoplasmic CFP-Jun/YFP-Fos interaction was also produced by ectopic HA-LMN A (Fig. 7A). In contrast, HA-Lamin B1 did not reduce FRET between YFP-Fos and CFP-Jun (Fig. 7A), and anti-c-Fos antibodies failed to immunoprecipitate endogenous lamin B1 in HeLa cells under conditions that yielded efficient immunoprecipitation of lamin A (and also immunoprecipitation of lamin B1 using anti-lamin A/C antibodies) (Fig. 7B). These findings are consistent with the notion that ectopic lamin A, but not lamin B1, interacts with c-Fos and inhibits the formation of c-Fos/c-Jun heterodimers in vivo.
Figure 7.

Ectopic lamin B1 does not impair c-Fos/c-Jun heterodimerization in vivo. (A) FRET was determined using the acceptor photobleaching method in transiently transfected COS-7 cells (see details in Materials and Methods). The graph shows the quantification of protein–protein interactions calculated as the percentage of pixels in the donor channel that increase their intensity value after photobleaching in 20–30 cells from two to four independent experiments. Cells were cotransfected with CFP-Jun + YFP-Fos (open bars) or CFP-Jun + YFP-Fos and either HA-LMN A or HA-LMN B1 (black bars). For negative controls, cells were cotransfected with YFP + CFP-Jun (striped bar). Results were analyzed using one-way ANOVA and Bonferroni's post hoc test. For simplicity, only comparisons with appropriate controls are shown. (NS) No significant difference (p > 0.05). (B) Lysates of COS-7 cells were subjected to immunoprecipitation with either anti-c-Fos or anti-lamin A/C antibodies as indicated, and the precipitated material was subjected to Western blot analysis (WB) to examine lamin A/C and lamin B1 expression.
Based on these findings, we hypothesized that A-type lamins would repress the transcriptional activation of AP-1 target genes. Transient transfection assays in HeLa cells demonstrated impaired serum-dependent transcriptional activation of the AP-1-responsive coll73-luciferase reporter gene by ectopic lamin A, which did not affect the activity of a synthetic E2F-responsive promoter (Fig. 6D). Taken together, our results suggest that lamin A/C suppresses AP-1 function (e.g., DNA-binding and transcriptional activation) via inhibition of ZIP-dependent c-Fos/c-Jun heterodimerization.
Lamin A/C dysregulation affects cell cycle activity
Since the results presented thus far suggested that lamin A/C interferes with AP-1 function, and considering the well-established role of AP-1 in cell cycle regulation (Angel and Karin 1991; Karin et al. 1997; Eferl and Wagner 2003), we sought to investigate the consequences of altering lamin A/C expression on cellular proliferation. We found a marked increase in [3H]-thymidine incorporation in serum-restimulated Lmna-/- MEFs compared with litter-matched wild-type Lmna+/+ MEFs, and 5-bromodeoxyuridine (BrdU) incorporation was higher in serum-starved Lmna-/- compared with wild-type MEFs (see Supplementary Fig. S6A,B). Conversely, ectopic HA-lamin A significantly reduced BrdU incorporation in COS-7 cells (Fig. 8A).
Figure 8.

Lamin A overexpression affects cell cycle dynamics. (A) Asynchronously growing COS-7 cells transfected with HA-lamin A expression vector were labeled for 4 h with 50 μM BrdU and then analyzed by indirect immunofluorescence microscopy to detect HA-lamin A expression (green) and BrdU incorporation (red). The graph shows the percentage of BrdU-positive cells in the indicated number of HA-lamin A-negative and HA-lamin A-positive cells from four independent transfections. (B–F) Flow cytometric analysis of asynchronously growing COS-7 cells cotransfected with HA-lamin A and either pYFP (control) (C–E) or pYFP-Fos (F). HA-lamin A was detected using monoclonal anti-HA antibody. The gray overlay in B represents HA-negative cells, and the empty overlay shows two subpopulations with low and high HA-lamin A expression that were gated separately for cell cycle distribution analysis. Graphs and percentage of cells in each phase of the cell cycle are shown as representative results of two experiments.
To further explore the effects of lamin A on cell cycle dynamics, we performed flow cytometric analysis of HA-lamin A-transfected cells. As shown in Figure 8B, we distinguished two subpopulations of transfected cells expressing either low or high HA-lamin A levels. When compared with HA-lamin A-negative cells (Fig. 8C), cell cycle distribution was essentially unchanged in cells expressing low levels of HA-lamin A (Fig. 8D), whereas cells expressing high HA-lamin A levels appeared arrested in late S or G2/M phase (Fig. 8E). Notably, YFP-Fos coexpression in high HA-lamin A-expressing cells shifted the cell cycle distribution toward mid-S phase (Fig. 8F), suggesting that inhibition of c-Fos may contribute to lamin A-dependent cell cycle arrest.
Discussion
Here we provide evidence for a novel function of A-type lamins as negative regulators of AP-1 function (e.g., DNA-binding and transcriptional activation) via their interaction with c-Fos at the NE in mammalian cells. Our in vitro and in vivo assays suggest that lamin A/C-dependent AP-1 suppression is mediated, at least in part, through inhibition of ZIP-dependent c-Fos/c-Jun heterodimerization. We have also shown that lamin A/C is necessary and sufficient to recruit c-Fos to the NE, and that ectopic lamin A causes growth arrest. Thus, we hypothesize that lamin A/C-dependent sequestration of c-Fos at the NE in mitogen-deprived cells contributes to reduced AP-1 DNA-binding activity and to the maintenance of the quiescent state. The rapid enhancement of AP-1 DNA-binding and transcriptional activation of AP-1 target genes induced by mitogen refeeding may be mostly driven by the massive accumulation of nucleoplasmic c-Fos protein, which is mostly free from the inhibitory interaction with lamin A/C. The subsequent suppression of AP-1 DNA-binding activity upon down-regulation of nucleoplasmic c-Fos protein at longer time points after serum refeeding might be facilitated by the sequestration of the residual c-Fos at the nuclear lamina. Further studies are warranted to address whether lamin A/C can also interfere with other transcription factor complexes containing AP-1 family members (e.g., Fos/NF-AT, Jun/ATF).
Our immunofluorescence studies showing c-Fos expression at the NE in MEFs and VSMCs appear to be in contrast with previous studies reporting lack of perinuclear c-Fos immunostaining in NIH 3T3 cells (Vriz et al. 1992). Several explanations may account for this apparent discrepancy. First, cell type-specific differences in c-Fos expression and localization should be considered. Indeed, c-Fos immunostaining in MEFs (Fig. 2C) and primary VSMCs (Fig. 5D–G) is almost exclusively nuclear, whereas HeLa cells express both cytoplasmic and nuclear c-Fos protein (Fig. 2A,B). Additionally, technical issues may be relevant. For example, perinuclear c-Fos detection in our studies required signal amplification; however, Vriz et al. (1992) examined c-Fos expression without any amplification step. Moreover, the anti-c-Fos antibody used in the Vriz study might not identify in immunofluorescence experiments its epitope in perinuclearly localized c-Fos. It is also noteworthy that our Western blot analyses confirmed the presence of endogenous (Fig. 5J) and ectopically expressed (Fig. 3C) c-Fos in the membrane-enriched extraction-resistant nuclear fraction.
Lamins differ from other intermediate filament proteins by having an additional six heptads in the rod domain that appear dispensable for homodimerization and filament formation but mediate heterotypic protein–protein interactions (Mical and Monteiro 1998; Schirmer et al. 2001). Our results show that three mutually exclusive leucine-rich regions of lamin A/C can interact in vitro with the bZIP of c-Fos. Likewise, the zinc finger motifcontaining transcription factor MOK2 can interact with both the coil 1b and coil 2 of lamin A/C (cf. Fig. 2C in Dreuillet et al. 2002). Because disruption of one LHR in the coil 1 of murine lamin A/C reduced binding to His6-bZIP-c-Fos (Fig. 1C), we suggest that different regions of lamin A/C can interact with the bZIP of c-Fos via hydrophobic contacts between leucine residues. Because lamin B1 failed to interact with c-Fos under conditions that yielded efficient c-Fos-lamin A/C interaction (Fig. 7) in spite of the presence of highly homologous LHRs in lamin A/C and lamin B1 (Supplementary Fig. S3), it is tempting to speculate that additional residues present between the leucines and/or structural requirements are essential for mediating the interaction between c-Fos and A-type lamins. This is, indeed, the case for the interaction among AP-1 factors (Angel and Karin 1991; Chinenov and Kerppola 2001). For example, c-Fos can heterodimerize with the various Jun proteins (c-Jun, Jun B, Jun D) but not with GCN4, Myc, or another Fos molecule, all of which contain highly homologous if not identical LHRs. Moreover, a comprehensive analysis of 2401 bZIP interactions with coiled-coil arrays revealed considerable partnering selectivity in spite of extensive sequence similarity among the bZIPs analyzed (Newman and Keating 2003). Additional studies are required to further characterize the sequence-related and structural needs for efficient c-Fos-lamin A/C heterodimer formation.
A-type lamins are mainly expressed in differentiated growth-arrested cells (Hutchison 2002), and Lmna-/- MEFs undergo growth arrest in response to DNA damage (Johnson et al. 2004). Consistent with these findings, we found that (1) c-Fos-lamin A/C colocalization at the NE predominates in starvation-synchronized quiescent cells (Fig. 2E); (2) DNA synthesis is enhanced in Lmna-/- MEFs (Supplementary Fig. S6), and lamin A overexpression inhibited cellular proliferation (Fig. 8). A high level of lamin A might hinder the disassembly of the nuclear lamina structure, a step necessary for NE breakdown and progression through mitosis (Nigg 1992; Moir et al. 2000). Because YFP-Fos coexpression in high HA-lamin A-expressing cells shifted the cell cycle distribution toward mid-S phase (Fig. 8F), we propose that inhibition of c-Fos may also contribute to lamin A-dependent cell cycle arrest. Furthermore, we cannot rule out the involvement of additional factors that might be dysregulated by elevated lamin A expression. For example, A-type lamins have been reported to interact with the transcription factors SREBP1, MOK2, BAF, and GCL and with the retinoblastoma gene product (Mancini et al. 1994; Ozaki et al. 1994; Kennedy et al. 2000; Markiewicz et al. 2002; Johnson et al. 2004; Zastrow et al. 2004; Gruenbaum et al. 2005; Mariappan and Parnaik 2005).
A growing body of evidence has highlighted a link between the nuclear lamina and a variety of human diseases termed laminopathies. Lmna mutations have been linked to the majority of laminopathies (Worman and Courvalin 2002; Gruenbaum et al. 2005), including Hutchinson-Gilford progeria syndrome (HGPS) (De Sandre-Giovannoli et al. 2003; Eriksson et al. 2003; Mounkes et al. 2003). Although A-type lamins are expressed in the majority of adult somatic cells, laminopathies are tissue-restricted diseases that affect mainly striated muscle, adipose tissue, and bone. Notably, mice with defective processing of prelamin A to lamin A as a result of homozygous Zmpste24 disruption, as well as mice lacking Lmna or carrying Lmna mutations associated with human laminopathies, show tissue-specific phenotypes markedly reminiscent of the symptoms observed in HGPS patients, including pathologies in muscle, skin, and bone (Sullivan et al. 1999; Bergo et al. 2002; Pendás et al. 2002; Nikolova et al. 2004). However, the molecular basis for this tissue specificity remains poorly defined. It therefore would be interesting to assess if any laminopathy-causing mutations can affect the interaction between lamin A/C and c-Fos or other interacting regulatory factors (e.g., SREBP1, MOK2, BAF, GCL, and the retinoblastoma gene product). Of note in this regard, alterations in both c-Fos and the A-type lamins in genetically modified mice are associated with bone defects. Overexpression of c-Fos in transgenic and chimeric mice specifically affects bone, cartilage, and hematopoietic cell development (Ruther et al. 1989). Likewise, c-Fos-null mice display altered hematopoiesis and develop severe osteopetrosis characterized by foreshortening of the long bones, ossification of the marrow space, and absence of tooth eruption (Johnson et al. 1992; Wang et al. 1992; Grigoriadis et al. 1994). Abnormal dentition and bone defects (e.g., malformation of scapulae, thin trabeculae, and decreased bone density) are also seen in homozygous knock-in mice carrying a mutation in the Lmna gene that causes the human autosomal dominant form of Emery-Dreifuss muscular dystrophy (Mounkes et al. 2003). Interestingly, bones of Lmna-/- mice appear to be more fragile (C.L. Stewart, pers. comm.), and defective processing of prelamin A to lamin A in Zmpste24-null mice correlates with spontaneous bone fractures and reduced cortical and trabecular bone volumes (Bergo et al. 2002). Whether deregulated c-Fos-lamin A/C interaction and disturbed bone development and homeostasis are causally linked merits further investigation. Future studies aimed at deciphering the structural and regulatory mechanisms governing the interaction between c-Fos and lamin A/C will not only improve our knowledge regarding the cross-talk between the nuclear architecture and the cell cycle and transcriptional machineries, but may also shed light on the tissue specificity of laminopathies.
Materials and methods
Plasmids
pBTM-c-Fos-bZIP (amino acids 123–230) was generated by subcloning the rat c-Fos cDNA encoding for the ZIP into pBTM116 (gift from Dr. P. Sanz, Instituto de Biomedicina de Valencia, Valencia, Spain). pGEX-lamin A (37–244), pGEX-lamin A (247–355), and pGEX-lamin A (356–571; gift from Dr. T. Ozaki, Chiba Cancer Center Research Institute, Chuo-Ku, Chiba, Japan) are described elsewhere (Ozaki et al. 1994). pGEX-lamin A (390–452) was generated by subcloning the PCR fragment encoding for amino acids 390–452 of rat lamin A into pGEX-4T (Pharmacia). HA-lamin A has been previously described (Raharjo et al. 2001). The AP-1-driven reporter plasmid coll73-luciferase (Webb et al. 1995) contains the sequence between -73 and +63 relative to the start of transcription of the human collagenase gene promoter (consensus AP-1 site located between -60 and -73 base pairs [bp]) (Angel et al. 1987). 4xE2F1bTATA-luciferase (gift from Dr. K. Walsh, Whitaker Cardiovascular Institute, Boston University School of Medicine, Boston, MA) was generated by insertion of a synthetic promoter containing four E2F consensus binding sites upstream of the luciferase reporter gene. pEYFP-c-Fos was generated by subcloning the full-length rat c-Fos cDNA into pEYFP-C1 (Clontech). pECFP-c-Jun was generated by subcloning the full-length human c-Jun cDNA into pECFP-C1 (Clontech). pECFP-lamin A was generated by subcloning the full-length lamin A (generated by PCR using HA-lamin A as a template) into pECFP-C1 (Clontech). The pECFP-YFP fusion plasmid was constructed by inserting YFP (generated by PCR amplification using pEYFP-C1 template) into pECFP-C1. To generate pcDNA3-HA-Lamin B1, the lamin B1 cDNA was amplified by PCR from an Invitrogen IMAGE clone and was subsequently subcloned into pcDNA3.1(-) containing an N-terminal HA tag between XhoI and AflII sites.
Antibodies
Antibodies directed against c-Fos (sc-052), lamin A/C (sc-636), GST (sc-138), p27 (sc-1641), cyclin A (sc-751), and CDK2 (sc-163-G) were purchased from Santa Cruz Biotechnology. Anti-hexahistidine antibody (clone BMG-His-1) was purchased from Roche, anti-lamin A/C (MCA-1429) from Serotec, anti-c-Fos (Ab-2) from Oncogen, anti-Nup50 (ab4005) and anti-lamin B1 (ab8982) from Abcam, and anti-BrdU conjugated to Alexa Fluor 594 from Molecular Probes. Isotype-specific HRP-coupled secondary antibodies were from Santa Cruz Biotechnology.
Yeast two-hybrid screening
The yeast strain TAT-7 (MATa ade2 his3 leu2 trp1 gal4 gal80 LYS2::lexAop-HIS3 URA3::lexAop-lacZ) was cotransformed with pBTM116-cFos-bZIP (amino acids 123–230) and a thymocyte cDNA library subcloned in pAD-Gal4-2.1 (gift from G. Gil, Institut Municipal d'Investigació Médica, Barcelona, Spain). Transformed cells were grown on SC medium lacking Leu-Trp-His. Selected colonies were transferred to plates with SC medium lacking Leu, and then a replica was made to nitrocellulose filter (Millipore type HATF 08525, 0.45 μm) to assay β-galactosidase activity. Inserts from the isolated pAD-Gal4-2.1 plasmids were sequenced on an ABIPrism 3100 Genetic Analyzer (Applied Biosystems) automated DNA sequencer using 5′-GAAGATACCCCACCAAACCC-3′ and 5′-GCGGGGTTTTTCAGTATCTA-3′ primers.
In vitro pull-down assays
Recombinant proteins were purified using glutathione-Sepharose 4B (Amersham) according to the manufacturer's instructions and were eluted with 50 mM Tris-Cl (pH 8.0). Hexahistidine-tagged c-Fos recombinant proteins (15 μg) (gift of T. Kerppola [Howard Hughes Medical Institute, Department of Biological Chemistry, University of Michigan Medical School, Ann Arbor, MI] and T. Curran [Department of Developmental Neurobiology, St. Jude's Children's Research Hospital, Memphis, TN]) were incubated in 10 mM HEPES (pH 7.6), 250 mM NaCl, 0.25% NP-40, and 5 mM EDTA (LSB buffer) supplemented with complete protease inhibitor cocktail (Roche). After 4 h of incubation on ice, glutathione-sepharose 4B was added to a final concentration of 10% and agitated at 4°C for 45 min. The beads were pelleted by centrifugation and washed three times with LSB buffer. Pellets were air-dried, resuspended in 2× Laemmli's buffer (Laemmli 1970), boiled for 5 min, and separated onto 12% SDS–polyacrylamide gels (SDS-PAGE).
Cell culture
COS-7, 293, and HeLa cells were obtained from the American Type Culture Collection. Lmna-null and littermate wild-type MEFs have been previously described (Sullivan et al. 1999). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 2 mmol/L L-glutamine (Life Technology). Primary VSMCs were isolated from adult rat aorta as previously described (Castro et al. 2003). Cells were incubated at 37°Cina5%CO2–95% O2 atmosphere. To render the cells quiescent, MEFs and VSMCs were maintained for 2–3 d in 0.5% FBS/DMEM and in mitogen-free insulin–transferrin–selenium media supplemented with 250 μM ascorbic acid and 10-11 M FeCl3 (ITC medium; Life Technology), respectively.
For proliferation assays, subconfluent serum-deprived cells were restimulated with 10% FBS to induce cell cycle re-entry. For [3H]-thymidine incorporation assays, cells were pulsed for 4 h with 1 mCi/L [3H]-thymidine (Amersham Biosciences). After washes with cold PBS, cells were lysed in 1% SDS. DNA was precipitated with 15% TCA, isolated using Whatman GF/C filter, and radioactivity incorporated into DNA was quantified in a scintillation counter (Wallac). For BrdU incorporation assays, cells were incubated for 4 h with 50 μM BrdU and analyzed by indirect immunofluorescence using Alexa Fluor 594-conjugated anti-BrdU antibody.
Subcellular fractionation
Subcellular fractionation was performed as described by Schreiber et al. (1989). Briefly, cells were washed with PBS and scraped into TEN buffer (150 mM NaCl, 1 mM EDTA, 40 mM TrisCl at pH 7.4). Cells were collected by brief centrifugation in microfuge tubes and resuspended in 10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, and 0.5 mM PMSF (Buffer A). After 15 min on ice, Nonidet NP-40 (Fluka) was added to a final concentration of 10%, and tubes were vortexed. Lysates were centrifuged at 4°C in a microfuge set at maximum speed to obtain the soluble cytoplasmic fraction (supernatant) and the nuclear pellet, which was resuspended in ice-cold Buffer C (20 mM HEPES at pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF) and agitated at 4°C for 15 min. The nuclear lysate was centrifuged for 5 min at 4°C to obtain the soluble nuclear fraction (supernatant) and the pellet containing the extraction-resistant fraction.
EMSA
Double-stranded oligonucleotides containing the AP-1 (5′-CGCTTGATGAGTCAG-3′, AP-1 site underlined) and the Sp1 (5′-ATTCGATCGGGGCGGGGCGAGC-3′, Sp1 site underlined) consensus sites were labeled with [γ-32P]dATP using polynucleotide kinase (New England Biolabs) and purified on a Sephadex G-50 column. For EMSA using soluble nuclear fractions from cultured cells (Figs. 5K, 6A,B), 10 μg of total protein was incubated with radiolabeled probe in 10 mM Tris-Cl (pH 7.5), 50 mM NaCl, 0.5 mM DTT, 0.5 mM EDTA, 4% glycerol, 1 mM MgCl2, and 50 μg/mL poly[d(I-C)]. For the EMSA of Figure 6C, 200 ng of each recombinant protein was used. His6-bZIP-c-Fos [rat amino acids 116–211; Fos(bZIP)] and His6-bZIP-c-Fos [rat amino acids 116–211; Jun(bZIP); gift of T. Curran and T. Kerppola], alone or in combination, were incubated in 50 mM TrisCl (pH 7.5), 100 mM KCl, 2 mM DTT, 0.1% NP-40, 4% glycerol, 1 mM MgCl2, and 50 μg/mL poly[d(I-C)] during 2 h on ice before addition of radiolabeled AP-1 probe. To assess the effect of recombinant lamin A, Fos(bZIP) was preincubated for 2 h with purified GST-lamin A/C 4–361 or GST. Reaction mixes were then incubated for 20 min with radiolabeled AP-1 probe followed by Jun(bZIP) addition. After 2 min, reaction mixtures were separated by native 5% PAGE and analyzed by autoradiography.
Immunoblot experiments
Purified proteins and cell lysates were separated by SDS-PAGE and transferred onto PVDF membranes (Immobilon-P; Millipore). Membranes were blocked in 5% dry milk and incubated with primary antibodies. Secondary HRP-coupled anti-isotype-specific antibodies were used for detection with enhanced chemiluminescence reagent (ECL plus; Amersham).
Coupled coimmunoprecipitation/Western blot analysis
HeLa whole cell extracts were prepared by sonication in ice-cold lysis buffer (20 mM Tris-HCl at pH 7.0, 1% NP-40, 150 mM NaCl, 10% glycerol, 10 mM EDTA, 20 mM NaF, 5 mM sodium pyrophosphate, 1 mM Na3VO4, 1 mM PMSF). Lysates were precleared with protein A agarose beads and incubated overnight with 3 μg of mouse monoclonal anti-lamin A/C (sc-636) or 3 μg of rabbit polyclonal anti-c-Fos (sc-052) antibodies. Antibody–protein complexes were isolated using 40 μL of 25% of protein A agarose beads (Sigma). Monoclonal anti-HA and normal rabbit IgG were used in negative controls for lamin A/C and c-Fos immunoprecipitations, respectively. Beads were washed twice with 1% NP-40/PBS and twice with TNE (10 mM Tris-HCl at pH 7.5, 500 mM NaCl, 1 mM EDTA). Immunoglobulin–protein complexes were eluted from beads by boiling in Laemmli's buffer (Laemmli 1970) and subjected to Western blot analysis.
Indirect immunofluorescence microscopy
Cells were grown on acid-treated glass coverslips (15–30 min incubation in 1% HCl/70% ethanol, extensive washes with tap water, two washes with 95% ethanol, air dry and autoclave). For single c-Fos and lamin A/C detection, cells were fixed for 10 min at room temperature with cold (-20°C) methanol or methanol:acetone (1:1), respectively. Cells were washed in 0.25% Tween 20 in PBS and blocked for 30 min at room temperature in 5% horse serum in PBS. Cells were incubated overnight at 4°C with anti-c-Fos antibody Ab-2 (1/100; Oncogene) or overnight at 37°C with anti-lamin A/C antibody MCA-1429 (Serotec). Detection of c-Fos and lamin A/C was achieved using biotinylated anti-rabbit or anti-mouse isotype-specific antibodies followed by Streptavidin-Texas red stain (Molecular Probes), respectively.
For double immunofluorescence microscopy, HeLa cells were fixed for 10 min at room temperature with cold methanol, washed in 0.25% Tween 20 in PBS, and blocked in 5% horse serum in PBS for 30 min at room temperature. An additional blocking step was performed using an avidin-biotin blocking kit according to the manufacturer's instructions (Master Diagnostica). Cells were incubated overnight at 4°C with anti-Fos antibody Ab-2 (1/10; Oncogene), washed extensively with PBS/0.25% Tween 20, and incubated at room temperature during 1 h with anti-lamin A/C antibody (1/100; sc-636). c-Fos was visualized with anti-rabbit biotin/streptavidine-FITC, and lamin A/C was detected using goat anti-mouse-TRITC. Nuclei were stained using Hoechst 33342. Images were acquired using a Radiance 2100 confocal microscope (Bio-Rad), with a 405-nm diode laser to excite Hoechst 33342, and 488-nm and 546-nm laser lines to excite FITC and TRITC, respectively. Fluorescence was detected through 440/20 BP, 530/15 BP, and 570 LP filters. Confocal microscope settings were adjusted to produce the optimum signal-to-noise ratio. The sequential mode was used to acquire fluorescence images to avoid any interference from overlapping fluorescence. Three-dimensional reconstructions were achieved by acquiring images with 1 μm step width (0.6 μm optical section width). Fluorescence analysis was performed using Laserpix software (Bio-Rad).
FRET confocal microscopy
COS-7 cells were cotransfected with FuGene6 (Roche) using the following combinations of plasmids to assess the level of c-Fos/lamin A interaction (Fig. 4A): pECFP-lamin A (1 μg) + pEYFP-c-Fos (1 μg) or pECFP-Lamin A (1 μg) + pEYFP (1 μg) as a negative control. To test if lamin A can inhibit the c-Fos/c-Jun interaction (Fig. 4B), cells were cotransfected with pECFP-c-Jun (0.5 μg) + pEYFP-c-Fos (0.5 μg) + pECFP-lamin A (1 μg). Negative controls consisted of cotransfections with pECFP-c-Jun (0.5 μg) + pEYFP (0.5 μg) or pECFP-lamin A (0.5 μg) + pEYFP (0.5 μg). The effect of both lamin A and lamin B1 on c-Fos/c-Jun interaction (Fig. 7A) was also tested in cells cotransfected with pECFP-c-Jun (0.5 μg) + pECFP-c-Fos (0.5 μg) in the absence or presence of either pHA-LMN A (1 μg) or pHA-LMNB1 (1 μg) (with pECFP-c-Jun [0.5 μg] + pEYFP-c-Fos [0.5 μg] as a negative control). Cotransfection of pECFP-YFP (0.5 μg) + pcDNA3 (0.5 μg) was used as positive control to calibrate the system. Images were acquired on a Leica TCS/SP2 confocal microscope with a 63× oil immersion objective (NA 1.4). An argon laser line of 458 nm was used to excite CFP (PMT window 465–505 nm) and a 514-nm line (20% laser intensity for acquisition and 65% for photobleaching) to excite YFP (PMT window 525–600 nm). FRET involves the nonradioactive transfer of energy from an excited-state donor fluorophore to a nearby acceptor. FRET studies were performed in 4% paraformaldehyde-fixed cells using the acceptor-photobleaching method (Kenworthy 2001), in which FRET is calculated as the relative increase in donor fluorescence as a result of the reduction or elimination of energy transfer when the acceptor is photobleached. Specifically, the percentage of pixels that increase their CFP fluorescence intensity after photobleaching was quantified in the region of interest using the following equation:
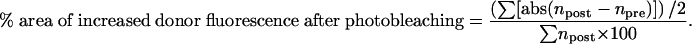 |
where abs is the absolute value, and npost and npre are the number of pixels in groups of 20 intensity values after and before photobleaching, respectively. For negative values, this parameter was considered 0.
Luciferase gene reporter assays
HeLa cells were transfected using the calcium phosphate method (Gorman et al. 1983). Transfection mixtures contained 5 μg of pMEX or pMEX-lamin A and 1.5 μg of luciferase reporter plasmid (either AP-1-dependent coll73-luciferase or E2F-dependent 4xE2F1bTATA-luciferase). Sixteen hours post-transfection, the medium was changed to 0.5% FBS. After 48 h of serum starvation, cells were left untreated or restimulated with 10% FBS. Cells were harvested in TEN buffer, and luciferase activity was measured using the luciferase gene reporter assay as recommended by the manufacturer (Roche).
Flow cytometry
Asynchronously growing COS-7 cells were cotransfected with HA-lamin A and either control pEYFP or pEYFP-c-Fos. Cells were trypsinized, washed two times in PBS, and collected by centrifugation for 10 min at 300 × g. After fixation in cold (-20°C) 70% ethanol for 2 h at 4°C, cells were washed with 1% BSA/PBS and incubated with anti-HA mouse monoclonal antibody (Sigma HA-5; diluted 1:1.000 in 1% BSA) at 4°C with agitation. After washes with 1% BSA/PBS, cells were incubated for 1 h with FITC-conjugated anti-mouse antibody (Santa Cruz Biotechnology, sc-2010; diluted 1:300 in 1% BSA), and finally with 50 μg/mL propidium iodode containing 0.1 mg/mL RNase A (for at least 10 min) to stain DNA. Labeled cells were analyzed in a Beckman-Coulter Epics XL-MCL flow cytometer. Two subpopulations with low and high HA-lamin A expression were detected and gated separately for cell cycle analysis. DNA histograms were fitted into cell cycle distributions by means of WinMDI (http://pingu.salk.edu/software.html) and Cylchred (http://www.cardiff.ac.uk/medicine/haematology/cytonetuk/documents/software.htm) public domain software.
Quantitative real-time RT–PCR (qRT–PCR)
Total RNA from primary rat VSMCs (passage 16) starved in 0.5% FBS/DMEM and stimulated with 10% FBS/DMEM was isolated using TriPure Isolation Reagent as recommended by the manufacturer (Roche). The SYBR Green RT–PCR Reagents Kit was used for qRT–PCR using a two-step protocol following the recommendations of the manufacturer (Applied Biosystems). In the first reaction, 1 μg of total RNA was reverse-transcribed into cDNA using random hexamer primers. In the second reaction, 5 μL of cDNA (undiluted for c-Fos amplification, or from a 1/10 dilution for amplification of the 18S rRNA internal control) was amplified using the following primers (designed with the Primer Express software from Applied Biosystems): c-Fos (forward), 5′-CTCAATGACCCTGAGCCCA-3′; c-Fos (reverse), 5′-TTGCTAATGTTCTTGACCGGC-3′; 18S rRNA (forward), 5′-TCGGAACTGAGGCCATGATT-3′; 18S rRNA (reverse), 5′-TGCGCCGGTCCAAGAAT-3′. Negative controls consisted of reactions without cDNA template.
Statistical analysis
Results are reported as mean ± SE. In experiments with two groups, differences were evaluated using a two-tail, unpaired Student's t-test. An analysis involving more than two groups was done using one-way ANOVA and Bonferroni's post hoc test.
Acknowledgments
We thank T. Kerppola and T. Curran for hexahistidine-tagged c-Fos and c-Jun proteins; T. Ozaki for GST-lamin A plasmids; K. Walsh for the 4xE2F1bTATA-luciferase reporter; G. Gil for the expression library used for the yeast two-hybrid screen; P. Sanz for the pBTM116 plasmid and for introducing us to the yeast two-hybrid technology; C.L. Stewart for sharing unpublished results; M. Baccarini, C. Caelles, A. Muñoz, and R. Foisner for critical reading of the manuscript; A. Navarro (supported by the Regional Government of Valencia, fellowship CTBFTS/2005/036) for technical help with this work; and M.J. Andrés-Manzano for the preparation of figures. This work was supported by grants from Instituto de Salud Carlos III (Red de Centros RE-CAVA, C03/01), from the Regional Government of Valencia (GV01-488), and from the Spanish Ministry of Education and Science and the European Regional Development Fund (SAF2004-03057). M.K. was supported by an European Union Marie Curie Programme fellowship, S.M.S.-G. by a fellowship of the Instituto de Salud Carlos III, and C.I. by fellowships from the Spanish Ministry of Science and Technology and from the CSIC-I3P Programme.
Supplemental material is available at http://www.genesdev.org.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.349506.
References
- Angel P. and Karin, M. 1991. The role of Jun, Fos, and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1072: 129-157. [DOI] [PubMed] [Google Scholar]
- Angel P., Baumann, I., Stein, B., Delius, H., Rahmsdorf, H.J., and Herrlich, P. 1987. 12-O-tetradecanoyl-phorbol-13-acetate induction of the human collagenase gene is mediated by an inducible enhancer element located in the 5′-flanking region. Mol. Cell. Biol. 7: 2256-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergo M.O., Gavino, B., Ross, J., Schmidt, W.K., Hong, C., Kendall, L.V., Mohr, A., Meta, M., Genant, H., Jiang, Y., et al. 2002. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. 99: 13049-13054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C., Díez-Juan, A., Cortés, M.J., and Andrés, V. 2003. Distinct regulation of mitogen-activated protein kinases and p27Kip1 in smooth muscle cells from different vascular beds. A potential role in establishing regional phenotypic variance. J. Biol. Chem. 278: 4482-4490. [DOI] [PubMed] [Google Scholar]
- Chinenov Y. and Kerppola, T.K. 2001. Close encounters of many kinds: Fos–Jun interactions that mediate transcription regulatory specificity. Oncogene 20: 2438-2452. [DOI] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A., Bernard, R., Cau, P., Navarro, C., Amiel, J., Boccaccio, I., Lyonnet, S., Stewart, C.L., Munnich, A., Le Merrer, M., et al. 2003. Lamin a truncation in Hutchinson-Gilford progeria. Science 300: 2055. [DOI] [PubMed] [Google Scholar]
- Dreuillet C., Tillit, J., Kress, M., and Ernoult-Lange, M. 2002. In vivo and in vitro interaction between human transcription factor MOK2 and nuclear lamin A/C. Nucleic Acids Res. 30: 4634-4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eferl R. and Wagner, E.F. 2003. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 3: 859-868. [DOI] [PubMed] [Google Scholar]
- Eriksson M., Brown, W.T., Gordon, L.B., Glynn, M.W., Singer, J., Scott, L., Erdos, M.R., Robbins, C.M., Moses, T.Y., Berglund, P., et al. 2003. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423: 293-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S. and Song, O. 1989. A novel genetic system to detect protein–protein interactions. Nature 340: 245-246. [DOI] [PubMed] [Google Scholar]
- Gorman C., Padmanabhan, R., and Howard, B.H. 1983. High efficiency DNA-mediated transformation of primate cells. Science 221: 551-553. [DOI] [PubMed] [Google Scholar]
- Grigoriadis A.E., Wang, Z.Q., Cecchini, M.G., Hofstetter, W., Felix, R., Fleisch, H.A. and Wagner, E.F. 1994. c-Fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 266: 443-448. [DOI] [PubMed] [Google Scholar]
- Gruenbaum Y., Margalit, A., Goldman, R.D., Shumaker, D.K., and Wilson, K.L. 2005. The nuclear lamina comes of age. Nat. Rev. Mol. Cell. Biol. 6: 21-31. [DOI] [PubMed] [Google Scholar]
- Hutchison C.J. 2002. Lamins: Building blocks or regulators of gene expression? Nat. Rev. Mol. Cell. Biol. 3: 848-858. [DOI] [PubMed] [Google Scholar]
- Johnson R.S., Spiegelman, B.M., and Papaioannou, V. 1992. Pleiotropic effects of a null mutation in the c-fos proto-oncogene. Cell 71: 577-586. [DOI] [PubMed] [Google Scholar]
- Johnson B.R., Nitta, R.T., Frock, R.L., Mounkes, L., Barbie, D.A., Stewart, C.L., Harlow, E., and Kennedy, B.K. 2004. A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc. Natl. Acad. Sci. 101: 9677-9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M., Zg, L., and Zandi, E. 1997. AP-1 function and regulation. Curr. Opin. Cell Biol. 9: 240-246. [DOI] [PubMed] [Google Scholar]
- Kennedy B.K., Barbie, D.A., Classon, M., Dyson, N., and Harlow, E. 2000. Nuclear organization of DNA replication in primary mammalian cells. Genes & Dev. 14: 2855-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy A.K. 2001. Imaging protein–protein interactions using fluorescence resonance energy transfer microscopy. Methods 24: 289-296. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685. [DOI] [PubMed] [Google Scholar]
- Lupas A. 1996. Coiled coils: New structures and new functions. Trends Biochem. Sci. 21: 375-382. [PubMed] [Google Scholar]
- Ma J. and Ptashne, M. 1988. Converting a eukaryotic transcriptional inhibitor into an activator. Cell 55: 443-446. [DOI] [PubMed] [Google Scholar]
- Mancini M.A., Shan, B., Nickerson, J.A., Penman, S., and Lee, W.H. 1994. The retinoblastoma gene product is a cell cycle-dependent, nuclear matrix-associated protein. Proc. Natl. Acad. Sci. 91: 418-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariappan I. and Parnaik, V.K. 2005. Sequestration of pRb by cyclin D3 causes intranuclear reorganization of lamin A/C during muscle cell differentiation. Mol. Biol. Cell 16: 1948-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markiewicz E., Dechat, T., Foisner, R., Quinlan, R.A., and Hutchison, C.J. 2002. Lamin A/C binding protein LAP2α is required for nuclear anchorage of retinoblastoma protein. Mol. Biol. Cell 13: 4401-4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mical T.I. and Monteiro, M.J. 1998. The role of sequences unique to nuclear intermediate filaments in the targeting and assembly of human lamin B: Evidence for lack of interaction of lamin B with its putative receptor. J. Cell Sci. 111 (Pt 23): 3471-3485. [DOI] [PubMed] [Google Scholar]
- Moir R.D., Spann, T.P., Lopez-Soler, R.I., Yoon, M., Goldman, A.E., Khuon, S., and Goldman, R.D. 2000. Review: The dynamics of the nuclear lamins during the cell cycle—Relationship between structure and function. J. Struct. Biol. 129: 324-334. [DOI] [PubMed] [Google Scholar]
- Mounkes L.C., Kozlov, S., Hernandez, L., Sullivan, T., and Stewart, C.L. 2003. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 423: 298-301. [DOI] [PubMed] [Google Scholar]
- Newman J.R. and Keating, A.E. 2003. Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science 300: 2097-2101. [DOI] [PubMed] [Google Scholar]
- Nigg E.A. 1992. Assembly–disassembly of the nuclear lamina. Curr. Opin. Cell Biol. 4: 105-109. [DOI] [PubMed] [Google Scholar]
- Nikolova V., Leimena, C., McMahon, A.C., Tan, J.C., Chandar, S., Jogia, D., Kesteven, S.H., Michalicek, J., Otway, R., Verheyen, F., et al. 2004. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Invest. 113: 357-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T., Saijo, M., Murakami, K., Enomoto, H., Taya, Y., and Sakiyama, S. 1994. Complex formation between lamin A and the retinoblastoma gene product: Identification of the domain on lamin A required for its interaction. Oncogene 9: 2649-2653. [PubMed] [Google Scholar]
- Pendás A.M., Zhou, Z., Cadiñanos, J., Freije, J.M., Wang, J., Hultenby, K., Astudillo, A., Wernerson, A., Rodríguez, F., Tryggvason, K., et al. 2002. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metal-loproteinase-deficient mice. Nat. Genet. 31: 94-99. [DOI] [PubMed] [Google Scholar]
- Raharjo W.H., Enarson, P., Sullivan, T., Stewart, C.L., and Burke, B. 2001. Nuclear envelope defects associated with LMNA mutations cause dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy. J. Cell Sci. 114: 4447-4457. [DOI] [PubMed] [Google Scholar]
- Ruther U., Komitowski, D., Schubert, F.R., and Wagner, E.F. 1989. c-fos expression induces bone tumors in transgenic mice. Oncogene 4: 861-865. [PubMed] [Google Scholar]
- Schirmer E.C., Guan, T., and Gerace, L. 2001. Involvement of the lamin rod domain in heterotypic lamin interactions important for nuclear organization. J. Cell Biol. 153: 479-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber E., Matthias, P., Muller, M.M., and Schaffner, W. 1989. Rapid detection of octamer binding proteins with `mini-extracts,' prepared from a small number of cells. Nucleic Acids Res. 17: 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan T., Escalante-Alcalde, D., Bhatt, H., Anver, M., Bhat, N., Nagashima, K., Stewart, C.L., and Burke, B. 1999. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 147: 913-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriz S., Lemaitre, J.M., Leibovici, M., Thierry, N., and Mechali, M. 1992. Comparative analysis of the intracellular localization of c-Myc, c-Fos, and replicative proteins during cell cycle progression. Mol. Cell Biol. 12: 3548-3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.Q., Ovitt, C., Grigoriadis, A.E., Mohle-Steinlein, U., Ruther, U., and Wagner, E.F. 1992. Bone and haematopoietic defects in mice lacking c-fos. Nature 360: 741-745. [DOI] [PubMed] [Google Scholar]
- Wang W., Chevray, P.M., and Nathans, D. 1996. Mammalian Sug1 and c-Fos in the nuclear 26S proteasome. Proc. Natl. Acad. Sci. 93: 8236-8240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb P., Lopez, G.N., Uht, R.M., and Kushner, P.J. 1995. Tamoxifen activation of the estrogen receptor/AP-1 pathway: Potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol. Endocrinol. 9: 443-456. [DOI] [PubMed] [Google Scholar]
- Worman H.J. and Courvalin, J.C. 2002. The nuclear lamina and inherited disease. Trends Cell Biol. 12: 591-598. [DOI] [PubMed] [Google Scholar]
- Zastrow M.S., Vlcek, S., and Wilson, K.L. 2004. Proteins that bind A-type lamins: Integrating isolated clues. J. Cell Sci. 117: 979-987. [DOI] [PubMed] [Google Scholar]