

Abstract
The control of deoxyribonucleotide levels is essential for DNA synthesis and repair. This control is exerted through regulation of ribonucleotide reductase (RNR). One mode of RNR regulation is differential localization of its subunits. In Saccharomyces cerevisiae, the catalytic subunit hererodimer, Rnr2/Rnr4, is localized to the nucleus while its regulatory subunit, Rnr1, is cytoplasmic. During S phase and in response to DNA damage, Rnr2-Rnr4 enters the cytoplasm, where it presumably combines with Rnr1 to form an active complex. The mechanism of its nuclear localization is not understood. Here, we report the isolation of the WTM (WD40-containing transcriptional modulator) proteins as regulators of Rnr2/Rnr4 localization. Overproduction of Wtm2 increased Rnr2/Rnr4. Deletion of WTM1, a homolog of WTM2, leads to the cytoplasmic localization of Rnr2/Rnr4, and increased hydroxyurea (HU)-resistance in mec1 mutants. Wtm1 binds Rnr2/4 complexes and release them to the cytoplasm in response to DNA damage. Forced localization of Wtm1 to the nucleolus causes Rnr2/Rnr4 complexes to relocalize to the nucleolus. Thus, Wtm1 acts as a nuclear anchor to maintain nuclear localization of Rnr2/4 complexes outside of S phase. In the presence of DNA damage this association is disrupted and Rnr2/Rnr4 become cytoplasmic, where they join with Rnr1 to form an intact complex.
Keywords: Cell cycle, DNA damage, checkpoint, regulated localization, ribonucleotide reductase
Faithful replication of DNA and repair after damage are critical to the survival of organisms. Both processes use deoxyribonucleotides (dNTPs) as building blocks to maintain genomic integrity. The rate-limiting enzyme in the production of dNTPs is the enzyme ribonucleotide reductase (RNR), which catalyzes the conversion of precursor ribonucleotide diphosphates into its deoxy-form (Elledge et al. 1993; Jordan and Reichard 1998; Eklund et al. 2001; Stubbe et al. 2001). The eukaryotic RNR is a tetrameric complex of two large regulatory subunits and two small subunits that house the di-iron center tyrosyl radical essential for biochemical activity. Mammalian cells have a single large subunit RRM1 (Brissenden et al. 1988) and two small subunits: RRM2, which is cell cycle regulated (Eriksson et al. 1984; Engstrom et al. 1985; Bjorklund et al. 1990), and p53R2, which is inducible in response to DNA damage (Nakano et al. 2000; Tanaka et al. 2000; Yamaguchi et al. 2001; Kimura et al. 2003). In Saccharomyces cerevisiae the RNR complex contains two large subunits of Rnr1 and one of each small subunit of Rnr2 and Rnr4 (Voegtli et al. 2001). Rnr4 diverged in several residues that are conserved from Escherichia coli to mammals and are important for coordinating the iron center in Rnr2 (Huang and Elledge 1997). Rnr1 (Elledge and Davis 1990), Rnr2 (Elledge and Davis 1987), and Rnr4 (Huang and Elledge 1997; Wang et al. 1997) are essential for mitotic growth. S. cerevisiae has an additional large subunit, Rnr3, whose level is highly induced after DNA damage (Elledge and Davis 1990).
Maintenance of the proper intracellular concentrations of dNTPs is important, and multiple complex regulatory mechanisms exist to achieve this homeostatic goal, including allosteric regulation, transcriptional regulation, post-translational regulation, and subcellular localization regulation. Allosteric regulation occurs through the large subunit, which alters the substrate specificity of the enzyme based on sensing individual dNTP concentrations and adjusting its substrate specificity to balance the pools as well as feedback inhibition by dATP to inhibit its activity to prevent dNTP levels from rising to inappropriate levels that are mutagenic (Mathews and Ji 1992; Chabes et al. 2003).
In response to low nucleotide levels, DNA replication stress, or DNA damage, the transcription rates of the RNR genes are rapidly elevated through activation of the DNA replication stress response pathway, which includes the MEC1-RAD53-DUN1 kinase cascade in budding yeast and the rad3-cds1-chk1 pathway in fission yeast (for reviews, see Osborn et al. 2002; Lambert and Carr 2005). Inactivation of the Crt1-Ssn6-Tup1 repressor complex is responsible for part of the transcriptional induction in S. cerevisiae (Zhou and Elledge 1992; Huang et al. 1998). Downstream effects of activation of the checkpoint pathway such as cell cycle arrest, stabilization of stalled forks, and inhibition of unfired origin helps to mitigate the toxicity of dNTP deficiencies (Elledge 1996; Desany et al. 1998; Santocanale and Diffley 1998; Lopes et al. 2001; Sogo et al. 2002).
Post-translational regulation in S. cerevisiae involves the Sml1 protein, which binds and inhibits the RNR complex. Sml1 is phosphorylated by Dun1 and degraded in S phase and also after DNA damage in a checkpoint-dependent manner to relieve RNR inhibition (Zhao et al. 1998, 2001; Zhao and Rothstein 2002; Uchiki et al. 2004). An indication of the intimate relationship between the checkpoint pathway and RNR function is revealed by the fact that overproduction of RNR1 by increasing its copy number (Desany et al. 1998; Vallen and Cross 1999) or deleting its repressor Crt1 (Huang et al. 1998) or deletion of the RNR1 inhibitor SML1 can suppress the lethality of RAD53 or MEC1 deletions (Zhao et al. 1998).
Recently it was discovered that RNR function is also regulated by subcellular localization. In both S. cerevisiae and Schizosaccharomyces pombe, the large subunit Rnr1 (SpCdc22) resides in the cytoplasm, and the small subunit complex Rnr2/4 (SpSuc22) is nuclear. In response to DNA damage or replication stress, the small subunit complex exits the nucleus and enters the cytoplasm, where it presumably joins the large subunit to form the active tetrameric complex. A small amount of small subunit complex exits the nucleus during a normal S phase, but most of this complex becomes cytoplasmic in the presence of DNA damage in S phase or G2 and this relocalization is dependent on the checkpoint signaling pathway (Yao et al. 2003).
In S. pombe, the localization is controlled through the Spd1 protein. Spd1 is degraded when cells enter S phase or in the presence of DNA damage outside of S phase. The latter is dependent on the rad3 checkpoint pathway. Deletion of spd1 causes constitutive cytoplasmic localization, while blocking its degradation leads to constitutive nuclear localization (Liu et al. 2003; Holmberg et al. 2005). There is no clear homolog of the Spd1 protein in S. cerevisiae; thus it is unclear if the mechanism is conserved. It is unclear if SpSuc22 is regulated by an anchoring mechanism or modulation of rates of import and export.
In this paper, we describe the detection of a genetic and physical association between RNR small subunits and members of the WTM (WD40-containing transcriptional modulator) family of WD40-repeat proteins previously implicated in transcriptional control. We found the WTM2 gene is a dosage suppressor of mec1Δ sml1Δ's hydroxyurea (HU) sensitivity. While overproduction of Wtm2 causes elevated levels of Rnr2/Rnr4 proteins, its homolog, WTM1, is essential for the nuclear localization of Rnr2 and Rnr4 during the G1 and G2/M stages of cell cycle, when the small subunits are normally localized in the nucleus in the absence of damage. Wtm1, like Wtm2, physically interacts with Rnr2 and Rnr4, and this interaction is decreased after cells experience genotoxic stress. We present evidence indicating that the Rnr2/Rnr4 complex is maintained in the nucleus through an anchoring mechanism in the absence of genotoxic stress and that Wtm1 is a key component of that anchor.
Results
WTM2 isolated as a dosage suppressor of mec1Δ sml1Δ's HU sensitivity
In an effort to identify additional downstream components of the MEC1 (ATR) pathway that influence recovery from replicational stress, we carried out a genetic selection to find high-expression suppressors of mec1Δ sml1Δ's HU sensitivity at 20 mM HU, a threshold concentration previously determined to give minimal background. A URA3 CEN GAL-cDNA library (Liu et al. 1992) was used to transform the mec1Δ sml1Δ strain. Transformants were plated on synthetic complete media lacking uracil (SC - URA), with glucose as the carbon source, and allowed to grow into small colonies. Colonies were then replica-plated to SC - URA media with galactose to induce the expression of the cDNA, then subsequently replica-plated to SC - URA galactose with 20 mM HU and incubated for 5 d to select for growing colonies. To ensure that the HUR phenotype of survivors were dependent on the resident plasmid and not random mutations, we tested the plasmid dependency by selecting for plasmid loss on 5-FOA plates and examining HUR. Colonies that lost their HUR phenotype upon plasmid loss had their plasmids recovered into E. coli. Rescued plasmids were retransformed into the parental mec1Δ sml1Δ strain, and shown to confer HUR. Of 33 plasmids that satisfied these criteria, all carried the gene WTM2, representing at least five independent clones based on differences in 3′ and 5′ ends. Double-mutant mec1Δ sml1Δ cells containing the GAL::WTM2 expression vector conferred HU resistance up to 30 mM HU in the presence of galactose (Fig. 1A,B).
Figure 1.

WTM2 is a dosage suppressor of mec1Δ sml1Δ's HU sensitivity. (A) mec1Δ sml1Δ cells transformed with vector alone (pRS416) or pGAL1-WTM2 CEN6 URA3 (pYDL5) plasmid were struck on galactose plates (YPGal) with 20 mM of HU. Two independent transformants are shown for each plasmid. (B) mec1Δ sml1Δ cells transformed with vector alone (pRS416) or the pGAL1-WTM2 CEN6 URA3 (pYDL5) plasmid were first grown in liquid media and serially diluted 10× and spotted on either glucose or galactose plates at the HU concentrations indicated.
Wtm2 binds Rnr2 and Rnr4
WTM2 was originally identified as a homolog of WTM1, along with the more distantly related WTM3 (UME1) gene (Pemberton and Blobel 1997). All three WTM genes encode nuclear proteins (Pemberton and Blobel 1997; Huh et al. 2003). Increased resistance to HU in mec1 mutants can be achieved in a number of ways including increasing Tel1 function (Sanchez et al. 1996), Rad53 function (Sanchez et al. 1996), Dun1 function (B. Desany and S.J. Elledge, unpubl.) or Rnr1 and Rnr3 function (Desany et al. 1998; Huang et al. 1998; Zhao et al. 1998; Vallen and Cross 1999). To look for connections to these pathways, we searched the existing databases for physical interactions with Wtm2. A search of yeast proteomic interactions at Biomolecular Interaction Network Database (BIND) revealed that Rnr2 was found associated with Wtm2 in immunoprecipitation of tagged and overproduced Wtm2 (Ho et al. 2002). The combination of genetic and physical interactions between Wtm2 and the Rnr pathway suggested that the WTM family might be regulators of the RNR pathway. To confirm that the physical interaction between Wtm2 and Rnr2 occurs with the proteins at endogenous levels, we epitope-tagged the WTM2 gene at its genomic locus with a triple-HA epitope. Immunoprecipitation of Wtm2-3HA pulled down both Rnr small subunits, Rnr2 and Rnr4, but not the large subunit Rnr1 (Fig. 2A).
Figure 2.

Wtm2 interacts with Rnr2 and Rnr4, and its overproduction leads to the activation of RNR pathway by increasing Rnr2 and Rnr4 levels. (A) Log-phase whole-cell extracts from a wild-type and WTM2-3HA-tagged strain were immunoprecipitated with or without (no-IgG) anti-HA antibodies. Input (10% of immunoprecipitation samples) and immunoprecipitation samples were separated by 10% SDS-PAGE and blotted for HA, Rnr1, Rnr2, and Rnr4. (B) mec1Δ cells containing a MEC1 URA3 plasmid (pBAD045) were transformed with empty vector (pRS415) or the pGAL1::WTM2 overexpression plasmid (pYDL6). Cells were struck on YPGal plates for 2 d, then struck onto Gal-5-FOA plates and incubated for 4 d. (C) mec1Δ sml1Δ cells carrying either an empty vector (pRS416) or the pGAL1::WTM2 overexpression vector (pYDL5) were grown to log phase in galactose SC - URA media. Total protein was precipitated by TCA and resolved by 10% SDS-PAGE and blotted with anti-tubulin, anti-Rnr2, and anti-Rnr4 antibodies. (D,E) Cells were grown as in C and processed for indirect immunofluorescence. Samples were stained with anti-Rnr2 (D) or anti-Rnr4 (E) primary antibodies and FITC-conjugated secondary antibodies. The arrow indicates the cytoplasmic rod-shaped structure. All photomicrographs comparing Rnr levels of wild-type and wtm1Δ cells were taken with the same exposure.
Wtm2 overproduction suppresses the lethality of mec1-null mutants
Several regulators of the RNR pathway have been previously shown to suppress mec1Δ lethality (Desany et al. 1998; Huang et al. 1998; Zhao et al. 1998; Vallen and Cross 1999). As Wtm2 overproduction seems to enhance the Rnr pathway as judged by enhancing HU resistance, we tested the ability of WTM2 overexpression to suppress mec1Δ lethality. A LEU2 CEN6 GAL-WTM2 plasmid, pYDL6, was transformed into a mec1Δ strain, kept alive by a copy of MEC1 on a URA3 CEN4 plasmid (pBAD045). The transformant was struck on YPGal to induce WTM2 transcription, then restruck onto 5-FOA plates with galactose to test the ability of pYDL6 to rescue mec1Δ lethality. As shown in Figure 2B, only the strain containing the GAL-WTM2 plasmid conferred growth on Gal-5-FOA media. Thus, Wtm2 overproduction suppresses the lethality of mec1Δ mutants.
Wtm2 overproduction alters the abundance and localizaton of Rnr2 and Rnr4
To investigate how the RNR pathway was activated, we examined Rnr protein levels and localization. Western blot analysis showed that mec1Δ sml1Δ cells carrying GAL-WTM2 URA3 plasmid (pYDL5) have higher levels of Rnr2 and Rnr4 relative to the same strain carrying vector alone (Fig. 2C). Rnr2 and Rnr4 reside primarily in the nucleus. Indirect immunofluorescence also supported these observations and showed elevated levels of Rnr2 and Rnr4 in both the nuclear and cytoplasmic compartments in cells overproducing WTM2 (Fig. 2D,E). In significant fractions of these cells, Rnr2 and Rnr4 staining appear to be cytoplasmic and rod-like structures (Fig. 2D, arrow; data not shown), indicating a possible novel cytoplasmic structure. Thus, Wtm2 overproduction alters both the levels and localization of Rnr2 and Rnr4 to confer HU-resistance to mec1Δ mutants.
Nuclear localization of Rnr2 and Rnr4 is defective in the wtm1Δ mutant
As Wtm2 acts as a positive regulator of Rnr2 and Rnr4 function, we anticipated that deletion of WTM2 might cause a reduction in Rnr function. Unexpectedly, deletion of WTM2 in haploid wild-type cells resulted in no observable phenotypes. These include any changes in the abundance or localization of Rnr2/Rnr4 or any detectable differences in sensitivity of wtm2Δ mutants to damaging agents such as HU, IR, or MMS (data not shown). Since Wtm2's homolog, Wtm1, shares 61% identity in protein sequence and is approximately fivefold to 10-fold higher in abundance (data not shown), we reasoned that Wtm1 might play a more dominant role and mask any significant phenotypes of wtm2-null mutants. Consistent with this possibility we observed that, unlike wild-type cells, which, when arrested in G1, showed the expected nuclear localization of Rnr2 and Rnr4 (Yao et al. 2003), the wtm1Δ mutant shows diffuse Rnr2 and Rnr4 localization throughout the cell (Fig. 3A,B). To independently confirm these findings we performed subcellular fractionation of the same yeast samples into nuclear and cytoplasmic fractions. These experiments yielded similar findings: The Rnr2 and Rnr4 subunits are predominantly nuclear in wild type, while primarily cytoplasmic in a wtm1Δ mutant (Fig. 3C). The integrity of the subcellular fractionation was confirmed by blotting for nuclear o-link polysaccharide pore protein and cytoplasmic phosphoglycerate kinase. The loss of nuclear Rnr2 and Rnr4 is not a cell cycle artifact since cells showed similar defects when examined in log phase or G2/M (data not shown). This defect is also not due to checkpoint activation driving the Rnr2 and Rnr4 out of the nucleus, since double mutants with the checkpoint kinase pathway such as mec1Δ wtm1Δ or the dun1Δ wtm1Δ also show the same defect (Fig. 3D). These observations are consistent with Wtm1 functioning as a nuclear anchor for Rnr2 and Rnr4.
Figure 3.

Nuclear localization of Rnr2 and Rnr4 is defective in wtm1Δ mutants. (A,B). Wild-type and wtm1Δ cells were first synchronized in G1 by α-factor, then stained with DAPI for DNA and anti-Rnr2 antibodies (A) and anti-Rnr4 antibodies (B) for immunofluorescence. (C) Wild-type and wtm1Δ cells were first synchronized in G1 by α-factor and fractionated into different subcellular compartments. Proteins extracted from whole-cell (W), cytoplasmic fraction (C), and nuclear fraction (N) were resolved by 10% SDS-PAGE and blotted with anti-Rnr2, anti-nuclear pore o-linked glycoprotein (Nuc Gly), and anti-phosphoglycerate kinase antibodies. (D) Log-phase wild-type, mec1Δ sml1Δ wtm1Δ, and dun1Δ wtm1Δ cells grown in YPD were stained with primary anti-Rnr2 and secondary FITC antibodies for immunofluorescence.
Deletion of WTM1 increases HU resistance in a mec1Δ sml1Δ background
It has been shown previously that Rnr1 is constitutively cytoplasmic while Rnr2 and Rnr4 are only cytoplasmic after DNA damage or during replication, when the demand for dNTPs increases (Yao et al. 2003). Given that the Rnr2 and Rnr4 are constitutively cytoplasmic in the wtm1Δ mutant, we predict that these cells should be more resistant to the effect of HU compared with wild type. Therefore, we deleted the WTM1 gene in a mec1Δ sml1Δ background in order to avoid checkpoint activation and to separate the effect of transcriptional induction of RNR genes from the localization of Rnr2 and Rnr4. As shown in Figure 4A, deletion of WTM1 has no effect on cell growth in the absence of HU, but significantly improves the viability of the mec1 strain on 2-mM HU plates, compared with a TRP1-integrated control or the wtm2Δ mutant. This phenotype is diminished as the concentration of HU increases to 3 mM, and completely lost at HU concentration of 5 mM or above (Fig. 4A; data not shown). The levels of Rnr2—the target of HU inhibition on Rnr function—are unchanged between the mec1 control, wtm1Δ, and wtm2Δ cells (Fig. 4B), ruling out the possibility that the increase in HU resistance in wtm1Δ is due to an increase in Rnr2 levels. In addition, the increase in viability of the mec1 mutant by WTM1 deletion is only observed in the presence of HU, and not with UV or IR treatment (data not shown), presumably because these other treatments require additional MEC1-dependent functions.
Figure 4.
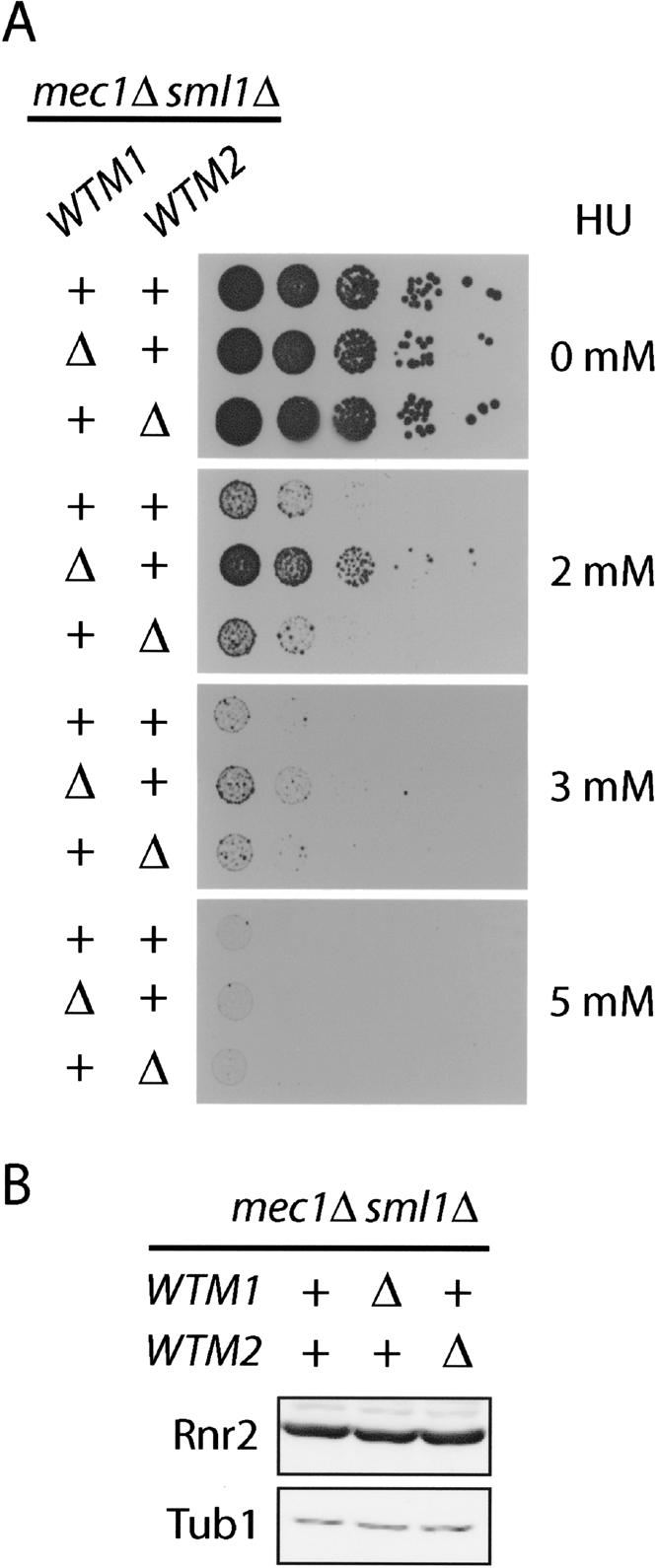
Deletion of WTM1 increases HU resistance of mec1Δ sml1Δ mutants. (A) mec1Δ sml1Δ strains deleted for the WTM1 locus (wtm1Δ::TRP1) or WTM2 locus (wtm2Δ::TRP1) were serially diluted 10-fold and spotted on YPD plates with increasing concentrations of HU and grown for 2 d. A mec1Δ sml1Δ strain serves as a control. (B) Log-phase protein extracts of strains used in part A were resolved by 10% SDS-PAGE and blotted with anti-Rnr2 and anti-Tub1 antibodies as a loading control.
Wtm1 interacts with Rnr2 and Rnr4 in a DNA damage-regulated manner
To further investigate the role of Wtm1 in the regulation of Rnr2 and Rnr4, we tested for a physical interaction between Wtm1 and the small subunits of RNR. To this end, we tagged the genomic copy of WTM1 under its endogenous promoter with 3MYC epitope at the N terminus (Schneider et al. 1995). The 3MYC-Wtm1 protein was immunoprecipited and blotted for Rnr2 and Rnr4. Rnr2 and Rnr4 coimmunoprecipitated with 3MYC-Wtm1 while control antibodies failed to bring down either protein (Fig. 5A).
Figure 5.

Wtm1 interacts with Rnr2 and Rnr4 in a DNA damage-regulated manner. (A) Protein extracts from log-phase wild-type and 3MYC-WTM1 strains were immunoprecipitated with anti-MYC or anti-HA (control) antibodies. Input (10% of immunoprecipitation) and immunoprecipitation samples were resolved by 10% SDS-PAGE and blotted with anti-MYC, anti-Rnr2, and anti-Rnr4 antibodies. (B) 3MYC-WTM1-tagged and wild-type cells were arrested in G1 by α-factor, then half of each sample was maintained in α-factor (αF), while the other half was release into 200 mM HU. Samples were processed for indirect immunofluorescence and stained with anti-MYC and anti-Rnr2 antibodies, followed by Alexor-488 and Cy3-conjugated secondary antibodies. (C) Cells were grown as in B. Protein extracts were immunoprecipitated with anti-MYC antibodies. Input (equivalent to 10% immunoprecipitation) and immunoprecipitation samples were resolved by 10% SDS-PAGE and blotted with anti-MYC and anti-Rnr2 antibodies. (D) Wild-type cells arrested in G2/M by nocodazole were treated with or without phleomycin (100 μg/mL) for 90 min while maintained in nocodazole, then were stained with anti-Rnr2 antibodies for immunofluorescence. (E) Cells were grown and treated as in D. Wild-type and tagged 3MYC-WTM1 protein extracts were prepared by TCA precipitation, resolved by 8% SDS-PAGE, and blotted with anti-Rad53 antibodies. (F) Cells were grown and treated as in D, then fixed with formaldehyde and immunoprecipitated with anti-MYC antibodies, followed by the reversal of cross-linking. Input (equivalent to 10% immunoprecipitation) and immunoprecipitation samples were resolved by 10% SDS-PAGE and blotted with anti-MYC, anti-Rnr2, and anti-Rnr4 antibodies.
To determine if the physical interactions between Rnr2 and Wtm1 are consistent with the observed nuclear-to-cytoplasmic translocation of Rnr2 and Rnr4 under genotoxic stress, we performed immunoprecipitations with protein extracts obtained from cells treated with HU. Cells were first arrested in G1 by α-factor. Half of the cells were released from G1 into HU for 90 min, while the other half were maintained in G1 arrest for the 90 min. Binding between Wtm1 and Rnr2/Rnr4 decreased in cells released from G1 into HU compared with those maintained in G1 even though Rnr2 levels were slightly higher in the HU-treated cells (Fig. 5C). This is consistent with our immunofluorescence observations, which showed Wtm1 and Rnr2 colocalized in the nucleus of the G1 cells, while Rnr2 translocated to the cytoplasm away from the nuclear Wtm1 in those cells released into HU (Fig. 5B).
To confirm that the regulation of Rnr2/Rnr4 localization and Wtm1 binding also occurs outside of S phase during DNA damage, we treated G2/M-arrested cells with phleomycin, which is known to cause double-stranded breaks and activate the DNA damage response. Cells were arrested and maintained in G2/M by nocodazole throughout the course of the experiment. Half of the samples were treated with phleomycin (100 μg/mL) for 90 min while the other half were maintained in G2/M without treatment. Coimmunoprecipitation data showed the interaction between Wtm1 and Rnr2/Rnr4 decreased for cells treated with phleomycin compared with the nontreated control (Fig. 5F), even though Rnr2 levels rose as expected due to the activation of Rad53 (Fig. 5E) and the transcriptional response to DNA damage (Allen et al. 1994; Nakada et al. 2003). This is consistent with the immunofluorescence data, which showed that Rnr2 becomes cytoplasmic after phleomycin treatment while Wtm1 stays nuclear (Fig. 5D; data not shown). The above results demonstrated that the physical interactions between Wtm1 and Rnr2/Rnr4 are consistent with the observed localization of these proteins and with the notion that Wtm1 may act as an anchor and regulator of Rnr function.
Relocating Wtm1 to the nucleolus can sequester Rnr2 and Rnr4 in the nucleolus
Although the above observations supported the role of Wtm1 as a nuclear anchor for Rnr2/Rnr4, we cannot rule out the possibility that Wtm1 functions as a nuclear importer for the RNR small subunits or as a transcriptional regulator of the actual anchor. To distinguish between these possibilities, we reasoned that if the nuclear anchor model is correct, then relocating Wtm1 to a new cellular compartment should recruit Rnr2 and Rnr4 into that compartment. To this end, we fused a 3MYC-WTM1 construct to the C terminus of the NOP1 gene, an essential fibrillarin protein that is exclusively localized to the nucleolus (Schimmang et al. 1989). This fusion was expressed in the wtm1Δ haploid background to ensure the only copy of WTM1 was the fusion protein (Fig. 6A). A truncated version of the fusion construct bearing the full-length Nop1 and 3MYC, but truncated for most of the WTM1 gene, was identified fortuitously by Western blotting during screening and served as a negative control (Fig. 6B). Indirect immunofluorescence confirmed that the Nop1-3MYC-Wtm1 protein is correctly localized to the nucleolus (Fig. 6C). Interestingly, by relocating Wtm1 from the nucleus to the nucleolus, Rnr2 becomes primarily localized to the nucleolus as well (Fig. 6C). The strain with truncation of the WTM1 gene fused to Nop1 did not recruit Rnr2 to the nucleolus. Furthermore, with the full-length NOP1-3MYC-WTM1 strain, after treatment with HU, Rnr2 disappeared from the nucleolus, while the Nop1-3MYC-Wtm1 fusion protein remained nucleolar, indicating that the regulation of the anchor is still intact even after the relocation to the nucleolus (Fig. 6C, bottom panel). For comparison, the Wtm1 and Rnr2 staining of a 3MYC-WTM1 strain are shown and appear to overlap with each other as well as with DAPI (Fig. 6C, top panel). Together these results argue that Wtm1 is a component of the nuclear anchor, rather than an importer or transcriptional regulator for the Rnr small subunits Rnr2 and Rnr4.
Figure 6.

Relocating Wtm1 to the nucleolus recruits Rnr2 and Rnr4 to the nucleolus. (A) A 3MYC-WTM1-his5+ fragment, constructed by fusion PCR, was transformed into the wtm1Δ haploid strain and recombined to the 3′ end of the endogenous NOP1 locus on chromosome IV to generate the NOP1-3MYC-WTM1 fusion. (B) Western blot analysis of protein extracts from the transformed cells blotted with anti-Nop1 antibodies shows the wild-type control (WT), truncated fusion (Trunc), and full-length fusion protein (FL). (C) For comparison, the top panel shows the localization of endogenous Wtm1 and Rnr2 relative to DAPI by immunofluorescence. wtm1Δ, NOP1-3MYC-WTM1, and truncation mutants were grown to log phase, and cells were stained with DAPI (blue), anti-Nop1/Alexor-488 (green), and anti-Rnr2/Cy3 (red) antibodies. The bottom panel shows immunofluorescence of log-phase NOP1-3MYC-WTM1 cells released into 200 mM HU for 2 h, then stained with DAPI, anti-Nop1/Alexor-488 (green), and anti-Rnr2/Cy3 (red) antibodies.
Discussion
Control of dNTP synthesis is critical for the fidelity of DNA replication and repair. To achieve this cells have evolved a multilayered regulatory strategy to control the activity of the key biosynthetic enzyme ribonucleotide reductase to ensure that dNTPs are made at the right time at the right levels. During a normal cell cycle the subunits of RNR are regulated transcriptionally and by regulated subcellular localization and inhibitor destruction. Outside of S phase, in the presence of DNA damage, checkpoint pathways regulate transcription, inhibitor stability, and cellular localization. Of particular interest has been the subcellular localization in which the small subunits are localized in the nucleus and are induced during S phase or in the presence of DNA damage to enter the cytoplasm, where they can combine with the large subunits to form an active tetrameric complex. Our study addresses the mechanism of this nuclear localization.
Wtm1 is part of a nuclear anchor for Rnr2 and Rnr4
We initially identified members of the Wtm (WD40-repeat transcriptional modulator) protein family as a suppressor of the sensitivity of mec1Δ sml1Δ deletion cells to HU. We found both Wtm1 and Wtm2 physically interact with the Rnr small subunits Rnr2 and Rnr4. Furthermore, deletion of WTM1 leads to the loss of nuclear localization of Rnr2 and Rnr4 in all stages of cell cycle. This observation could be explained by three distinct models. First, deletion of Wtm1 could result in a constitutive DNA damage signal to activate the delocalization mechanism for Rnr2 and Rnr4. Second, Wtm1 could act as part of the nuclear import/export machinery to increase the nuclear import of the Rnr small subunits or decrease their export from the nucleus. Third, Wtm1 could act as a component of an anchor that binds and sequesters Rnr2 and Rnr4 inside the nucleus.
We eliminated the first model because deletion of WTM1 did not activate the checkpoint constitutively (our unpublished data) and the WTM1 deletion still mislocalized Rnr2 and Rnr4 in a checkpoint-defective mutant. Evidence against the second model was the fact that we had observed no change in Wtm1 localization after genotoxic stress caused by HU or phleomycin treatment, suggesting that it is at least unlikely to be a nuclear shuttle in the import process of Rnr2 and Rnr4. This alone does not rule out model 2. However, we obtained very strong support for model 3 from the observation that relocalization of Wtm1 to a new subcellular compartment, the nucleolus, caused relocalization of the Rnr2/Rnr4 complex to the nucleolus. Importantly, nucleolar localized Wtm1 maintained its ability to allow Rnr2/Rnr4 relocalization to the cytoplasm in response to DNA damage. Thus, based on this data, we propose Wtm1 to be a nuclear anchor for Rnr small subunit Rnr2 and Rnr4, although we cannot rule out a role in nuclear import. Consistent with this, deletion of WTM1 significantly increases the viability of mec1 mutants in HU.
The role of Wtm2
The role of WTM2, the gene originally isolated as a high-dosage suppressor of mec1Δ sml1Δ's HU sensitivity, is less clear. Previous studies have identified Wtm1 as an abundant component of a nuclear protein complex, and Wtm2 as a closely related but less abundant homolog with physical interactions with Wtm1 (Pemberton and Blobel 1997). This suggests that Wtm2 is likely to play a supportive role compared with the more abundant and possibly functionally dominant Wtm1, explaining the lack of obvious change in nuclear localization of Rnr small subunits in a wtm2-null. Alternatively, Wtm2 may function independently as a nuclear anchor for the Rnr small subunits, releasing them under an as yet unidentified condition when dNTPs are required. Since Wtm2 can physically associate with Rnr2 and Rnr4, we believe its fortuitous isolation is likely due to the overexpressed Wtm2 binding and sequestering Rnr2 and Rnr4, increasing their overall and cytoplasmic abundance by affecting their stability. Why Wtm2 overproduction causes cytoplasmic localization and increased levels of Rnr2 and Rnr4 is not understood, but preliminary experiments indicate that the increased protein levels do not appear to be due to increased transcription (Y. Lee and S. Elledge, unpubl.).
Regulation of Rnr-Wtm1 association
The precise nature of the physical interactions between Wtm1 and Rnr2/Rnr4 is not yet known. Although recombinant Wtm proteins and Rnr small subunits failed to bind in vitro, this may be due to problems of folding of WD40 protein. Many WD40 proteins require the function of the CCT chaperone (Valpuesta et al. 2002; Siegers et al. 2003), which has also been shown to bind overproduced Wtm1 in yeast extracts (Ho et al. 2002). CCT is absent in the systems we employed to produce the recombinant protein. We speculate that the WD40-repeat domain may provide a scaffold for protein-protein interaction, and the Rnr small subunits may then bind to this scaffold directly. However, we cannot rule out the possibility that there are additional proteins missing in the system that are required to facilitate binding between Wtm1 and the Rnr2/Rnr4 complex.
How the regulation in response to DNA damage impinges on the Rnr2/4 localization remains to be determined. We have observed that Wtm1 remains localized in the nucleus after genotoxic stress. We have further observed that after HU treatment or phleomycin treatment, the physical interaction between Wtm1 and the Rnr small subunits is significantly diminished, consistent with the release of Rnr2 and Rnr4 into the cytoplasm. Among many possible modifications, we believe that the most likely mechanism is through protein phosphorylation by one of the checkpoint kinases such as Dun1, the most distal kinase in the checkpoint cascade required for damage-induced delocalization. This modification would then result in the release of the Rnr small subunits into the cytoplasm, where they can then combine with cytoplasmic Rnr1 to form an active complex. How this is accomplished is not known and it will be important to identify the relevant substrate(s) of Dun1 in this system.
Conservation of RNR localization as a strategy for controlling dNTP levels
While the mechanisms seem to vary, all organisms in which it has been examined in detail control the localization of Rnr subunits in response to DNA damage. In mammals, damage-induced p53R2 seems to move to the nucleus in response to damage. However, its fungal counterparts in S. pombe and S. cerevisiae move in the opposite direction to join the large subunits in the cytoplasm. The differential localization between species might reflect the different amounts of DNA each has to replicate. In yeast, sufficient dNTPs may be able to enter the nucleus from the cytoplasm at a rate sufficient to replicate their chromosomes, while their larger cousins might require a more direct nuclear source of dNTPs to accomplish optimal replication rates. However, all three species control localization to optimize it in response to damage.
Despite the apparently similar strategies employed in the two yeasts, it is unclear at this point whether they perform this regulation in the same fashion mechanistically. The Wtm gene family in S. cerevisiae appears to be unique, and other than the more closely related Candida glabrata and Kuveromyces lactis species, no clear homologs are identified by BLAST analysis in other species including S. pombe. Similarly, Spd1, a Rnr inhibitor shown to be involved in maintaining nuclear localization of the Rnr small subunits in S. pombe, does not appear to have a readily identifiable structural homolog in S. cerevisiae. Furthermore, despite the fact that S. cerevisiae has two small proteins involved in Rnr regulation, Sml1 and Hug1, deletion of either of these does not result in the loss of Rnr2/4 nuclear localization as seen in the S. pombe spd1 deletion. Thus the overall conservation of the mechanism in budding and fission yeast is in question.
Consistent with the absence of a clear Spd1 homolog, the machinery for degradation of Spd1 in response to DNA damage, Cul4 and Ddb1, does not appear to be conserved in S. cerevisiae. However, it is interesting to note that two Ddb1-interacting proteins, Ddb2 and Csa, contain WD40 repeats like the Wtm family. Whether this is purely coincidental or has functional significance remains to be seen. Since the control of the Rnr pathway is quite complex, it is possible that organisms may use unique regulatory mechanisms fine-tuned to their specific life cycle and environmental needs. Yet it is also possible that this anchor system is conserved and that counterparts like the Wtm family in S. pombe and Spd1 in S. cerevisiae have yet to be uncovered, and are simply parts of a larger, currently unknown, anchoring complex. These questions and the mechanism of both S-phase delocalization and DNA damage-induced delocalization remain important areas of future investigation.
Materials and methods
Media and growth condition
Cells were grown at 30°C unless stated otherwise. SC media were made according to Kaiser et al. (1994), and the carbon sources used were either glucose or galactose as indicated. G1 arrest with αF and G2/M arrest with nocodazole were done as previously described (Desany et al. 1998). The phleomycin concentration used was 100 μg/mL and the treatment was as described by Nakada et al. (2003).
Isolation of the WTM2 gene
The strain YDL110 (mec1Δ::his5+ sml1Δ::his5+) is grown in YPD and transformed with a URA CEN cDNA library (Liu et al. 1992) using the lithium acetate method. Transformants were first plated on SC-URA GLU (glucose) for 2 d, then replica plated to SC-Ura GAL (galactose) for 1 d to induce the expression of transcript. Transformants were subsequently replica plated to SC-URA GAL 20-mM HU plates, and surviving colonies were picked after 5 d. Positive clones that were confirmed on 20-mM HU plates were tested for plasmid dependency by the loss of HU resistance after 5-FOA selection and by the ability of the rescued plasmids to confer the HU-resistance phenotype to newly transformed YDL110 (mec1Δ::his5+ sml1Δ::his5+).
Indirect immunofluorescence
The protocol for immunofluorescence is as described by Yao et al. (2003). Cells on slides were incubated with primary antibodies (1:5000 to 1:10,000 for rabbit anti-Rnr2 and anti-Rnr4 [Yao et al. 2003]; 1:100 to1:500 for anti-MYC; 1:100 to 1:500 for anti-Ha in PBS with 5 mg/mL BSA) at room temperature for 3 h, washed, then incubated with secondary antibodies at room temperature for 2 h at 1:1000 for Cy3-conjugated goat anti-rabbit antibodies, 1:1000 for Cy3-conjugated goat anti-mouse antibodies, and 1: 1000 for Alexor-488-conjugated goat anti-mouse antibodies. Slides were examined and photographed on a Zeiss Axioplan II microscope.
Subcellular fractionation
The fractionation protocol employed was adapted and modified from Rieder and Emr (2000). Yeast cells (100 OD600) were harvested, washed, and resuspended into 2 mL of cell suspension buffer (100 mM PIPES-KOH at pH 9.4, 10 mM DTT) and incubated at 30°C for 10 min. Cells are then washed and resuspended in 2 mL of zymolyase buffer (50 mM Tris-Cl at pH 7.5, 10 mM MgCl2, 1.2 M sorbitol, 1 mM DTT), and digested with 40 μL of zymolyase 100T (5 mg/mL) at 30°C until >90% of cells lysed in fresh water (∼20-30 min). Cells were washed twice with zymolyase buffer, then resuspended in 4 mL of ficoll buffer (18% w/v Ficoll-400, 10 mM Tris-Cl at pH 7.5, 20 mM KCl, 5 mM MgCl2, 3 mM DTT, 1 mM EDTA, 1 mM PMSF, protease inhibitor cocktail; Roche) at room temperature. Cells were broken with 30 moderate to slow strokes in a Dounce homogenizer with a loose pestle. Nonlysed cells were removed by spinning at 3000g for 15 min. The lysate was then spun at 20,000g for 15 min. The resulting supernatant was labeled as cytosol and the pellet was labeled as nuclei.
Western blotting
Samples were prepared using the TCA precipitation method as described by Longhese et al. (1997). Proteins pellets were resuspended in Tris buffer and SDS loading buffer and boiled, and the solubilized fraction was resolved by SDS-PAGE. Proteins were transferred to Protran membrane (Schleicher and Schuell) and detected using primary polyclonal anti-Rnr2, anti-Rnr4, or monoclonal anti-HA, anti-MYC antibodies. Proteins were detected with secondary HRP antibodies.
Test for suppression of mec1Δ lethality
Overexpressed genes were tested for their ability to suppress mec1Δ lethality essentially as described by Desany et al. (1998). Briefly, a LEU2 CEN4 plasmid carrying the gene of interest is transformed into Y2352, plated on SC-LEU GAL media, and tested for the ability to form colonies on plates of SC GLU media supplemented with 5-FOA.
Immunoprecipitation and protein cross-linking and reversal
Immunoprecipitations were performed as describe in Bonifacino et al. (1999). The protein cross-linking protocol was modified from Parsons et al. (2005). Briefly, yeast cells were fixed with 1% formaldehyde for 10 min at 37°C, then the formaldehyde was quenched by addition of 125 mM glycine. Cells were washed twice with 25 mM HEPES (pH 7.5) buffer with 1% Triton, 200 mM NaCl, 1 mM PMSF, 0.5 mM Na3VO4, 30 mM NaF, and protease inhibitor cocktail (Roche), then processed for immunoprecipitation.
Strains and plasmids
For a complete list of yeast strains and plasmids used for this work, see Table 1.
Table 1.
Strains and plasmids used in this study
| Strain | Genotype | Source |
|---|---|---|
| Y300 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-100 | Allen et al. 1994 |
| Y2350 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-101 mec1Δ::his5+ sml1Δ::his5+ | This study |
| Y2351 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-100 WTM2-3HA::his5+ | This study |
| Y2352 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-100 mec1Δ::his5+ + pBAD045 | This study |
| Y2353 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-100 mec1Δ::his5+ + pBAD045 + pRS415 | This study |
| Y2354 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-100 mec1Δ::his5+ + pBAD045 + pYDL5 | This study |
| Y2355 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-101 mec1Δ::his5+ sml1Δ::his5+ + pRS416 | This study |
| Y2356 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-101 mec1Δ::his5+ sml1Δ::his5+ + pYDL5 | This study |
| Y2357 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-100 wtm1Δ::his5+ | This study |
| Y2358 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-101 mec1Δ::his5+ sml1Δ::his5+ wtm1Δ::TRP1 | This study |
| Y2359 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-100 dun1Δ::HIS3 wtm1Δ::TRP1 | This study |
| Y2360 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-100 3MYC-WTM1 | This study |
| Y2361 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-100 NOP1-3MYC-WTM1::his5+ | This study |
| Y2362 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-100 NOP1-3MYC-WTM1(truncated)::his5+ | This study |
| Y2363 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-101 mec1Δ::his5+ sml1Δ::his5+ wtm1Δ::TRP1 | This study |
| Y2364 | MATa trp1-1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-101 mec1Δ::his5+ sml1Δ::his5+ wtm2Δ::TRP1 | This study |
| Y2365 | MATa TRP1 ura3-1 his3-11,15 leu2-3,112 ade2-1 can1-101 mec1Δ::his5+ sml1Δ::his5+ | This study |
| Plasmid | Relevant markers | Source |
|---|---|---|
| pRS416 | AprURA3 | Sikorski and Hieter 1989 |
| pRS415 | AprLEU2 | Sikorski and Hieter 1989 |
| pBAD045 | AprURA3 MEC1 | Desany et al. 1998 |
| pYDL5 | AprGAL1-WTM2 URA3 | This study |
| pYDL6 | AprGAL1-WTM2 LEU2 | This study |
Acknowledgments
We thank members of the Elledge laboratory for reagents and helpful discussion, M. Huang for sharing unpublished results, and the Stubbe laboratory for their generous gift of Rnr1, Rnr2, and Rnr4 antibodies. This work was supported by a grant from the NIH to S.J.E. S.J.E. is an investigator with the Howard Hughes Medical Institute.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.1380506.
References
- Allen J.B., Zhou, Z., Siede, W., Friedberg, E.C., and Elledge, S.J. 1994. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes & Dev. 8: 2401-2415. [DOI] [PubMed] [Google Scholar]
- Bjorklund S., Skog, S., Tribukait, B., and Thelander, L. 1990. S-phase-specific expression of mammalian ribonucleotide reductase R1 and R2 subunit mRNAs. Biochemistry 29: 5452-5458. [DOI] [PubMed] [Google Scholar]
- Bonifacino J.S., Dell'Angelical, E.C., and Springer, T.A. 1999. Immunoprecipitation. In Current protocols in protein science (eds. J.E. Coligan et al.), Unit 9.8. Wiley Interscience, Hoboken, NJ. [DOI] [PubMed]
- Brissenden J.E., Caras, I., Thelander, L., and Francke, U. 1988. The structural gene for the M1 subunit of ribonucleotide reductase maps to chromosome 11, band p15, in human and to chromosome 7 in mouse. Exp. Cell Res. 174: 302-308. [DOI] [PubMed] [Google Scholar]
- Chabes A., Georgieva, B., Domkin, V., Zhao, X., Rothstein, R., and Thelander, L. 2003. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 112: 391-401. [DOI] [PubMed] [Google Scholar]
- Desany B.A., Alcasabas, A.A., Bachant, J.B., and Elledge, S.J. 1998. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes & Dev. 12: 2956-2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund H., Uhlin, U., Farnegardh, M., Logan, D.T., and Nordlund, P. 2001. Structure and function of the radical enzyme ribonucleotide reductase. Prog. Biophys. Mol. Biol. 77: 177-268. [DOI] [PubMed] [Google Scholar]
- Elledge S.J. 1996. Cell cycle checkpoints: Preventing an identity crisis. Science 274: 1664-1672. [DOI] [PubMed] [Google Scholar]
- Elledge S.J. and Davis, R.W. 1987. Identification and isolation of the gene encoding the small subunit of ribonucleotide reductase from Saccharomyces cerevisiae: DNA damage-inducible gene required for mitotic viability. Mol. Cell. Biol. 7: 2783-2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 1990. Two genes differentially regulated in the cell cycle and by DNA-damaging agents encode alternative regulatory subunits of ribonucleotide reductase. Genes & Dev. 4: 740-751. [DOI] [PubMed] [Google Scholar]
- Elledge S.J., Zhou, Z., Allen, J.B., and Navas, T.A. 1993. DNA damage and cell cycle regulation of ribonucleotide reductase. Bioessays 15: 333-339. [DOI] [PubMed] [Google Scholar]
- Engstrom Y., Eriksson, S., Jildevik, I., Skog, S., Thelander, L., and Tribukait, B. 1985. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J. Biol. Chem. 260: 9114-9116. [PubMed] [Google Scholar]
- Eriksson S., Graslund, A., Skog, S., Thelander, L., and Tribukait, B. 1984. Cell cycle-dependent regulation of mammalian ribonucleotide reductase. The S phase-correlated increase in subunit M2 is regulated by de novo protein synthesis. J. Biol. Chem. 259: 11695-11700. [PubMed] [Google Scholar]
- Ho Y., Gruhler, A., Heilbut, A., Bader, G.D., Moore, L., Adams, S.L., Millar, A., Taylor, P., Bennett, K., Boutilier, K., et al. 2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415: 180-183. [DOI] [PubMed] [Google Scholar]
- Holmberg C., Fleck, O., Hansen, H.A., Liu, C., Slaaby, R., Carr, A.M., and Nielsen, O. 2005. Ddb1 controls genome stability and meiosis in fission yeast. Genes & Dev. 19: 853-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M. and Elledge, S.J. 1997. Identification of RNR4, encoding a second essential small subunit of ribonucleotide reductase in Saccharomyces cerevisiae. Mol. Cell. Biol. 17: 6105-6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M., Zhou, Z., and Elledge, S.J. 1998. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 94: 595-605. [DOI] [PubMed] [Google Scholar]
- Huh W.K., Falvo, J.V., Gerke, L.C., Carroll, A.S., Howson, R.W., Weissman, J.S., and O'Shea. E.K. 2003. Global analysis of protein localization in budding yeast. Nature 425: 686-691. [DOI] [PubMed] [Google Scholar]
- Jordan A. and Reichard, P. 1998. Ribonucleotide reductases. Annu. Rev. Biochem. 67: 71-98. [DOI] [PubMed] [Google Scholar]
- Kaiser C., Michaelis, S., and Mitchell, A. 1994. Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Kimura T., Takeda, S., Sagiya, Y., Gotoh, M., Nakamura, Y., and Arakawa, H. 2003. Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat. Genet. 34: 440-445. [DOI] [PubMed] [Google Scholar]
- Lambert S. and Carr, A.M. 2005. Checkpoint responses to replication fork barriers. Biochimie 87: 591-602. [DOI] [PubMed] [Google Scholar]
- Liu H., Krizek, J., and Bretscher, A. 1992. Construction of a GAL1-regulated yeast cDNA expression library and its application to the identification of genes whose overexpression causes lethality in yeast. Genetics 132: 665-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Powell, K.A., Mundt, K., Wu, L., Carr, A.M., and Caspari, T. 2003. Cop9/signalosome subunits and Pcu4 regulate ribonucleotide reductase by both checkpoint-dependent and -independent mechanisms. Genes & Dev. 17: 1130-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese M.P., Paciotti, V., Fraschini, R., Zaccarini, R., Plevani, P., and Lucchini, G. 1997. The novel DNA damage checkpoint protein ddc1p is phosphorylated periodically during the cell cycle and in response to DNA damage in budding yeast. EMBO J. 16: 5216-5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M., Cotta-Ramusino, C., Pellicioli, A., Liberi, G., Plevani, P., Muzi-Falconi, M., Newlon, C.S., and Foiani, M. 2001. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557-561. [DOI] [PubMed] [Google Scholar]
- Mathews C.K. and Ji, J. 1992. DNA precursor asymmetries, replication fidelity, and variable genome evolution. Bioessays 14: 295-301. [DOI] [PubMed] [Google Scholar]
- Nakada D., Shimomura, T., Matsumoto, K., and Sugimoto, K. 2003. The ATM-related Tel1 protein of Saccharomyces cerevisiae controls a checkpoint response following phleomycin treatment. Nucleic Acids Res. 31: 1715-1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K., Balint, E., Ashcroft, M., and Vousden, K.H. 2000. A ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene 19: 4283-4289. [DOI] [PubMed] [Google Scholar]
- Osborn A.J., Elledge, S.J., and Zou, L. 2002. Checking on the fork: The DNA-replication stress-response pathway. Trends Cell. Biol. 12: 509-516. [DOI] [PubMed] [Google Scholar]
- Parsons J.L., Dianova, I.I., Allinson, S.L., and Dianov, G.L. 2005. DNA polymerase β promotes recruitment of DNA ligase IIIα-XRCC1 to sites of base excision repair. Biochemistry 44: 10613-10619. [DOI] [PubMed] [Google Scholar]
- Pemberton L.F. and Blobel, G. 1997. Characterization of the Wtm proteins, a novel family of Saccharomyces cerevisiae transcriptional modulators with roles in meiotic regulation and silencing. Mol. Cell. Biol. 17: 4830-4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder S.E. and Emr, S.D. 2000. Isolation of subcellular fractions from the yeast Saccharomyces cerevisiae. In Current protocols in cell biology (eds. J. Bonifacino et al.), Unit 3.8. Wiley Interscience, Hoboken, NJ. [DOI] [PubMed]
- Sanchez Y., Desany, B.A., Jones, W.J., Liu, Q., Wang, B., and Elledge, S.J. 1996. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271: 357-360. [DOI] [PubMed] [Google Scholar]
- Santocanale C. and Diffley, J.F. 1998. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395: 615-618. [DOI] [PubMed] [Google Scholar]
- Schimmang T., Tollervey, D., Kern, H., Frank, R., and Hurt, E.C. 1989. A yeast nucleolar protein related to mammalian fibrillarin is associated with small nucleolar RNA and is essential for viability. EMBO J. 8: 4015-4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider B.L., Seufert, W., Steiner, B., Yang, Q.H., and Futcher, A.B. 1995. Use of polymerase chain reaction epitope tagging for protein tagging in Saccharomyces cerevisiae. Yeast 11: 1265-1274. [DOI] [PubMed] [Google Scholar]
- Siegers K., Bolter, B., Schwarz, J.P., Bottcher, U.M., Guha, S., and Hartl, F.U. 2003. TRiC/CCT cooperates with different upstream chaperones in the folding of distinct protein classes. EMBO J. 22: 5230-5240. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sikorski R.S. and Hieter, P.A. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo J.M., Lopes, M., and Foiani, M. 2002. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297: 599-602. [DOI] [PubMed] [Google Scholar]
- Stubbe J., Ge, J., and Yee, C.S. 2001. The evolution of ribonucleotide reduction revisited. Trends Biochem. Sci. 26: 93-99. [DOI] [PubMed] [Google Scholar]
- Tanaka H., Arakawa, H., Yamaguchi, T., Shiraishi, K., Fukuda, S., Matsui, K., Takei, Y., and Nakamura, Y. 2000. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 404: 42-49. [DOI] [PubMed] [Google Scholar]
- Uchiki T., Dice, L.T., Hettich, R.L., and Dealwis, C. 2004. Identification of phosphorylation sites on the yeast ribonucleotide reductase inhibitor Sml1. J. Biol. Chem. 279: 11293-11303. [DOI] [PubMed] [Google Scholar]
- Vallen E.A. and Cross, F.R. 1999. Interaction between the MEC1-dependent DNA synthesis checkpoint and G1 cyclin function in Saccharomyces cerevisiae. Genetics 151: 459-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valpuesta J.M., Martin-Benito, J., Gomez-Puertas, P., Carrascosa, J.L., and Willison, K.R. 2002. Structure and function of a protein folding machine: The eukaryotic cytosolic chaperonin CCT. FEBS Lett. 529: 11-16. [DOI] [PubMed] [Google Scholar]
- Voegtli W.C., Ge, J., Perlstein, D.L., Stubbe, J., and Rosenzweig, A.C. 2001. Structure of the yeast ribonucleotide reductase Y2Y4 heterodimer. Proc. Natl. Acad. Sci. 98: 10073-10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.J., Chabes, A., Casagrande, R., Tian, X.C., Thelander, L., and Huffaker, T.C. 1997. Rnr4p, a novel ribonucleotide reductase small-subunit protein. Mol. Cell. Biol. 17: 6114-6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T., Matsuda, K., Sagiya, Y., Iwadate, M., Fujino, M.A., Nakamura, Y., and Arakawa, H. 2001. p53R2-dependent pathway for DNA synthesis in a p53-regulated cell cycle checkpoint. Cancer Res. 61: 8256-8262. [PubMed] [Google Scholar]
- Yao R., Zhang, Z., An, X., Bucci, B., Perlstein, D.L., Stubbe, J., and Huang, M. 2003. Subcellular localization of yeast ribonucleotide reductase regulated by the DNA replication and damage checkpoint pathways. Proc. Natl. Acad. Sci. 100: 6628-6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. and Rothstein, R. 2002. The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. Proc. Natl. Acad. Sci. 99: 3746-3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Muller, E.G., and Rothstein, R. 1998. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell 2: 329-340. [DOI] [PubMed] [Google Scholar]
- Zhao X., Chabes, A., Domkin, V., Thelander, L., and Rothstein, R. 2001. The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J. 20: 3544-3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z. and Elledge, S.J. 1992. Isolation of crt mutants constitutive for transcription of the DNA damage inducible gene RNR3 in Saccharomyces cerevisiae. Genetics 131: 851-866. [DOI] [PMC free article] [PubMed] [Google Scholar]