

Abstract
Despite the presence of over 3 million transposons separated on average by ∼500 bp, the human and mouse genomes each contain almost 1000 transposon-free regions (TFRs) over 10 kb in length. The majority of human TFRs correlate with orthologous TFRs in the mouse, despite the fact that most transposons are lineage specific. Many human TFRs also overlap with orthologous TFRs in the marsupial opossum, indicating that these regions have remained refractory to transposon insertion for long evolutionary periods. Over 90% of the bases covered by TFRs are noncoding, much of which is not highly conserved. Most TFRs are not associated with unusual nucleotide composition, but are significantly associated with genes encoding developmental regulators, suggesting that they represent extended regions of regulatory information that are largely unable to tolerate insertions, a conclusion difficult to reconcile with current conceptions of gene regulation.
The mammalian genome contains only slightly more protein-coding genes than the simple nematode worm Caenorhabditis elegans (C. elegans Sequencing Consortium 1998; International Human Genome Sequencing Consortium 2004; Waterston et al. 2002; Imanishi et al. 2004) and yet programs the ontogeny of a far more complex animal. While the numbers of protein isoforms may be increased by alternative splicing, the regulatory architecture of the genome also appears to increase with complexity (Mattick and Gagen 2005; Siepel et al. 2005). In insects and worms, genes with complex functions are surrounded by larger noncoding regions than those with simple functions (Nelson et al. 2004), and in vertebrates the Gene Ontology categories of genes adjacent to stable gene deserts (large regions of nonprotein-coding DNA) or having large introns containing multispecies conserved sequence elements are strongly biased toward development and transcriptional regulation (Ovcharenko et al. 2005; Sironi et al. 2005).
Bioinformatic detection of regulatory elements on a genome scale is difficult, and it is not known what proportion of the genome is functional. A common approach is to search for sequences that are conserved between species on the reasonable thesis that conservation over significant evolutionary distances indicates selection, which in turn indicates function. Indeed, the absolute amount of DNA under negative (purifying) selection is greater in genomes of complex organisms and from yeast to vertebrates in order of increasing genome size and general biological complexity; increasing fractions of conserved bases are found to lie outside of the exons of known protein-coding genes (Siepel et al. 2005). A comparison of human and mouse genomes indicates that at least 5% is under purifying selection (an amount of DNA that is similar to that in the entire C. elegans or Drosophila melanogaster genomes), two-thirds of which is nonprotein-coding (Waterston et al. 2002), which may be an underestimate (Smith et al. 2004).
Clearly, conserved regions represent only a fraction of the functional elements in the genome. By definition, there must be some relevant and divergent functional sequences that dictate the differences between species, many of which would be expected to be regulatory. Moreover, functional regulatory sequences may also drift in sequence. For example, histone methylation patterns are strongly conserved between human and mouse at orthologous loci even though many methylated sites do not show underlying DNA sequence conservation higher than the background (Bernstein et al. 2005).
Recently it was reported that there are large numbers of ultraconserved elements in the human genome whose sequences have remained essentially frozen throughout much of vertebrate evolution (Bejerano et al. 2004). During further analysis of these sequences, we noticed that many were associated with much larger nonconserved regions that lacked any recognizable transposons, suggesting that such regions may be a more general feature of the genome.
Transposable elements make up ∼45% of the human genome, a result of over 3 million SINE-, LINE-, DNA-, and LTR-transposon insertion events (Lander et al. 2001). Their average length is about 400 bp (range 0.1–8 kb) (Lander et al. 2001) with an average distance between them of 476 bp (see Methods). A similar transposon density occurs in mouse, although most transposons in human and mouse have entered these lineages independently since their divergence (Waterston et al. 2002). Although some extended regions of low-transposon density have previously been noted, particularly in the vicinity of HOX genes (Lander et al. 2001; Waterston et al. 2002; Wagner et al. 2003), no comprehensive analysis of the incidence and distribution of such regions in the mammalian genome has been reported.
Results
Identification of transposon-free regions
We undertook a structured analysis to identify the longest segments of the human and mouse genomes that lacked any recognizable transposon insertions that were not comprised of significant amounts of satellite or other highly repetitive sequences. We excluded any regions that contained >20% of nontransposon repeat sequences, or homology to the mitochondrial genome (see Methods). To exclude regions that have undergone recent expansion (e.g., tandem repeats of complex DNA), we also removed all regions that contain >20% self homology using BLASTZ (see Methods). Our final data set comprises 860 transposon-free regions (TFRs) over 10 kb in length covering over 12 Mb of the human genome, the longest is over 81 kb in length and a similar number (993) covering over 13 Mb of the mouse genome (see Table 1). TFRs are primarily comprised of complex unique sequences (<4% repeats on average). The list of human and mouse TFRs are given in Supplemental Tables S1 and S2, respectively.
Table 1.
Summary of TFR number and total size in three mammalian species
|
≥15 kb
|
≥10 kb
|
≥5 kb
|
||||
|---|---|---|---|---|---|---|
| Number | Size | Number | Size | Number | Size | |
| Human | 223 | 4.7 Mb | 860 | 12.2 Mb | 9249 | 65.7 Mb |
| Mouse | 206 | 4.1 Mb | 993 | 13.3 Mb | 12,313 | 85.1 Mb |
| Opossum | 137 | 3.1 Mb | 559 | 8.2 Mb | 7025 | 50.0 Mb |
The presence of TFRs is not explained by random transposon integration
Given the genome-wide density of transposons, we estimated the probability (P) of the chance occurrence of this number of TFRs over 10 kb to be <10-300, and the expected number of such TFRs to be close to zero (0.002) if transposons were randomly distributed in these genomes (see Methods). It has been reported that the distribution of some transposon families correlates with varying regional G+C content across the genome (Lander et al. 2001). To examine whether the occurrence of TFRs may simply be a function of varying G+C content affecting transposon density, we divided the genome into nonoverlapping 100-kb windows and grouped them based on their G+C content, which ranged from 31% to 64%. TFRs are present in all fractions ranging from 33% to 64% G+C (see Supplemental Table S3). Using the same assumption of random transposon insertion, we estimated the expected number of TFRs based on the observed transposon density in each fraction of the genome (i.e., independently within each G+C range). In 32/34 G+C fractions, the expected number of TFRs is significantly lower than the observed number (P-values between 10-10 and 10-270) (Supplemental Tables S3). The exceptions contain only seven TFRs in the two highest G+C fractions (0.04 > P > 10-4). Similar results were observed using window sizes of 50 and 200 kb (results not shown), indicating that regional transposon density has little impact on the observed number of TFRs, although a proportion of TFRs (∼25%) do contain G+C rich sequence (see below).
Many TFRs have been maintained throughout mammalian evolution
We next examined whether the TFRs in human and mouse might occur in orthologous regions, which would provide further evidence of selective constraint. We mapped mouse TFR sequences to the human genome and found that 401 (47%) of the human TFRs overlap a mouse TFR, and that on average, 44% of their length is covered by the overlapping mouse TFR. Using a stringent bootstrap analysis (see Methods) we estimated the probability of observing this overlap by chance to be <10-8. It should be noted that the apparent length of mouse TFRs is significantly compromised by numerous gaps in the genome assembly (see, e.g., Fig. 1B). Accounting for this, we found that 85% of all human TFRs overlap a mouse TFR ≥5 kb, covering on average 69% of bases in these TFRs. Thus, a large proportion of TFRs in the human genome occur in the same syntenic position as in mouse, despite the fact that the majority of identifiable transposons in mouse and human have entered their respective lineages independently after their divergence from their common ancestor (Waterston et al. 2002). This suggests that these regions have been resistant to transposon interruption for at least 85 million years, despite these genomes being bombarded with transposon insertions in the interim.
Figure 1.

Transposon-free regions surrounding the NR2F1 gene or its closest homolog in six amniote species. Each panel shows a modified screenshot displaying a 90-kb genomic region from the UCSC Genome Browser (http://genome.ucsc.edu/). Horizontal red bars indicate TFRs, the NR2F1 gene is in blue, brown ticks indicate transposons, black bars in the Gaps track indicate gaps in the genome assembly, and dashed red boxes highlight large regions free of annotated transposons. (A) Human (chr5:92,910–93,000 kb, May 2004). (B) Mouse (chr13:74,743–74,833 kb, May 2004) showing five TFRs ≥ 5 kb (separated by one SINE and several small assembly gaps). (C) Rat (chr2:5,757–5,847 kb, June 2003). (D) Dog (chr3:18,489–18,569 kb, July 2004). The NR2F1 homolog is the closest match to the human mRNA (Non-Dog RefSeq Genes track). (E) Opossum (scaffold_19227:2,391–2,481 kb, October 2004). The NR2F1 homolog is the closest match to the human NR2F1 protein (Human Proteins track). (F) Chicken (chrW:2,350–2,440 kb, February 2004). The NR2F1 homolog is the closest match to the human mRNA (Non-Chicken RefSeq Genes track).
In many cases it was also possible to identify large TFRs in syntenic locations of other species in addition to mouse. For example, the human gene NR2F1 lies within a 57-kb TFR (chr5.354) and the orthologous genes in mouse, rat, dog, opossum, and chicken are all also associated with large TFRs covering the same region (Fig. 1).
To gain further insight into the conservation of TFRs over longer evolutionary periods, we identified all TFRs in the draft marsupial opossum genome (http://www.broad.mit.edu). The opossum is evolutionarily distant from human and mouse but contains a similar load of transposable elements (∼30% of bases compared with 40% and 46% in mouse and human, respectively). Due to the large number of gaps remaining in the opossum genome assembly, we used a slightly different set of criteria to identify all opossum TFRs (see Methods). We identified 559 regions of the opossum genome of at least 10 kb that lacked any recognizable transposon sequences (see Table 1). We found that 27% of human TFRs ≥10 kb overlap an orthologous opossum TFR ≥10 kb and that, on average, 25% of their length is covered by the overlapping opossum TFRs. We also found that 52% of human TFRs ≥10 kb overlap an opossum TFR ≥5 kb, covering on average 40% of bases in these TFRs (P < 10-8; see Methods). This suggests that many TFRs are evolutionarily conserved features that existed prior to, and have been largely maintained since the divergence of eutherian mammals and marsupials ∼170 million years ago (Hedges and Kumar 2003). Although we considered broadening our analysis to more evolutionarily distant organisms such as chicken or fish, these genomes do not have a sufficient transposon density to distinguish between regions that have been selectively maintained transposon free and those that are simply free of transposons by chance.
The majority of human TFRs overlap annotated genes
On average, 15% of bases covered by TFRs are annotated as exonic, and 8% as protein coding. This contrasts to 2.2% and 1.2% of the human genome as a whole annotated as exonic and protein coding, respectively, indicating a sevenfold enrichment of exonic sequence within TFRs. We identified a limited number of cases where a TFR is comprised almost entirely of exonic sequence. However, only 6% of TFRs contain >50% exonic sequence. In the remainder, exons occupy on average only 12% of the bases, and one-third of TFRs are <5% exonic. Thus, although exons are enriched in TFRs, most of the TFR sequences are intronic (43%) and intergenic (42%) (Fig. 2).
Figure 2.

Breakdown of nucleotide annotations of 12.2 Mb of human TFRs. The pie chart illustrates the fraction of TFR bases that are annotated by the UCSC Known Genes track (Karolchik et al. 2003) as protein coding (coding), 5′ and 3′-untranslated regions (UTR), intronic and intergenic regions.
The majority of human TFRs (85%) overlap one or more annotated genes, usually (80%) including the 5′- and/or 3′-UTR. However, the presence of TFRs is not simply a function of genes per se being refractory to transposon insertions, as the average distance between transposons within human genes is ∼525 bp, not dissimilar to that of the genome as a whole (∼480 bp), and the number of TFRs estimated to occur by chance within genes is close to zero (expected = 0.007, P < 10-324) (see Methods). In addition, almost all TFRs (94%, covering 24% of TFR bases) overlap at least one spliced EST or mRNA. This suggests that many of the apparent nongenic TFRs may in fact be associated with nonannotated transcripts. However, a small number of TFRs occur deep within so-called gene deserts. For example, a 14.5-kb TFR (chr5.295) is found within an 800-kb gene desert located between the genes CCNH and MGC33214 on chromosome 5. Although there is little evidence for transcription in this area, there is also an orthologous 16-kb TFR in the syntenic region of the mouse genome.
Regulatory genes and miRNAs are enriched in TFRs
Table 2 shows the 10 longest human TFRs (Supplemental Table S5 gives a similar list for mouse). The longest human TFR (chr7.119) is over 81 kb and resides within the HOXA cluster. Surprisingly, nine of the 10 longest TFRs overlap genes encoding transcription factors. The only exception, TFR chr5.305, does not overlap any annotated protein-coding genes although it does harbor a microRNA (miRNA) precursor, mir-9-2. Indeed, of the 321 human miRNAs in the miRNA registry (Griffiths-Jones 2004), 29 (9%) are contained within TFRs. As TFRs cover only 0.4% of the genome, this represents a 23-fold enrichment of miRNAs in TFRs.
Table 2.
List of the 10 longest human TFRs
| TFR ID | Genomic position | Length (bp) | P valuea | % G + C | % bases conservedb | Overlapping genes | % exonic bases |
|---|---|---|---|---|---|---|---|
| chr7.119 | chr7:26938206 | 81208 | <10-75 | 52 | 45 | HOXA4-11 | 17 |
| chr5.354 | chr5:92928424 | 57661 | <10-54 | 46 | 37 | NR2F1 | 14 |
| chr2.598 | chr2:176777785 | 53918 | <10-48 | 52 | 38 | HOXD8-13 | 18 |
| chr11.129 | chr11:31759920 | 46909 | <10-45 | 49 | 35 | PAX6 | 5 |
| chr17.162 | chr17:43994277 | 46224 | <10-41 | 52 | 42 | HOXB4-6 | 19 |
| chr13.226 | chr13:99405192 | 43687 | <10-42 | 52 | 43 | ZIC2, ZIC5 | 12 |
| chr5.305 | chr5:87986303 | 40148 | <10-33 | 44 | 35 | MIRN9-2 | 0 |
| chr2.316 | chr2:104910522 | 39265 | <10-39 | 49 | 34 | POU3F3 | 4 |
| chr15.252 | chr15:94658960 | 37951 | <10-37 | 48 | 53 | NR2F2 | 8 |
| chr7.319 | chr7:96264636 | 37555 | <10-34 | 49 | 37 | DLX5 | 4 |
Probability of finding one or more TFRs of this length in the subset of the genome with the same GC content (see Methods)
Percent of bases that are covered by conserved regions described by Siepel et al. (2005)
We looked for categories of molecular function as defined in the Gene Ontology (GO) database that are significantly enriched in genes associated with TFRs. Of the 641 genes with an assigned GO annotation overlapping human TFRs, 167 are annotated as having the molecular function “transcription-factor activity”, a 4.4-fold enrichment (P < 10-66). This enrichment increases to 7.7-fold (108/235 genes, P < 10-70) in the case of TFRs larger than 15 kb. Notably, TFRs that overlap a gene annotated transcription-factor activity contain, on average, only 12% exonic sequence, less than the 15% average for all TFRs. Other GO categories that are also significantly enriched are DNA binding, regulation of transcription and other biological processes, and development (Table 3).
Table 3.
Gene ontology (GO) categories of human genes overlapping TFRs
| GO | Description | Na | Expb | Obsc | Foldd | Pe |
|---|---|---|---|---|---|---|
| Molecular function: | ||||||
| GO:0003700 | transcription factor activity | 786 | 38 | 167 | 4.4 | <10-66 |
| GO:0030528 | transcription regulator activity | 1106 | 53 | 195 | 3.6 | <10-63 |
| GO:0003677 | DNA binding | 1755 | 85 | 235 | 2.8 | <10-55 |
| GO:0003676 | nucleic acid binding | 2764 | 134 | 286 | 2.1 | <10-45 |
| Biological process: | ||||||
| GO:0045449 | regulation of transcription | 1738 | 83 | 244 | 2.9 | <10-64 |
| GO:0019219 | regulation of nucleic acid metabolism | 1762 | 85 | 244 | 2.9 | <10-62 |
| GO:0006355 | regulation of transcription, DNA-dependent | 1665 | 80 | 233 | 2.9 | <10-60 |
| GO:0006350 | transcription | 1818 | 87 | 244 | 2.8 | <10-59 |
| GO:0031323 | regulation of cellular metabolism | 1874 | 90 | 246 | 2.7 | <10-58 |
| GO:0006351 | transcription, DNA-dependent | 1720 | 83 | 234 | 2.8 | <10-58 |
| GO:0019222 | regulation of metabolism | 1927 | 92 | 247 | 2.7 | <10-56 |
| GO:0050789 | regulation of biological process | 2929 | 141 | 301 | 2.1 | <10-50 |
| GO:0050794 | regulation of cellular process | 2731 | 131 | 287 | 2.2 | <10-49 |
| GO:0050791 | regulation of physiological process | 2682 | 129 | 283 | 2.2 | <10-48 |
| GO:0051244 | regulation of cellular physiological process | 2599 | 125 | 277 | 2.2 | <10-48 |
| GO:0007275 | development | 1723 | 83 | 201 | 2.4 | <10-37 |
| GO:0009653 | morphogenesis | 1088 | 52 | 130 | 2.5 | <10-24 |
| GO:0009887 | organogenesis | 850 | 41 | 103 | 2.5 | <10-19 |
| GO:0048513 | organ development | 903 | 43 | 104 | 2.4 | <10-18 |
| GO:0007399 | neurogenesis | 389 | 19 | 61 | 3.3 | <10-16 |
Total number of genes in genome assigned to this GO category
Expected number of genes overlapping with TFRs
Observed number of genes overlapping with TFRs
Fold enrichment of observed over expected
P-values
We also identified several sets of paralogous genes involved in developmental regulation in which several members are each associated with TFRs (Supplemental Table S6). For example, the IRX family of homeobox transcription factors contains six members (IRX1–IRX6), and all but IRX6 are associated with TFRs between 10 and 22 kb. Other examples include HOX, PAX, FOX (forkhead box), SOX (HMG1/2 box), LHX (LIM/homeobox), POU, SIX, TBX, and ZIC gene families, which play key roles in development, as well as multiple members of the COL (collagen), EPHA (EPH-like receptor protein-tyrosine kinase, implicated in nervous system development), SLITRK (integral membrane proteins expressed predominantly in neural tissue), FGF (fibroblast growth factor), and PCDH (cadherins involved in cell adhesion) gene families. This suggests that the mechanism responsible for TFR maintenance was established early in evolution before these gene duplication events occurred, and has been maintained despite the subsequent divergence in their primary sequence.
Conservation of TFR sequences
There are 481 ultraconserved regions of the genome that contain 100% sequence identity between human, mouse, and rat genomes over a minimum of 200 bp (Bejerano et al. 2004). Although these ultraconserved elements cover only 126 kb (0.004%) of the genome, we found that 87 (18%) of them overlap with TFRs (P < 10-8). The average length of TFRs associated with one or more ultraconserved elements is 19 kb. This suggests that ultraconserved elements are often associated with transposon-free regions many times their own length (see, e.g., Supplemental Fig. S1).
We next compared the average conservation of the sequences within human TFRs to that of the genome as a whole. Of the 12.2 Mb contained in TFRs, 10 Mb is present within the UCSC human–mouse genome alignment (Karolchik et al. 2003). The average identity of the alignable sequence within TFRs is 74.8% in comparison with the average for the entire genome of 69.3%.
In a recent study, Siepel and coworkers (Siepel et al. 2005) conducted a comprehensive search that aimed to identify all conserved elements in vertebrate genomes, using genome-wide multiple alignments of five vertebrate species. The resulting set of conserved elements is available on the UCSC genome browser (phastConsElements) and covers 4.8% of the human genome. We found that 29% of TFR bases correspond to the conserved elements, suggesting that the remaining 71% of TFR bases have little evidence for selective constraint on primary sequence. In a small subset of TFRs (6%), conserved elements occupy >50% of bases, the most extreme case being TFR chr13.12, in which almost 84% of bases correspond to conserved elements. Nevertheless, 277 TFRs contain more than 10 kb of nonconserved bases (see, e.g., Supplemental Fig. S2). Even under a very strict assumption that transposon insertions are prohibited in conserved elements, observing this number of regions devoid of transposons by chance is still close to zero (see Supplemental Table S4). This indicates that primary sequence conservation may not be the only constraint on transposon insertion in TFRs. Furthermore, it is important to note that of the 242 genes with an assigned GO annotation overlapping these 277 TFRs, 103 are annotated as having transcription-factor activity, a 7.1-fold enrichment (P < 10-63), demonstrating that the association between TFRs and regulatory genes is not simply a byproduct of the enrichment for conserved regions within TFRs.
Murine retroviral insertions associated with cancer are enriched in TFRs
Further evidence that TFRs are associated with regulatory regions come from molecular genetic studies. The Mouse Retroviral Tagged Cancer Gene Database catalogs the integration sites of genomic retroviral insertions found in mouse tumors, and lists ∼350 common integration sites (CIS) where insertions have been identified in a minimum of two independent tumors (Akagi et al. 2004). A total of 22 TFRs contain one or more CIS insertions in nonexonic sequences (P < 10-8). In addition, insertion of two artificial transposons into the TFR around the Evx2 gene caused limb defects and affected transcription of the downstream Hoxd genes (Monge et al. 2003). These observations suggest that TFRs are associated with extended regions that are important in the control of differentiation and development.
G+C content within TFRs
Although the distribution of most TFRs is independent of regional G+C content, we investigated whether the TFRs themselves have unusual nucleotide composition, which might be a barrier to transposon insertion. The G+C content of the human TFRs ranges from 29% to 69%, and exhibits a broadly bimodal distribution with a minimum between the peaks at 57% G+C content (Supplemental Fig. S3). A similar distribution also occurs in the mouse (Supplemental Fig. S4). We therefore arbitrarily divided the human data set and repeated the analyses described above. A total of 245 human TFRs contain 57% or greater G+C bases (high G+C) with an average size of 13.7 kb, with the majority (615, 72%) containing 56% or less G+C with an average size of 14.4 kb. Both groups are highly associated with known genes, although the high G+C TFRs are more enriched for exonic sequence (see Table 4). Known genes that overlap with high G+C TFRs show no significant enrichment for any particular GO term, whereas the lower G+C TFRs have a 4.9-fold enrichment for genes annotated transcription-factor activity (P < 10-50). Both groups are often located in regions syntenic with TFRs in the mouse genome, but the relative level of synteny drops sharply in the high G+C TFRs when compared with the opossum genome (see Table 4). The enrichment of ultraconserved elements within TFRs also occurs primarily within lower G+C TFRs (see Table 4). Conversely, high G+C TFRs are more enriched for CpG islands that cover 20% of their length, whereas on average, CpG islands cover 11% of the length of lower G+C TFRs, which is not necessarily surprising given their overlap with genes. We also observed that a high proportion of the high G+C TFRs are located near the ends of chromosomes, 50% are located within 2 Mb of annotated chromosome ends, and 71% within 10 Mb (compared with 2% and 14%, respectively, of lower G+C TFRs). These observations suggest that TFRs may be comprised of more than one type. TFRs with mid-range G+C content are characterized by strong association with transcription factors, whereas TFRs with high G+C content are largely associated with subtelomeric regions of the genome.
Table 4.
Annotations associated with human TFRs
| High GC (≥57%) | Lower GC (<57%) | All TFRs | |
|---|---|---|---|
| TFRs ≥ 10 kb | 245 | 615 | 860 |
| Fraction of TFRs associated with known genes | 93% | 81% | 85% |
| Fraction TFR bases exonic | 22% | 12% | 15% |
| Fraction of TFRs that overlap with mouse TFRs | 38% | 50% | 47% |
| Fraction of TFRs that overlap with opossum TFRs | 6% | 36% | 27% |
| Percent of ultraconserved elements within TFRs | 1% | 17% | 18% |
| Fraction of TFR bases covered by CpG islands | 20% | 11% | 13% |
| Fraction of TFRs within 2 Mb of chromosome ends | 50% | 2% | 16% |
| Fraction of TFRs within 10 Mb of chromosome ends | 70% | 14% | 20% |
Shorter TFRs may also be under functional constraint
The decision to use a threshold length of 10 kb in this analysis was purely arbitrary and does not exclude the possibility that shorter TFRs may possess similar properties as those described above. When we repeated our analysis using the lower threshold length of TFRs ≥5 kb many of the same characteristics were apparent. Using the same criteria as above, we identified 9294 TFRs ≥5 kb covering 66 Mb of the human genome (similar in mouse), significantly more than expected by a model of random transposon insertion (expected = 87, P-value < 10-324). As with the TFRs ≥10 kb, TFRs ≥5 kb are associated with known genes, miRNAs, ultraconserved elements, and CpG islands (see Supplemental text, section S1), suggesting that at least a subset of these shorter TFRs may also be constrained.
Discussion
In this work we identified 860 regions ≥10 kb of the human genome that appear to have been maintained transposon-free over a large period of evolutionary time. These regions are comprised of mainly nonrepetitive, complex, nonprotein-coding DNA, and many of these regions are associated with regulatory genes. There are two plausible general mechanisms by which a region may be maintained transposon free; the underlying sequence may be resistant to transposon integration or transposon insertion in the region is deleterious and is therefore subject to strong negative selection. However, considering the heterogeneity of TFR sequences and that most TFRs coincide with genes, we favor the latter explanation.
What could be the molecular and genetic basis of the existence of extended genomic regions that are refractory to transposon insertion over long periods of evolution? Transposons abound in, and indeed dominate the human and mouse genomes. Although some regions within TFRs are evidently under heavy constraint at the primary sequence level, there are also large regions with little or no apparent sequence conservation, and the lack of transposons in these sequences cannot be explained on this basis. Our observations are also consistent with the recent findings that genes encoding proteins involved in development and transcriptional regulation are associated with extended flanking noncoding regions and large introns that are enriched for conserved sequences (Ovcharenko et al. 2005; Sironi et al. 2005), and that the density of evolutionarily constrained sequences (the majority of which are noncoding) is inversely correlated with the density of mobile elements (Cooper et al. 2005). The association of TFRs with particular types of genes, notably those encoding transcription factors/developmental control proteins, and the fact that TFRs are largely comprised of nonprotein-coding sequences, strongly suggests that TFRs represent extended regions of complex regulatory information important to the control of the expression of particular types of genes during differentiation and development, which is also supported by the fact that a significant number of retroviral insertions into TFRs are associated with cancer.
However, it is difficult to explain mechanistically the requirement of 10 kb (let alone 80 kb) of uninterrupted sequence in terms of the current paradigm of cis-regulatory regions containing multiple protein-binding sites (clustered or otherwise) that control the transcription of the adjacent gene(s). This in turn suggests that TFRs might be the passive signatures of one or more as yet poorly understood mechanisms of gene regulation that operate in higher organisms.
One possibility is that that the regions of low-sequence conservation act simply as spacers to separate regulatory elements that must occur in a precise positional and distance relationship for proper function. However, why this might be the case over such extended distances is difficult to reconcile with current conceptions of regulatory mechanisms, including the proposed looping of long-range enhancer elements (Ogata et al. 2003). Another possibility is that TFRs are critical regulatory regions that are methylation sensitive, and that insertion of transposons may disrupt regional chromatin structure (Arnaud et al. 2000). It has recently been shown that methylation patterns at orthologous loci are strongly conserved between human and mouse in the absence of obvious sequence conservation (Bernstein et al. 2005).
An alternative (and not mutually exclusive) explanation is that these regions encode critical regulatory RNAs, including miRNA clusters (Altuvia et al. 2005), interruption of which affects their subsequent processing and/or function. Many functional noncoding RNAs, such as H19 and XIST, are not highly conserved between species (Juan et al. 2000; Chureau et al. 2002) and, indeed, H19 lies within a 17-kb TFR. In addition, some TFRs overlap imprinted regions, which in turn are more broadly characterized by low density of short interspersed transposable elements (SINEs) (Greally 2002). This includes the IGF2-H19 and Gnas loci, both of which are known to contain multiple ncRNAs (Amarger et al. 2002; Holmes et al. 2003). Interestingly, the IGF2 region in platypus, which in contrast to marsupials and eutherian mammals is not imprinted, contains 88 SINE insertions (Weidman et al. 2004).
Whatever the molecular mechanisms involved, the presence of TFRs identifies large, presumably regulatory regions that are important to mammalian ontogeny, but that would otherwise be difficult to detect using traditional computational approaches based on primary sequence conservation. Such approaches have indicated that at least 5% of the human and mouse genome is under common purifying selection (Waterston et al. 2002). It should be noted, however, that different (albeit overlapping) subsets of noncoding sequences are conserved between human and other species (Thomas et al. 2003; Frazer et al. 2004). This and our data suggest that the extent of nonprotein-coding regulatory sequences under functional selection (both negative for essential functions, and positive for adaptive radiation), depending on the strength of the selection forces and the underlying mechanistic constraints on the drift of different types of sequences, may be much greater than previously thought (Smith et al. 2004; Mattick and Makunin 2005).
Methods
Identification of TFRs
A mirror of the UCSC genome browser and database (http://genome.ucsc.edu/) (Karolchik et al. 2003) was created and hg17 (human May 2004) (Lander et al. 2001), mm5 (mouse May 2004) (Waterston et al. 2002), and monDom1 (opossum October 2004, The Broad Institute, MA, USA; http://www.broad.mit.edu; E. Landers, pers. comm.) genome databases were loaded. We extracted all regions between adjacent DNA, LINE, SINE, and LTR transposons in human, using the RepeatMasker track (http://www.repeatmasker.org) (Karolchik et al. 2003). Gaps in the assembly were treated the same as transposons. Random chromosomes were excluded. We identified 3.2 million regions in the human genome between recognizable transposons with an average size of 476 bp. All regions >10 kb were identified and are subsequently referred to as Transposon Free Regions (TFRs). The same procedures were used for identifying TFRs in mouse.
Due to the large number of assembly gaps in the recently released opossum genome (89,000 fragment gaps within 19,000 scaffolds) it was not practical to delimit opossum TFRs by gaps in addition to transposon sequence. Two forms of gap exist in the October 2004 opossum genome assembly, scaffold gaps, where the relative order and orientation of scaffolds are not known, and fragment gaps; these are gaps in the assembly within the scaffolds where the relative order and orientation of the sequence either side of the gap is known. To overcome this limitation, we identified all regions between adjacent DNA, LINE, SINE, and LTR transposons. The gap-free length of each element was calculated as its length minus the length of any fragment gaps within the TFR. All regions with a gap-free length of at least 10 kb were identified. The sizes of any opossum TFR given in this study refer to the gap-free length. As the draft opossum sequence is completed and the gaps are filled, it is possible that some gaps within a given TFR will be found to contain transposon sequence; however, it is also likely that as the scaffolds are joined, some shorter TFRs will be joined to form larger TFRs.
Filtering of TFRs
TFRs were filtered in three steps as follows: (1) all TFRs that contained >20% nontransposon repetitive sequence as identified by either the RepeatMasker (http://www.repeatmasker.org) or Simple repeats tracks (Benson 1999) were removed from the analysis. (2) The BLASTZ program (Schwartz et al. 2003) was then used to compare the soft masked sequence of each TFR to itself (default command line parameters were used with the addition of C = 3). Ignoring the trivial self hit, any TFR where >25% of bases could be mapped to other regions of the same TFR were removed from the analysis. (3) Large fragments of the mitochondrial genome can be found integrated in a number of locations across the nuclear genome (Richly and Leister 2004). The BLASTZ program was then used to compare the soft masked sequence of each TFR to the mitochondrial genome (default parameters with the addition of C = 3). Any TFR where >20% of bases could be mapped to the mitochondrial genome were removed from the analysis. In total, these filters removed 511 TFRs ≥10 kb, including several large repetitive exons of genes such as titin, as well as a number of repetitive snoRNA clusters. For example, Cavaillé et al. (2000) identified two snoRNA clusters of 72 and 46 kb located in the region of chromosome 15 involved with Prader-Willi syndrome that contain no transposons.
Estimation of expected number of TFRs in the genome
To estimate the expected number (μ) of TFRs in the human genome, we used the following formula (Karlin and Macken 1991):
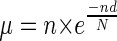 |
Where N = the total number of bases between transposons, n = the total number of transposons, and d = the minimum size of TFR (e.g., 10,000 bp). The probability of finding the observed number of TFRs was estimated using the Poisson distribution with parameter μ.
An independent estimate of the expected number of TFRs in the genome was made with 100,000 iterations of a bootstrap procedure and the results were identical to the formula above, to one significant figure (data not shown).
Estimation of expected number of TFRs in different G+C content fractions of the genome
The genome was divided into nonoverlapping windows of 100 kb. Any window that consisted of >40% sequence gap was excluded. For each window, the G+C content was measured and the number of TFRs that overlapped the window by >50% was counted. The windows were then grouped by G+C content (1% bins). The total number of transposons (counted as blocks of masked DNA, LINE, SINE, or LTR transposon sequences) and the total length of nontransposon sequence were counted for each G+C bin. The statistical model described above was then used to estimate the expected number of TFRs for each G+C bin, and the probability of generating the observed count given the assumption that transposons will be randomly distributed within each G+C bin.
Estimation of expected number of TFRs in different G+C content fractions of the genome, assuming all conserved sequence is resistant to transposon insertion
For the purposes of this analysis, we required each base within the genome to be defined as either conserved or nonconserved. We divided all bases in the genome into one of these two categories using the conserved regions identified in the phastConsElements track available on UCSC Genome Browser (Siepel et al. 2005). This data was then used to identify all TFRs that contain a minimum 10 kb of nonconserved sequence. Expected number of TFRs and probability of generating observed results by chance were calculated as above with the exception that the total length of nontransposon, nonconserved bases was used for the parameter N.
Estimation of the probability of observing a TFR of a given length
The regional G+C content for each of the 10 longest TFRs was calculated by extending a window 50 kb either side of the midpoint of the TFR. This G+C content was used in conjunction with the transposon density information given in Supplemental Table S3 and the Poisson distribution described above to estimate the probability of observing one or more TFRs of that size in the fraction of the genome of that G+C content.
Estimation of expected number of TFRs in the genic fraction of the genome
The UCSC Known Genes track was used to identify all genic regions of the genome, including all introns, exons, and UTRs. Within this fraction, the total number of transposons (counted as blocks of masked DNA, LINE, SINE, or LTR sequence) and the total length of nontransposon sequence were calculated. These values were used in the formula above to estimate the expected number of TFRs. The observed number of TFRs was calculated by identifying all regions of at least 10 kb that lack any recognizable transposons.
Estimation of the probability of miRNAs overlapping TFRs
A bootstrap method was used to estimate the chance to observe 29 of 321 annotated miRNAs overlapping the human 10-kb TFRs. The TFRs were randomly assigned new chromosomal locations, and the number of elements intersecting with miRNAs was recorded. This process was repeated for 10,000,000 iterations. The 67% confidence interval (CI) for number overlapping was [1,2]; the 99% CI was [0,10]. The maximum number of overlaps observed was 27.
Estimation of the probability of ultraconserved elements overlapping TFRs
A bootstrap method was used to estimate the chance to observe 87 of 481 ultraconserved elements overlapping the human 10-kb TFRs. The TFRs were randomly assigned new chromosomal locations and the number of elements intersecting with ultraconserved elements was recorded. This process was repeated for 100,000,000 iterations. The 67% confidence interval (CI) for number overlapping was [1,3]; the 99% CI was [0,6]. The maximum number of overlaps observed was 13.
Estimation of the probability of Conserved Integration Site (CIS) elements overlapping TFRs
A bootstrap method was used to estimate the chance to observe 22 of 993 mouse TFRs overlapping a CIS. The TFRs were randomly assigned new chromosomal locations, and the number of elements intersecting with the CIS elements was recorded. This process was repeated for 100,000,000 iterations. The 67% confidence interval (CI) for number overlapping was [1,5]; the 99% CI was [0,8]. The maximum number of overlaps observed was 18.
Coding/noncoding/intergenic annotations
The fraction of TFRs annotated as coding, UTR, intronic, and intergenic was taken from the UCSC Known Genes track (Karolchik et al. 2003).
Human and mouse genome size
We used the human genome size of 2.85 Gb and a mouse genome size of 2.62 Gb, which excludes all gaps in the genome assemblies.
Mapping human sequences to the mouse genome
To map the human TFRs to the mouse genome, we used the UCSC hg17/mm5 “nets” and “chains” files (Kent et al. 2003), and “orthoMap” tool (Karolchik et al. 2003). As this tool is optimized to work on sequences smaller than 5 or 10 Kb, we first split our sequences into 10 equal-sized blocks, then required that eight of 10 blocks mapped to mouse and that the total size of the mapped region in mouse was 80%–150% of the size in human. The same procedure was used to map mouse and opossum TFRs to the human genome.
Bootstrap estimate for human/mouse, human/opossum overlap by chance
To simulate human and mouse TFRs overlapping by random chance, we used an artificial genome, randomly placed mouse TFRs, not allowing overlaps, then randomly placed human TFRs, not allowing overlaps with other human TFRs. We then counted the number of human TFRs overlapping mouse and the length of the overlapping regions. The genome size was chosen as 400 Mb for two reasons: (1) this is much smaller than the actual 1.1 Gb of alignable human–mouse genome; and (2) to account for the enrichment of exons in TFRs. The human genome has ∼64 Mb of exonic sequence, and a hypothetical 427-Mb genome with 15% exons would also have ∼64 Mb of exonic sequence, so we believe this size would adjust for the enrichment of exonic sequence. The simulation of TFRs ≥ 10 kb human and TFRs ≥ 10 kb mouse was repeated 100,000,000 times; no simulation exceeded the observed number or size of overlaps (P < 10-8).
Gene Ontology enrichment and P-values
GO annotations were taken from the July 2004 EMBL GOA UniProt database (Camon et al. 2004) and the July 2004 GO schema (Harris et al. 2004). Known Isoforms identifiers for UCSC Known Genes were used to make sure one gene was only counted once where there were multiple isoforms. A Perl script and SQL code were created to calculate enrichment of terms and Fisher's Exact P-values against a background of all GO annotated genes in the UCSC Known Genes database. Any GO term with less than twofold enrichment, or a P-value > 10-15, or <10 associated genes, was discarded. While we did not directly correct for multiple-hypothesis testing, in practice we performed <120 individual tests deeming the reported P-values highly significant.
Conservation of primary sequence within TFRs
UCSC hg17 vs. mm5 human/mouse “axt” alignments (Kent et al. 2003) (“chained” and “netted”) and the “axtAndBed” and “axtCalcMatrix” (Karolchik et al. 2003) programs were used for conservation analyses.
Ultraconserved elements
Ultraconserved elements (Bejerano et al. 2004) from hg16 were mapped to hg17 coordinates using the “liftOver” (Karolchik et al. 2003) feature of the UCSC genome browser.
CpG islands
CpG islands were identified from the CpG Islands track of the UCSC Genome Browser (Karolchik et al. 2003).
MicroRNAs
A GFF file of known human miRNAs (Release 7.0, June 2005) was downloaded from miRBase (http://microrna.sanger.ac.uk/) (Griffiths-Jones 2004).
Acknowledgments
We thank David Haussler, Jim Kent, and the members of the UCSC browser team for providing many of the programs, alignments, and annotations on the UCSC Genome Browser that were used in this analysis, as well as the excellent support from the mailing list. We thank the Genome Sequencing Consortia for the human, mouse, opossum, and other genome sequences used in this analysis. We also thank the other members of the Mattick group for helpful discussions and Martin Frith for his assistance in statistical analyses. This research was supported by the Australian Research Council and the Queensland State Government.
Article published online ahead of print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.4624306.
Footnotes
[Supplemental material is available online at www.genome.org.]
References
- Akagi, K., Suzuki, T., Stephens, R.M., Jenkins, N.A., and Copeland, N.G. 2004. RTCGD: Retroviral tagged cancer gene database. Nucleic Acids Res. 32 D523-D527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altuvia, Y., Landgraf, P., Lithwick, G., Elefant, N., Pfeffer, S., Aravin, A., Brownstein, M.J., Tuschl, T., and Margalit, H. 2005. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 33 2697-2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarger, V., Nguyen, M., Laere, A.S., Braunschweig, M., Nezer, C., Georges, M., and Andersson, L. 2002. Comparative sequence analysis of the INS-IGF2-H19 gene cluster in pigs. Mamm. Genome 13 388-398. [DOI] [PubMed] [Google Scholar]
- Arnaud, P., Goubely, C., Pelissier, T., and Deragon, J.M. 2000. SINE retroposons can be used in vivo as nucleation centers for de novo methylation. Mol. Cell. Biol. 20 3434-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W.J., Mattick, J.S., and Haussler, D. 2004. Ultraconserved elements in the human genome. Science 304 1321-1325. [DOI] [PubMed] [Google Scholar]
- Benson, G. 1999. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27 573-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, B.E., Kamal, M., Lindblad-Toh, K., Bekiranov, S., Bailey, D.K., Huebert, D.J., McMahon, S., Karlsson, E.K., Kulbokas III, E.J., Gingeras, T.R., et al. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120 169-181. [DOI] [PubMed] [Google Scholar]
- Camon, E., Magrane, M., Barrell, D., Lee, V., Dimmer, E., Maslen, J., Binns, D., Harte, N., Lopez, R., and Apweiler, R. 2004. The Gene Ontology Annotation (GOA) Database: Sharing knowledge in UniProt with Gene Ontology. Nucleic Acids Res. 32 D262-D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaillé, J., Buiting, K., Kiefmann, M., Lalande, M., Brannan, I., Horsthemke, B., Bachellerie, J., Brosius, J., and Hüttenhofer, A. 2000. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc. Natl. Acad. Sci. 97 14311-14316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. elegans Sequencing Consortium. 1998. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 282 2012-2018. [DOI] [PubMed] [Google Scholar]
- Chureau, C., Prissette, M., Bourdet, A., Barbe, V., Cattolico, L., Jones, L., Eggen, A., Avner, P., and Duret, L. 2002. Comparative sequence analysis of the X-inactivation center region in mouse, human, and bovine. Genome Res. 12 894-908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, G.M., Stone, E.A., Asimenos, G., Green, E.D., Batzoglou, S., and Sidow, A. 2005. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15 901-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer, K.A., Tao, H., Osoegawa, K., de Jong, P.J., Chen, X., Doherty, M.F., and Cox, D.R. 2004. Noncoding sequences conserved in a limited number of mammals in the SIM2 interval are frequently functional. Genome Res. 14 367-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greally, J.M. 2002. Short interspersed transposable elements (SINEs) are excluded from imprinted regions in the human genome. Proc. Natl. Acad. Sci. 99 327-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones, S. 2004. The microRNA Registry. Nucleic Acids Res. 32 D109-D111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, M.A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., Eilbeck, K., Lewis, S., Marshall, B., Mungall, C., et al. 2004. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32 D258-D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges, S.B. and Kumar, S. 2003. Genomic clocks and evolutionary timescales. Trends Genet. 19 200-206. [DOI] [PubMed] [Google Scholar]
- Holmes, R., Williamson, C., Peters, J., Denny, P., and Wells, C. 2003. A comprehensive transcript map of the mouse Gnas imprinted complex. Genome Res. 13 1410-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi, T., Itoh, T., Suzuki, Y., O'Donovan, C., Fukuchi, S., Koyanagi, K.O., Barrero, R.A., Tamura, T., Yamaguchi-Kabata, Y., Tanino, M., et al. 2004. Integrative annotation of 21,037 human genes validated by full-length cDNA clones. PLoS Biol. 2 856-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2004. Finishing the euchromatic sequence of the human genome. Nature 431 931-945. [DOI] [PubMed] [Google Scholar]
- Juan, V., Crain, C., and Wilson, C. 2000. Evidence for evolutionarily conserved secondary structure in the H19 tumor suppressor RNA. Nucleic Acids Res. 28 1221-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin, S. and Macken, C. 1991. Assessment of inhomogeneities in an E. coli physical map. Nucleic Acids Res. 19 4241-4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik, D., Baertsch, R., Diekhans, M., Furey, T.S., Hinrichs, A., Lu, Y.T., Roskin, K.M., Schwartz, M., Sugnet, C.W., Thomas, D.J., et al. 2003. The UCSC Genome Browser Database. Nucleic Acids Res. 31 51-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J., Baertsch, R., Hinrichs, A., Miller, W., and Haussler, D. 2003. Evolution's cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. 100 11484-11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409 860-921. [DOI] [PubMed] [Google Scholar]
- Mattick, J.S. and Gagen, M.J. 2005. Accelerating networks. Science 307 856-858. [DOI] [PubMed] [Google Scholar]
- Mattick, J.S. and Makunin, I.V. 2005. Small regulatory RNAs in mammals. Hum. Mol. Genet. 14 R121-R132. [DOI] [PubMed] [Google Scholar]
- Monge, I., Kondo, T., and Duboule, D. 2003. An enhancer-titration effect induces digit-specific regulatory alleles of the HoxD cluster. Dev. Biol. 256 212-220. [DOI] [PubMed] [Google Scholar]
- Nelson, C.E., Hersh, B.M., and Carroll, S.B. 2004. The regulatory content of intergenic DNA shapes genome architecture. Genome Biol. 5 R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata, K., Sato, K., Tahirov, T.H., and Tahirov, T. 2003. Eukaryotic transcriptional regulatory complexes: Cooperativity from near and afar. Curr. Opin. Struct. Biol. 13 40-48. [DOI] [PubMed] [Google Scholar]
- Ovcharenko, I., Loots, G.G., Nobrega, M.A., Hardison, R.C., Miller, W., and Stubbs, L. 2005. Evolution and functional classification of vertebrate gene deserts. Genome Res. 15 137-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly, E. and Leister, D. 2004. NUMTs in sequenced eukaryotic genomes. Mol. Biol. Evol. 21 1081-1084. [DOI] [PubMed] [Google Scholar]
- Schwartz, S., Kent, W.J., Smit, A., Zhang, Z., Baertsch, R., Hardison, R.C., Haussler, D., and Miller, W. 2003. Human–mouse alignments with BLASTZ. Genome Res. 13 103-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel, A., Bejerano, G., Pedersen, J.S., Hinrichs, A.S., Hou, M., Rosenbloom, K., Clawson, H., Spieth, J., Hillier, L.W., Richards, S., et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15 1034-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi, M., Menozzi, G., Comi, G.P., Cagliani, R., Bresolin, N., and Pozzoli, U. 2005. Analysis of intronic conserved elements indicates that functional complexity might represent a major source of negative selection on non-coding sequences. Hum. Mol. Genet. 14 2533-2546. [DOI] [PubMed] [Google Scholar]
- Smith, N.G., Brandstrom, M., and Ellegren, H. 2004. Evidence for turnover of functional noncoding DNA in mammalian genome evolution. Genomics 84 806-813. [DOI] [PubMed] [Google Scholar]
- Thomas, J.W., Touchman, J.W., Blakesley, R.W., Bouffard, G.G., Beckstrom-Sternberg, S.M., Margulies, E.H., Blanchette, M., Siepel, A.C., Thomas, P.J., McDowell, J.C., et al. 2003. Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424 788-793. [DOI] [PubMed] [Google Scholar]
- Wagner, G.P., Amemiya, C., and Ruddle, F. 2003. Hox cluster duplications and the opportunity for evolutionary novelties. Proc. Natl. Acad. Sci. 100 14603-14606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420 520-562. [DOI] [PubMed] [Google Scholar]
- Weidman, J.R., Murphy, S.K., Nolan, C.M., Dietrich, F.S., and Jirtle, R.L. 2004. Phylogenetic footprint analysis of IGF2 in extant mammals. Genome Res. 14 1726-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]