

Abstract
Objectives
Mutations in LGI1 cause autosomal dominant partial epilepsy with auditory features (ADPEAF), a form of familial temporal lobe epilepsy with auditory ictal manifestations. The authors aimed to determine what proportion of ADPEAF families carries a mutation, to estimate the penetrance of identified mutations, and to identify clinical features that distinguish families with and without mutations.
Methods
The authors sequenced LGI1 in 10 newly described ADPEAF families and analyzed clinical features in these families and others with mutations reported previously.
Results
Three of the families had missense mutations in LGI1 (C42R, I298T, and A110D). Penetrance was 54% in eight families with LGI1 mutations the authors have identified so far (five reported previously and three reported here). Excluding the original linkage family, the authors have found mutations in 50% (7/14) of tested families. Families with and without mutations had similar clinical features, but those with mutations contained significantly more subjects with auditory symptoms and significantly fewer with autonomic symptoms. In families with mutations, the most common auditory symptom type was simple, unformed sounds (e.g., buzzing and ringing). In two of the newly identified families with mutations, some subjects with mutations had idiopathic generalized epilepsies.
Conclusions
LGI1 mutations are a common cause of autosomal dominant partial epilepsy with auditory features. Current data do not reveal a clinical feature that clearly predicts which families with autosomal dominant partial epilepsy with auditory features have a mutation. Some families with LGI1 mutations contain individuals with idiopathic generalized epilepsies. This could result from either an effect of LGI1 on risk for generalized epilepsy or an effect of co-occurring idiopathic generalized epilepsy-specific genes in these families.
Autosomal dominant partial epilepsy with auditory features (ADPEAF) is a form of idiopathic temporal lobe epilepsy with auditory ictal manifestations.1–4 In 1995, we localized the gene to a 10-cM region on chromosome 10q22–24 in a single large family with autosomal dominant inheritance of epilepsy.1 Among 11 individuals with idiopathic epilepsy in the family, 55% described auditory symptoms with their seizures. These included unformed sounds such as buzzing, ringing, or clicking; distortions such as volume changes or muffling; and complex sounds such as a specific radio jingle or singer.3 One person described seizures precipitated by sounds.
In 1999, linkage was reported to an overlapping interval in a large Basque family with similar symptoms, which the authors called autosomal dominant lateral temporal lobe epilepsy.5 This report narrowed the minimal genetic region to approximately 3 cM, assuming the causative gene was the same in the two families. In 2002, positional cloning efforts focused on this 3-cM interval led to the identification of the causative gene, leucine-rich glioma-inactivated 1 (LGI1).6,7 Subsequently, a number of other mutations have been reported (figure 1).8–13
Figure 1.
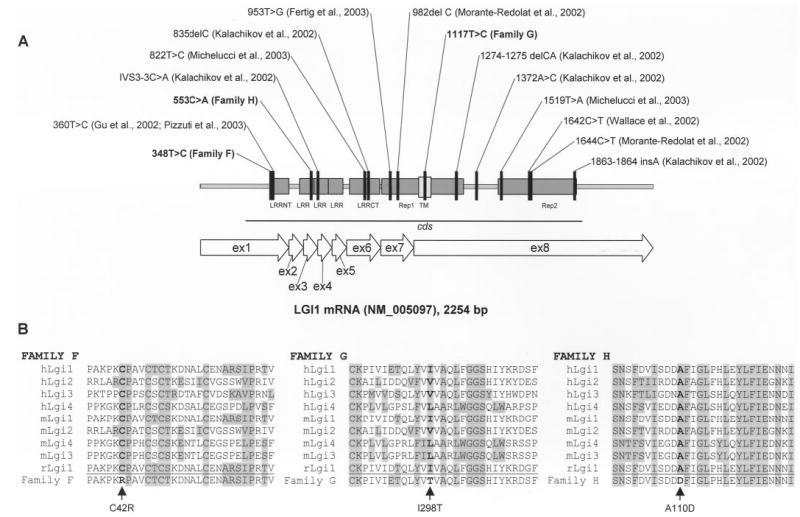
(A) Positions of LGI1 mutations in published families with autosomal dominant partial epilepsy with auditory features, counted from the first nucleotide of the LGI1 mRNA sequence from GenBank accession no. NM_005097. In the reports of the European collaborative group,7,13 the mutations were counted from the initiation codon; these have been renumbered from the first nucleotide. In addition, the 1320C>T mutation was corrected to 1420C>T (counting from the initiation codon; J. Perez-Tur, personal communication); this is shown as 1644C>T counting from the first nucleotide. (B) Positions of the amino acid substitutions on the multiple alignments of Lgi proteins in human, mouse, and rat.
LGI1 is not homologous to any known ion channel, and the mechanism by which it causes epilepsy is unknown. Protein homology suggests it is likely to be involved in CNS development.6 The encoded protein, Lgi1, contains three leucine-rich repeats (LRRs) and aligns closely with other LRRs that bind nerve growth factor and other neurotrophins.6 LGI1 is a member of a subfamily of LRR-encoding genes, denoted LGI1, LGI2, LGI3, and LGI4.14
Expression of LGI1 is absent or significantly reduced in many high-grade, but not low-grade, gliomas, suggesting that it may play a role in progression of glial tumors.15–17 However, the gene is expressed primarily in neurons rather than glia,6,7 and no definite cases of glioblastoma have been reported in families with epilepsy caused by LGI1 mutations.
This article addresses three questions of importance with respect to LGI1 mutations: 1) how common are LGI1 mutations in families containing multiple individuals with ictal auditory symptoms; 2) what is the penetrance of LGI1 mutations in families with ADPEAF; and 3) what is the range of phenotypic manifestations of mutations in LGI1? Answers to these questions will help identify patients likely to carry mutations and may also provide clues to the mechanism by which mutations increase risk for epilepsy.
Methods.
Clinical data collection.
The current study included 10 newly tested families containing 2 or more subjects with idiopathic epilepsy with ictal auditory symptoms. Three of the 10 families were referred specifically because of auditory symptoms. The remaining 7 families were drawn from our database of 94 families collected for genetic linkage analysis. All families containing two or more subjects with ictal auditory symptoms were selected from the linkage database for inclusion. The linkage families were ascertained without regard to epilepsy syndrome or occurrence of auditory symptoms; hence, we also used them to estimate the proportion of familial focal epilepsy that meets criteria for ADPEAF.
Both sets of families were ascertained through a variety of mechanisms, including physician referrals from neurologists at our institution and elsewhere, self-referrals solicited through a study web site, flyers mailed to voluntary organizations, and presentations at meetings for people with epilepsy. The Columbia University Medical Center Institutional Review Board approved the study.
The protocol for clinical diagnosis and classification has been described in detail previously.18 Briefly, we collected information on each family member using a set of validated semistructured interviews, usually administered by telephone.19,20 Whenever possible, we also collected medical records from the patients’ treating physicians. These frequently contained additional seizure descriptions, histories of etiologic factors, and EEG and neuroimaging data. Some patients were also given a brief neurologic examination and a study EEG. We did not perform EEGs on unaffected individuals except when EEG data might have helped to confirm a suspected epilepsy diagnosis.
To derive a final diagnosis, the information collected on each subject was reviewed by experienced epileptologists (W.A.H. and T.A.P.), who were blinded to information about other family members. Interpretation of the data from medical records, especially EEG and neuroimaging reports, included an assessment of the quality of the information received. When an actual EEG tracing was included in the medical record, we reviewed it directly. When only an EEG report was available, we assessed its likely validity by determining whether it was performed at an established epilepsy center, the reporting electroencephalographer was board certified, and the description and clinical-electrographic correlation were consistent and appropriate. Reports of questionable validity were not used to make a diagnosis.
Epilepsy was defined as a lifetime history of two or more unprovoked seizures. Subjects with epilepsy whose first unprovoked seizure was preceded by an insult to the CNS were classified as remote symptomatic, and those with no identified cause were classified as idiopathic. Seizures precipitated by acute alterations in homeostasis or insults to the CNS (including febrile seizures) were excluded from the definition of epilepsy and classified as acute symptomatic. Seizures were classified according to the 1981 criteria of the International League Against Epilepsy (ILAE),21 and epilepsies were classified according to the 1989 ILAE criteria for classification of epilepsy syndromes.22
Because of our special interest in auditory symptoms, we added a question to the diagnostic interview to ask about them systematically, both preceding generalized tonic-clonic seizures and during small spells: “Do you usually hear any unusual sounds or have any change in your hearing? (If yes) What is the sound or change in your hearing?” In families in which two or more individuals with partial seizures responded “yes” to this question, we reviewed all of the information collected for each individual a second time to classify the semiology of partial seizures. We used five categories of semiology—sensory, motor, autonomic, psychic, and reflex—and subclassified sensory symptoms as auditory, visual, vertiginous, olfactory, gustatory, somatosensory, and cephalic. We also subclassified auditory symptoms into five categories: simple (e.g., buzzing, ringing, and humming), complex (e.g., voices or music), distortion (e.g., volume changes), cognitive (receptive aphasia), and reflex (seizures precipitated by sounds; see supplementary data, classification of auditory symptoms and tables E1 to E3, on the Neurology Web site for additional details; go to www.neurology.org).
Molecular analysis.
Whole blood samples were collected in sodium acid citrate dextrose tubes and processed within 3 days of collection. To detect sequence variants in LGI1, we sequenced the gene’s eight coding exons in DNA extracted from blood or EBV-transformed lymphoblastoid cell lines. PCR primers were placed at the exon boundaries to amplify complete exonic sequences and corresponding intron–exon junctions. PCR amplification products were purified over 96-well glass fiber plates (Whatman, Kent, UK) and sequenced in both directions using dye-terminator chemistry and ABI 373 automated sequencers (Applied Biosystems, Weiterstadt, Germany). Sequence variants were identified using the Sequencher3 program (Gene Codes Corporation, Ann Arbor, MI) and verified by manual inspection. Interpretation of the sequence analysis data was performed blind to disease status.
Statistical analysis.
All analyses were carried out with SPSS statistical software (Chicago, IL). We used Fisher’s exact tests for categorical variables and t-tests for continuous variables.
Results.
The 94 families collected for genetic linkage analysis contained 322 individuals with idiopathic epilepsy, of whom 169 had partial seizures. Thirty-four percent (58/169) of those with partial seizures had auditory symptoms. Fifty-one percent (48/94) of the families contained 2 or more subjects with idiopathic focal epilepsy, and of those, 19% (9/48) met our criteria for ADPEAF (i.e., 2 or more subjects with ictal auditory symptoms). Two of the nine families had been tested previously and found to have mutations in LGI1 (Families 6610 and C).6 The remaining seven linkage families that met criteria for ADPEAF are reported here.
Given our known interest in this syndrome, we explored the possibility that subjects with auditory symptoms had been referred to us selectively, leading to an overestimate of the proportion of familial focal epilepsy meeting criteria for ADPEAF. For this purpose, we examined separately the subset of 31 families that had been ascertained in our original familial aggregation study, between 1985 and 1988, before publication of the article describing the syndrome in 1995.1 These 31 families were virtually identical to the remaining families, both in the proportion containing two or more subjects with idiopathic focal epilepsy (52%) and in the proportion of those meeting criteria for ADPEAF (19%). The proportion of individuals with partial seizures who had ictal auditory symptoms was slightly lower in these 31 families than in the others (27% vs 38%), but the difference was not significant.
All 10 families included in the current study (7 from our linkage database and 3 others ascertained because of auditory symptoms) were of European descent. They contained 43 individuals with idiopathic epilepsy (4 primary generalized; 31 focal; 3 focal and primary generalized; and 5 unclassifiable). Seventy-nine percent (27/34) of those with partial seizures had ictal auditory symptoms. Age at onset averaged 14 ± 1.95 years (SE). None of the families contained an individual with glioblastoma.
We sequenced LGI1 in one affected subject from each family and identified mutations in three of them (two from the linkage database and one referred because of auditory symptoms). In the families of these three subjects, we sequenced the gene in all remaining sampled individuals and confirmed that the mutations cosegregated with epilepsy (figure 2).
Figure 2.
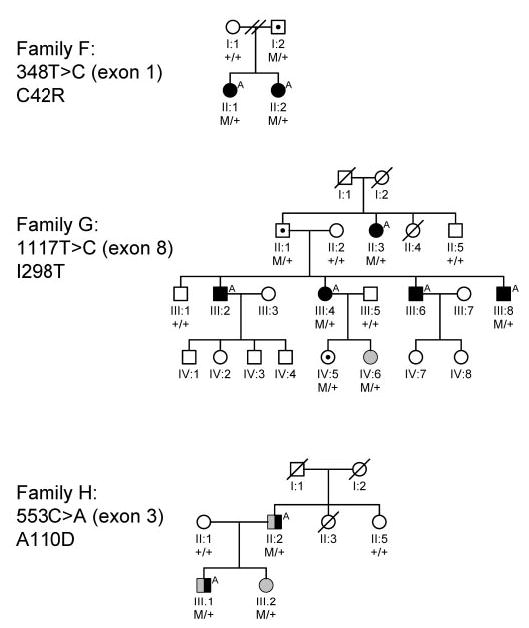
Families with newly identified mutations in LGI1. M/+indicates mutation carrier; +/+ indicates no mutation. Symbol definitions: □ ○ = Unaffected; ▪A •A = Idiopathic partial epilepsy with auditory features; ⊡ ⋅ = Unaffected mutation carrier; ▥ ◍ = Idiopathic generalized epilepsy; ◨ ◑ = Idiopathic partial and generalized epilepsy.
Family F had a missense mutation in exon 1 (348T>C, counting from the first nucleotide), resulting in a cysteine-toarginine substitution in amino acid residue 42 (C42R; see table and figure 2). Both affected sisters carried the mutation, as did their unaffected father.
Table.
Clinical features in three newly identified ADPEAF families with LGI1 mutations
| Family | Patient | Age at onset | Seizure type(s); and syndrome, if any* | EEG findings (source) | Auditory aura(s) | Other aura(s) |
|---|---|---|---|---|---|---|
| F | II:1 | 10 | SP, SGTC | Left frontal-temporal transients (EEG report from treating physician) | Complex, cognitive, reflex | Visual blurring, “odd smell” |
| II:2 | 12 | SP, CP, SGTC | Normal EEG (EEG report from treating physician) | Simple, complex, distortion, cognitive | Right eye and face twitching/tingling; colorful visual aura | |
| G | II:3 | 8 | CP, SGTC | No data available | Simple | Difficulty with motor control, dissociated feeling |
| III:2 | 22 | CP, SGTC | No data available | Simple, distortion, cognitive, reflex | Déjà vu, aphasia | |
| III:4 | 28 | SP, SGTC | No data available | Simple, distortion, cognitive | None | |
| III:6 | 18 | SP, SGTC | No data available | Distortion | Fear | |
| III:8 | 6 | CP, possible SP, SGTC | Intermitted theta and sharp transients in left temporal region (EEG report from treating physician) | Simple, cognitive | Aphasia, epigastric sensation | |
| IV:6 | 10 | Nocturnal GTC, IGE NOS | Generalized 2.5–3 hz spike-slow wave activity (EEG report from treating physician) | None | None | |
| H | II:2 | 13 | CP, SGTC, MYO; JME | 3 hz generalized spike-wave during hyperventilation and photic stimulation (EEG tracing reviewed directly) | Simple | Lightheadedness |
| III:1 | 11 | CP, SGTC, MYO; JME | Bilateral sharp and slow-wave activity with right sided predominance (EEG report from treating physician) | Distortion | Flowery olfactory aura, visual distortions and flickering lights | |
| III:2 | 3 | ABS, nocturnal GTC; pyknolepsy | 4 hz generalized spike-wave and polyspike wave (EEG tracing and report from our institution) | None | None |
Seizure types and syndromes: SP = simple partial; CP = complex partial; SGTC = secondarily generalized tonic-clonic; nocturnal GTC = generalized tonic-clonic, nocturnal only; MYO = myoclonic; ABS = absence; JME = juvenile myoclonic epilepsy; IGE NOS = idiopathic generalized epilepsy, not otherwise specified.
ADPEAF = autosomal dominant partial epilepsy with auditory features; LGI1 = leucine-rich glioma-inactivated 1.
Family G had a missense mutation in exon 8 (1117T>C), resulting in an isoleucine-to-threonine substitution in amino acid residue 298 (I298T). Five of the six affected individuals in the family had focal epilepsy with auditory symptoms. The remaining affected subject (IV:6) had two nocturnal generalized tonic-clonic seizures (GTCs) at age 10 and 11 years. No myoclonic or absence seizures were noted. An EEG report from the treating physician indicated generalized bursts of 2.5 to 3-Hz spike- and slow-wave activity. She was treated successfully with valproic acid. We classified her syndrome as idiopathic generalized epilepsy (IGE), not otherwise specified (NOS), a category we used for IGE-like syndromes that do not fit clearly into existing IGE categories (e.g., because of atypical age at onset or seizure type constellations, or isolated GTCs that do not occur on awakening).
All four tested affected family members (including the one with IGE NOS) had the mutation. The remaining two affected subjects declined to give blood samples. Three unaffected individuals in the family (one of whom was married in) did not carry the mutation. Two other unaffected subjects did carry the mutation, consistent with reduced penetrance: II:1, aged 76 years, and IV:5, aged 17 years at the time of study.
Family H had a missense mutation in exon 3 (553C>A), resulting in an alanine-to-aspartate substitution in residue 110 (A110D). Three individuals had idiopathic epilepsy, and all had the mutation. Two were classified as having focal epilepsy with auditory symptoms, but both also had generalized-onset (myoclonic) seizures. II:2 had secondarily generalized seizures beginning at age 13 years, preceded by a warning of feeling light-headed and hazy, and complex partial seizures with similar symptoms, also including a high-pitched ringing in his right ear. He also had myoclonic seizures while watching television, beginning at age 21 years, and witnessed by his wife. An EEG tracing from his treating physician, which we reviewed directly, showed 3-Hz generalized spike-wave activity during hyperventilation and photic stimulation. He was classified as having auditory partial epilepsy and juvenile myoclonic epilepsy.
Subject III:1 had complex partial and secondarily generalized seizures preceded by auditory symptoms, and also myoclonic seizures. GTCs began at age 11 years and were preceded by a combination of auditory, visual, olfactory, and autonomic symptoms. In addition to these partial seizures, he had myoclonic seizures, beginning at age 15 years, with rapid jerks of the arms and legs while watching television and occurring most often after sleep deprivation. He also described occasional eyelid twitching with these episodes and had no alteration of consciousness or confusion during or after these events. An EEG report from his treating physician indicated sharp and slow-wave activity with right-sided predominance. He was also classified as having auditory partial epilepsy and juvenile myoclonic epilepsy.
Subject III:2, who was treated at our institution, had pyknolepsy, onset at age 3 years, with 4-Hz generalized spike-wave and polyspike-wave activity on EEG. Absence seizures remitted at age 10 years. At age 22 years, she had two GTCs, the first ~8 months after a moderately severe head injury and the second ~2 months later. Both were nocturnal, with no warning or prodrome, and could not be classified further.
The positions of the three newly identified mutations within LGI1 and the alignments of the mutated Lgi1 proteins with other members of the LGI gene family are shown in figure 1. The C42R mutation in Family F affects the beginning of the cysteine-rich N-terminal LRR domain, which has cysteine in this position in all known Lgi proteins. The A110D mutation in Family H changes a highly conserved alanine to aspartate in the second LRR domain. The I298T mutation in Family G localizes to the protein’s putative transmembrane domain and substitutes a polar amino acid (threonine) at a highly conserved position normally occupied by aliphatic amino acids isoleucine or valine.
We estimated penetrance in all eight families in which we have identified mutations so far (five reported previously and three reported here).6 We excluded the probands and their first-degree relatives from this analysis because their epilepsy histories had led to selection of the families for study. The analysis included 76 remaining family members (excluding married-in subjects) aged >20 years with unambiguous diagnoses (15 with idiopathic epilepsy and 61 unaffected). In this group of relatives, the proportion tested for mutations was 87% (13/15) in affected subjects but only 39% (24/61) in unaffected subjects. To correct for this bias, we estimated the number of carriers among the 37 unaffected subjects who were not tested. Among unaffected family members who were tested, 21% (5/24) were carriers. Assuming the same proportion applies to those who were not tested, 7.7 (37 × 0.21) would be expected to carry a mutation. Similarly, we assumed that both of the untested affected subjects had a mutation because all of the tested affected subjects did. Based on these calculations, we estimated that 27.7 relatives had a mutation (i.e., 5 + 15 + 7.7), of whom 15 were affected, giving a penetrance estimate of 54% with an approximate 95% CI of 35 to 73%.
After excluding the family used to define the syndrome initially,1 we compared the clinical features in the seven families with mutations and seven families without (see supplementary data on the Neurology Web site for additional details). The two sets of families were similar in the number of individuals with idiopathic epilepsy, age at onset, and distribution of seizure type. Subjects with partial seizures were more likely to have auditory symptoms in families with mutations than in those without (96% vs 72%; p = 0.049; Fisher’s exact test). Visual symptoms were less common in families with mutations than in families without (29% vs 48%), but this difference was not significant. Autonomic symptoms were less common in families with mutations than in those without (17% vs 56%; p = 0.007; Fisher’s exact test). The two sets of families did not differ significantly in the types of auditory symptoms. In families with mutations, the most common type of auditory symptom was simple, unformed sounds such as buzzing and ringing (often accompanied by other auditory symptom types).
Discussion.
Mutations in LGI1 are a common cause of ADPEAF. Among 15 tested families, we have found mutations in 8 (5 reported earlier and 3 reported here). If we exclude the original family used to describe the syndrome and localize the gene, the proportion of tested families with mutations is 50% (7/14). The proportion of families with mutations will clearly depend on the criteria used to define ADPEAF. We have used a simple definition: two or more subjects with idiopathic focal epilepsy with ictal auditory symptoms. Our results suggest that families who meet this definition are likely to carry mutations in LGI1.
In parallel with our findings, a European collaborative group has found LGI1 mutations in 5 of 10 families they have analyzed so far, assembled from Spain, Italy, and Germany.7,13 They stated that families were selected based on autosomal dominant inheritance, seizures whose semiology indicated a lateral temporal lobe onset (not just auditory symptoms), absence of neurologic signs, and absence of known structural brain pathology. However, it is not straightforward to evaluate either consistency with autosomal dominant inheritance or consistency with lateral temporal lobe onset, and details of the specific criteria used were not given. Although auditory symptoms were not required for selection, all but one of their families had auditory symptoms; the remaining family had ictal aphasia without other auditory symptoms and was found to carry a mutation.
Our penetrance estimate of 54% (95% CI, 35 to 73%) is lower than that reported previously (71%).1 This lower estimate is probably explained, at least in part, by our correction for two biases that would have inflated the penetrance estimate. First, the families were ascertained because of multiple affected individuals, automatically inflating the proportion of tested individuals with idiopathic epilepsy. We corrected for this bias by excluding from the analysis probands and their first-degree relatives, whose epilepsy status defined eligibility of the families for inclusion. This approach reduced the number of relatives available for analysis and consequently also reduced the precision of our estimate. Second, affected family members were more likely to be tested for mutations than were unaffected family members (mostly because more unaffected subjects declined to give blood samples); therefore, a disproportionate number of unaffected mutation carriers were “missing” from the calculation. We corrected for this bias by estimating the number of untested family members likely to have a mutation. Although we corrected for these two biases, we could not correct for an additional selection bias inherent in the types of families we were studying. Given that these families contain a high proportion of affected individuals, they are automatically selected for high-penetrance mutations and for other genes or environmental factors that may influence expression.23 We are currently working on other methods to evaluate penetrance that address potential biases and consider the age at onset of the disorder.
Clarification of the clinical features of epilepsy associated with LGI1 mutations is important to facilitate identification of families with mutations and to provide clues to the mechanism of action of the gene. However, current information does not point to a clinical feature that clearly predicts which families have mutations among those containing two or more subjects with auditory symptoms. The proportion of individuals with auditory symptoms is inflated in the families included here because they were selected to contain two or more such individuals. However, despite our use of the same criteria to ascertain all of the families, auditory symptoms were more common in families with mutations than in those without. With the present sample size, we did not detect any significant differences in the types of auditory symptoms in families with vs without. Additional investigation of auditory symptom types with a larger sample size is warranted.
The significantly lower prevalence of autonomic symptoms in families with mutations than in those without (16% vs 56%) may indicate less mesial temporal involvement. Autonomic symptoms, particularly epigastric or visceral sensations, are characteristic of mesial rather than lateral temporal lobe seizure onset, although they may occur because of spread from other brain regions.
The prevalence of auditory symptoms in the families in our linkage study was surprisingly high. Overall, 34% of subjects with partial seizures in these families reported auditory symptoms, and among families containing two or more subjects with focal epilepsy, 19% met our criteria for ADPEAF. Selective referral of families with these symptoms because of our known interest in ADPEAF is not a likely explanation. Families ascertained before recognition of the syndrome were just as likely to contain two or more subjects with auditory symptoms as were those ascertained later. The proportion of individuals with partial seizures who had auditory symptoms was somewhat lower in families ascertained before syndrome recognition than in those ascertained later (27% vs 38%), but the difference was not significant. Also, in the families ascertained before symptom recognition, some of the data were collected before we added a specific question about auditory symptoms to the interview. The lower proportion of subjects with auditory symptoms in these earlier families may indicate that auditory symptoms are likely to be missed unless a specific question is asked, which is rarely done in clinical practice.
Our linkage families were not sampled systematically from a known population; therefore, the frequency of auditory symptoms, or of ADPEAF, remains to be determined in an unselected sample of epilepsy patients or families containing multiple affected persons. In any given collection of families, the proportion with ADPEAF depends on the proportion containing two or more subjects with idiopathic focal epilepsy (because this is a minimum requirement for ADPEAF). Further, among families containing two or more individuals with idiopathic focal epilepsy, the proportion with ADPEAF probably increases with increasing number of affected individuals. In our linkage database, the proportion of families meeting criteria for ADPEAF was 13% (5/39) in families containing 2 or 3 individuals with idiopathic focal epilepsy, but 44% (4/9) in families containing 4 or more individuals with idiopathic focal epilepsy.
The three new missense mutations reported here are all likely to be pathogenic. None was found in a set of 123 control subjects (246 chromosomes) of European ancestry we studied previously; all segregated with disease in the families in which they were found, and all affect conserved amino acid residues.6 The mutations in Families F and H are in the extra-cellular portion of the protein and probably change the properties of the block of LRR domains known to be involved in protein–protein interactions.24 The mutation in Family G is the first found to localize to Lgi1’s putative transmembrane region (see figure 1). Functional studies of the effects of all three mutations on neuronal excitability would be of great interest but are not possible at this time because of limited understanding of the gene’s effect.
The mutation in Family F, C42R, is only four amino acids away from the C46R mutation identified in two families, one Norwegian8 and the other Italian.9,10 Haplotype analysis of the two families with the C46R mutation is needed to explore their ancestral relationship; if they are truly unrelated, the finding of two closely located mutations in three families may indicate a mutation hotspot. Both families with the C46R mutation had sensory aphasia as an important part of the phenotype.9,25 In Family F, both affected individuals also reported difficulty in understanding language at the onset of their seizures. The occurrence of aphasia in all three families raises the possibility that these mutations have similar functional effects. However, aphasia has also been found in two families with mutations in exon 8: Family G, in which three of the five subjects with focal epilepsy had aphasia, and another Italian family reported recently with the 1519T>A mutation.13
We have found no definite cases of glioblastoma in the families with mutations, although one person in the original linkage family died of a brain tumor of unspecified type.6 This does not prove risk is not increased in these families because glioblastoma is rare, and few cases would be expected with such a small sample size. However, it does argue against a dramatic increase in risk.
Finally, one of our most interesting findings is the occurrence of IGEs in four subjects with mutations in two families with LGI1 mutations. In Family G, the subject with IGE NOS had only two seizures, both while asleep, and was classified as having generalized epilepsy based on her EEG. Her epilepsy may have been focal, and the generalized epilepti-form EEG abnormality may have been coincidental, or the original EEG may have been misinterpreted (although the EEG report appeared likely to be valid). In Family H, two subjects (II:2 and III:1) had generalized (myoclonic) seizures and focal epilepsy with auditory symptoms, and the other (III:2) had pyknolepsy followed by unclassifiable GTCs after a head injury of uncertain significance. The constellation of clinical features in Family H may be caused by two genotypes (LGI1 and an unidentified IGE genotype) segregating within the same family, each of which increases risk for a different type of epilepsy. Alternatively, LGI1 mutations may increase risk for generalized and focal epilepsy, with phenotypic expression determined by other interacting genes or environmental factors. Molecular investigation of LGI1’s function is needed to clarify how mutations increase risk for epilepsy and what phenotypes they are likely to produce. Analysis of the phenotypes associated with mutations, such as that reported here, is essential to interpret the functional data and may provide clues to function before molecular data are available.
Supplementary Material
Acknowledgments
The authors thank the families for their participation in this research. They also thank Wanda Garcia, Walkiria Jimenez, MS, and Carmen Liriano, MD, for their assistance with data collection.
Footnotes
Additional material related to this article can be found on the Neurology Web site. Go to www.neurology.org and scroll down the Table of Contents for the April 13 issue to find the title link for this article.
References
- 1.Ottman R, Risch N, Hauser WA, et al. Localization of a gene for partial epilepsy to chromosome 10q [see comments] Nat Genet. 1995;10:56–60. doi: 10.1038/ng0595-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ottman R, Barker-Cummings C, Lee JH, Ranta S. Genetics of autosomal dominant partial epilepsy with auditory features. In: Berkovic SF, Genton P, Hirsch E, Picard F, eds. Genetics of Focal Epilepsies: Clinical Aspects and Molecular Biology. London: John Libbey, 1999:95–102.
- 3.Winawer MR, Ottman R, Hauser WA, Pedley TA. Autosomal dominant partial epilepsy with auditory features: defining the phenotype. Neurology. 2000;54:2173–2176. doi: 10.1212/wnl.54.11.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Winawer MR, Martinelli Boneschi F, Barker-Cummings C, et al. Four new families with autosomal dominant partial epilepsy with auditory features: clinical description and linkage to chromosome 10q24. Epilepsia. 2002;43:60–67. doi: 10.1046/j.1528-1157.2002.45001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poza JJ, Saenz A, Martinez-Gil A, et al. Autosomal dominant lateral temporal epilepsy: clinical and genetic study of a large Basque pedigree linked to chromosome 10q. Ann Neurol. 1999;45:182–188. doi: 10.1002/1531-8249(199902)45:2<182::aid-ana8>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 6.Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335–341. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morante-Redolat JM, Gorostidi-Pagola A, Piquer-Sirerol S, et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet. 2002;11:1119–1128. doi: 10.1093/hmg/11.9.1119. [DOI] [PubMed] [Google Scholar]
- 8.Gu W, Brodtkorb E, Steinlein OK. LGI1 is mutated in familial temporal lobe epilepsy characterized by aphasic seizures. Ann Neurol. 2002;52:364–367. doi: 10.1002/ana.10280. [DOI] [PubMed] [Google Scholar]
- 9.Pizzuti A, Flex E, Di Bonaventura C, et al. Epilepsy with auditory features: a LGI1 gene mutation suggests a loss-of-function mechanism. Ann Neurol. 2003;53:396–399. doi: 10.1002/ana.10492. [DOI] [PubMed] [Google Scholar]
- 10.Pizzuti A, Giallonardo AT. Correction. Ann Neurol. 2003;54:137. [Google Scholar]
- 11.Fertig E, Lincoln A, Martinuzzi A, Mattson RH, Hisama FM. Novel LGI1 mutation in a family with autosomal dominant partial epilepsy with auditory features. Neurology. 2003;60:1687–1690. doi: 10.1212/01.wnl.0000063324.39980.4a. [DOI] [PubMed] [Google Scholar]
- 12.Wallace RH, Izzillo P, Macintosh AM, Mulley JC, Berkovic SF. Mutations in LGI1 in an Australian family with familial temporal lobe epilepsy with auditory features [abstract] Am J Hum Genet. 2002;71:s472. [Google Scholar]
- 13.Michelucci R, Poza JJ, Sofia V, et al. Autosomal dominant lateral temporal epilepsy: clinical spectrum, new epitempin mutations, and genetic heterogeneity in seven European families. Epilepsia. 2003;44:1289–1297. doi: 10.1046/j.1528-1157.2003.20003.x. [DOI] [PubMed] [Google Scholar]
- 14.Gu W, Wevers A, Schroder H, et al. The LGI1 gene involved in lateral temporal lobe epilepsy belongs to a new subfamily of leucine-rich repeat proteins. FEBS Lett. 2002;519:71–76. doi: 10.1016/s0014-5793(02)02713-8. [DOI] [PubMed] [Google Scholar]
- 15.Chernova OB, Somerville RP, Cowell JK. A novel gene, LGI1, from 10q24 is rearranged and downregulated in malignant brain tumors. Oncogene. 1998;17:2873–2881. doi: 10.1038/sj.onc.1202481. [DOI] [PubMed] [Google Scholar]
- 16.Somerville RP, Chernova O, Liu S, Shoshan Y, Cowell JK. Identification of the promoter, genomic structure, and mouse ortholog of LGI1. Mamm Genome. 2000;11:622–627. doi: 10.1007/s0033500101280. [DOI] [PubMed] [Google Scholar]
- 17.Cowell JK. Epilepsy research gets new guidance. Nat Med. 2002;8:219–220. doi: 10.1038/nm0302-219. [DOI] [PubMed] [Google Scholar]
- 18.Winawer MR, Rabinowitz D, Barker-Cummings C, et al. Evidence for distinct genetic influences on generalized and localization-related epilepsy. Epilepsia. 2003;44:1176–1182. doi: 10.1046/j.1528-1157.2003.58902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ottman R, Hauser WA, Stallone L. Semistructured interview for seizure classification: agreement with physicians’ diagnoses. Epilepsia. 1990;31:110–115. doi: 10.1111/j.1528-1157.1990.tb05368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ottman R, Lee JH, Hauser WA, et al. Reliability of seizure classification using a semistructured interview. Neurology. 1993;43:2526–2530. doi: 10.1212/wnl.43.12.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Commission on Classification and Terminology ILAE. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia. 1981;22:489–501. doi: 10.1111/j.1528-1157.1981.tb06159.x. [DOI] [PubMed] [Google Scholar]
- 22.Commission on Classification and Terminology ILAE. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989;30:389–399. doi: 10.1111/j.1528-1157.1989.tb05316.x. [DOI] [PubMed] [Google Scholar]
- 23.Begg CB. On the use of familial aggregation in population-based case probands for calculating penetrance. J Natl Cancer Inst. 2002;94:1221–1226. doi: 10.1093/jnci/94.16.1221. [DOI] [PubMed] [Google Scholar]
- 24.Scheel H, Tomiuk S, Hofmann K. A common protein interaction domain links two recently identified epilepsy genes. Hum Mol Genet. 2002;11:1757–1762. doi: 10.1093/hmg/11.15.1757. [DOI] [PubMed] [Google Scholar]
- 25.Brodtkorb E, Gu W, Nakken KO, Fischer C, Steinlein OK. Familial temporal lobe epilepsy with aphasic seizures and linkage to chromosome 10q22-q24. Epilepsia. 2002;43:228–235. doi: 10.1046/j.1528-1157.2002.32001.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.