

Abstract
Herpes simplex virus 1 mutants lacking the gene encoding glycoprotein D (gD) and the gD normally present in the envelope of the virus (gD−/− stocks) or mutants lacking the gD gene but containing trans-induced gD in their envelopes (gD−/+) cause apoptosis in human SK-N-SH cells. The gD−/− virions are taken up by endocytosis and are degraded, whereas gD−/+ viruses replicate but produce gD−/− virus. Apoptosis is blocked by delivery of the gD gene in trans. Studies designed to test several hypotheses concerning the role of gD in apoptosis revealed the following. (i) gD−/− and gD−/+ stocks induce fragmentation of cellular DNA in SK-N-SH, HEp-2, HeLa, and Vero cell lines. (ii) Chloroquine blocks apoptosis induced by gD−/− stocks but not by gD−/+ stocks. The drug also rescues gD−/− from degradation. (iii) Cells transduced with cation-independent mannose 6-phosphate receptor (CI-MPR) block apoptosis induced by either gD−/− or gD−/+ virus. (iv) Expression of sequences antisense to the cloned CI-MPR gene induced apoptosis by themselves. Wild-type virus but not gD−/− or gD−/+ stocks of mutant virus blocked apoptosis induced by the expression of CI-MPR antisense sequences. These results exclude the possibility that to block apoptosis, gD must interact with its HveA receptor, a member of the tumor necrosis factor alpha receptor family. Instead, the data suggest that gD blocks the influx of lysosomal enzymes into the endosomal compartment by binding to CI-MPR. This conclusion is consistent with published reports that phosphorylated gD interacts with CI-MPR.
In the course of studies of herpes simplex virus 1 (HSV-1) mutants which induce apoptosis, workers in this laboratory found that mutants lacking glycoprotein D (gD) induce programmed cell death in human SK-N-SH cells, as measured by translocation of annexin V and DNA fragmentation (29). The details and especially the requirements for blocking apoptosis by these mutants depend on the nature of the stocks. gD− mutants grown in cells expressing gD incorporate the glycoprotein in their envelopes and hence have been designated gD−/+. The gD−/+ virus stocks can infect gD− cells, but the progeny of this infection is cell associated, lacks gD in its envelope (and hence is designated gD−/−), and cannot productively infect cells.
Electron microscopic studies done at short intervals after infection with gD−/− stocks have revealed the presence of partially degraded viral particles in endosome-like vesicles, but no trace of virus particles or viral gene expression was detected past 1 h after infection. Cells transduced with baculoviruses expressing gD or gJ driven by the human cytomegalovirus immediate-early promoter blocked apoptosis induced by gD−/− or gD−/+ viruses (29). However, detailed analyses of the requirements of gD to block apoptosis suggest that the mechanisms by which the two virus stocks induce apoptosis are different. Specifically, whereas the ectodomain of gD is sufficient to block apoptosis induced by gD−/+ stocks, the structure required to block apoptosis induced by gD−/− stocks is the entire gD gene or a combination of two macromolecules, of which one consists of the gD ectodomain plus transmembrane domain and the other consists of the gD transmembrane domain and the cytoplasmic carboxyl-terminal domain (30). The conclusion of these studies is that while the blocking activity is in the ectodomain, the cellular compartment in which the ectodomain functions depends on the virus stock that triggers the apoptosis.
To block apoptosis, gD must interact with cellular proteins by itself or in association with other viral proteins. The functions of gD are well documented in numerous publications. In essence, gD mediates viral entry into cells by its interaction with one of several cellular receptors (27). The interaction with an entry receptor is followed by mobilization of additional viral glycoproteins which cause the fusion of the viral envelope with the plasma membrane. The major protein receptors involved in entry are HveA, a member of the tumor necrosis factor alpha (TNF-α) receptor family (20), and members of a family of surface proteins known as nectins (6). The cation-independent mannose 6-phosphate receptor (CI-MPR) has also been reported to interact with gD, albeit it does not appear to play a role in entry (4, 5). Conceivably, each of these receptors could, in the absence of the gD ligand, activate a cascade of events leading to cell death. Specifically:
(i) HveA and lymphotoxin β receptor are known to bind LIGHT, a member of a superfamily of TNF ligands. Lymphotoxin β receptor has been reported to be both necessary and sufficient for the induction of apoptosis in tumor cells (25). HveA together with its ligand has also been reported to activate NF-κB and AP-1 (18). It is conceivable that in uninfected cells in the absence of gD, unengaged HveA could serve a role similar to that of lymphotoxin β.
(ii) The nectin family of proteins is a key and integral component of intercellular adhesion complexes (24, 28). Although there is no direct evidence of association of nectin with programmed cell death, it is conceivable that morphological changes in the cell surface structure disrupt the nectin-nectin interactions between adjacent cells that, in the absence of a gD, could lead to apoptosis.
(iii) CI-MPR has been implicated in numerous regulatory and transport functions. CI-MPR has been postulated to be a tumor suppressor protein (14); CI-MPR is frequently mutated in human breast and colon cancers (2). CI-MPR also regulates the targeting of lysosomal enzymes. Thus, CI-MPR binds newly synthesized lysosomal enzymes which are then packaged in transport vesicles and transported with CI-MPR to the endosomal compartment from which CI-MPR is recycled and the lysosomal enzymes are transported to lysosomes (13, 31). CI-MPR is the receptor of granzyme B, a serine protease released into targeted cells by cytotoxic T lymphocytes (21). Conceivably, granzyme B interacts with CI-MPR to block the excessive release of lysosomal enzymes that induce cell death.
The objective of the studies described in this report was to begin to unravel the role of gD in blocking apoptosis induced in the infected cells in its absence. The key findings reported here are as follows.
(i) Earlier studies were done with SK-N-SH cells derived from a malignant glioma. In this report we show that gD−/− and gD−/+ induce apoptosis in HEp-2, HeLa, and Vero cell lines.
(ii) Chloroquine blocks apoptosis induced by gD−/− but not by gD−/+ stocks and prevents the degradation of gD−/− virus. The results suggest that acidification of the endosomal compartment is an essential step to the degradation of the gD−/− virions and cell death and are consistent with the hypothesis that the events critical to apoptosis in cells infected with gD−/− and gD−/+ viruses take place in different cellular compartments.
(iii) Cloned CI-MPR blocks apoptosis induced by either gD−/− or gD−/+ viruses in SK-N-SH cells.
MATERIALS AND METHODS
Cells and viruses.
SK-N-SH, HEp-2, HeLa, and Vero cells were obtained from the American Type Culture Collection (Rockville, Md.) and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. Insect cell line sf9 (Spodoptera frugiperda) was obtained from PharMingen (San Diego, Calif.). Unless indicated, cultures were seeded less than 20 h prior to infection and assayed at 60 to 70% confluence. gD−/− and gD−/+ stocks of mutant viruses were produced as described elsewhere (29). The HSV-1(KOS) d120 mutant, a kind gift of N. DeLuca (University of Pittsburgh), lacks both copies of the α4 gene and was grown in a Vero-derived cell line (E5) expressing the α4 gene. The d120 mutant induces programmed cell death in all cell lines tested to date, although the mechanism by which this is accomplished is cell line dependent (8-10).
Infection of cell lines with gD−/− or gD−/+ and assays for fragmentation of cellular DNA.
Subconfluent cultures of HEp-2, HeLa, SK-N-SH, or Vero cells in 25-cm2 flasks were mock infected or exposed to HSV-1(F) or gD−/− or gD−/+ mutant virus stocks for 2 h at PFU/cell ratios as stated in Results. The inocula were then replaced with fresh medium and incubation at 37oC was continued for an additional 18 to 24 h as stated in Results. The cells were harvested and assayed for the presence of fragmented cell DNA as described in detail elsewhere (10).
Construction of baculovirus recombinants expressing sense and antisense CI-MPR.
The baculovirus transfer vector pAc-CMV, containing the cytomegalovirus immediate-early promoter-enhancer sequences in the XhoI-BamHI sites of pAc-SG2, was described elsewhere (29). The plasmid including full-length CI-MPR was a kind gift provided from R. G. MacDonald, University of Nebraska, Omaha.
To construct the hemagglutinin (HA)-tagged CI-MPR into pAc-CMV, the plasmid was digested with SalI and XbaI to release 8.6 kb of the CI-MPR gene (23). This fragment was further digested with NdeI to release the 600-bp fragment from the 3′ terminus. This fragment was used as a template to amplify the HA-tagged carboxyl-terminal fragment with the forward primer GGAGCCAACATATGCCAGGTGAAGCCCAACGATC and the reverse primer GCTCTAGATCAAGCATAATCTGGCACATCATAGATGTGTAAGAGGTCCTCGTCGCTG. The HA tag was inserted in front of the stop codon. The PCR fragment was inserted back into the NdeI/XbaI fragment to yield the HA-tagged CI-MPR. The HA-tagged CI-MPR was blunted by Klenow and inserted into the transfer vector pAc-CMV predigested with EcoRI and BglII and blunted by Klenow. The resulting clones were screened for both sense and antisense orientations of CI-MPR sequence relative to the cytomegalovirus promoter-enhancer sequence.
The procedures for generation of recombinant baculovirus and infection of mammalian cells were as described elsewhere (29).
Immunoblot assays.
Polyclonal antibody against HA was purchased from Santa Cruz Biotechnology. Protein concentration in whole-cell lysates was determined with a protein assay reagent (Bio-Rad Laboratories, Hercules, Calif.). Infected or uninfected cell lysates (50 mg of protein per lane) were electrophoretically separated in 7% denaturing polyacrylamide gels, electrically transferred to a nitrocellulose sheet, blocked for 2 h in 5% milk in phosphate-buffered saline (PBS) at room temperature, and then reacted with the primary antibody as indicated. After a thorough rinse, the nitrocellulose sheets were reacted at room temperature for 1 h with the secondary antibody (Bio-Rad) conjugated to alkaline phosphatase diluted 1:3,000 in PBS and containing 1% bovine serum albumin and 0.05% Tween 20, rinsed in AP buffer (100 mM Tris [pH 9.5], 100 mM NaCl, and 5 mM MgCl2), and developed by the addition of AP buffer containing 5-bromo-4-chloro-3-indolylphosphate and nitroblue tetrazolium. The reaction was stopped by the addition of stop buffer (100 mM Tris [pH 7.6], and 10 mM EDTA). All rinses were done with PBS containing 0.05% Tween 20.
Chloroquine treatment.
Subconfluent (70%) cultures of SK-N-SH cells in 25-cm2 flasks were exposed to 10 μM, 50 μM, 100 μM, or 200 μM chloroquine (Sigma, St. Louis, Mo.) for 1 h. The cells were rinsed with mixture 199 supplemented with 1% calf serum (medium 199V) three times and maintained in DMEM supplemented with 10% fetal bovine serum.
X-Gal staining.
The cells were rinsed twice with PBS, fixed for 10 min at 4°C in 2% paraformaldehyde-0.05% glutaraldehyde solution, rinsed four times with PBS, and overlaid with X-Gal staining solution (5 mM potassium ferricyanide, 5 mM potassium ferricyanide·3H2O, 2 mM MgCl2, and 1 mg of X-Gal per ml) for 3 to 6 h at 37°C. Rinsing the cells twice with PBS stopped the X-Gal reaction.
Total RNA extraction and reverse transcription (RT)-PCR detection of antisense CI-MPR.
Subconfluent cultures of SK-N-SH cells in 25-cm2 flasks were infected with 10 PFU of recombinant baculovirus carrying CI-MPR in the antisense orientation. After 24 h of incubation, the medium was replaced with 1 ml of Trizol (Gibco-BRL #15596-018). The cell lysates were scraped, collected, and extracted once with chloroform. The RNA was precipitated from the aqueous phase with isopropyl alcohol, collected by centrifugation at 10,000 rpm for 20 min, rinsed with 75% ethanol, dried, and dissolved in 18 μl of diethyl pyrocarbonate (DEPC)-treated water. The RNA was treated with 0.5 U of RNase-free DNase (Gibco-BRL #18068-015) for 1 h at 37°C and extracted with phenol-chloroform, precipitated, and finally dissolved in DEPC-treated water.
For RT-PCR, 1 μg of total RNA was mixed with 120 ng of forward primer (for antisense CI-MPR detection) or reverse primer (for endogenous CI-MPR detection) in a 16-μl total volume and heated at 70°C for 10 min and chilled in ice for 3 to 5 min. The PCR was then carried out with forward and reverse primers according to the instructions contained in the RT-PCR kit (Promega, Madison).
Double infection.
Subconfluent cultures of SK-N-SH cells in 25-cm2 flasks were first exposed at 37°C for 2 h to 10 PFU of recombinant baculovirus per cell and then to either 100 PFU equivalents of gD−/− or 10 PFU of gD−/+ mutant viruses per cell or wild-type HSV-1(F) as indicated in Results. The cell cultures were then maintained for an additional 18 h at 37°C in medium containing 2.5 mM sodium butyrate.
RESULTS
gD−/− and gD−/+ induce apoptosis in several cell lines.
Earlier studies of the apoptosis induced by gD−/− and gD−/+ virus stocks were done primarily with SK-N-SH cells (29). In order to determine whether apoptosis was associated with a unique gD receptor, it was of interest to survey several cell lines for their susceptibility to apoptosis on exposure to either gD−/− or gD−/+ virus. In this series of experiments, replicate cultures of HEp-2, HeLa, Vero, or SK-N-SH cells were exposed to gD−/− or gD−/+ viruses in ratios ranging from 10 to 150 PFU equivalents of gD−/− or 5 to 20 PFU of gD−/+ per cell.
In the earlier study we showed that gD−/− stocks required a higher multiplicity of infection then gD−/+ stocks to induce apoptosis (29). Wild-type virus does not induce apoptosis at any multiplicity tested. The cells were harvested at 18 h after infection and processed as described in Materials and Methods. The results (Fig. 1) were as follows. Cellular DNA fragmentation was observed in all cell lines exposed to at least 100 PFU equivalents of gD−/− and 15 PFU of gD−/+ stocks per cell. The DNA fragment ladders were clearly delineated in electrophoretically separated DNA fragments in lysates of SK-N-SH, Vero, and HeLa cells. The DNA fragments contained in lysates of infected HEp-2 cells were more random in size distribution, suggesting the possibility that the degradation of DNA in HEp-2 cells occurs by a process different from that occurring in other cell lines.
FIG. 1.

gD−/− and gD−/+ stocks of gD null mutants of HSV-1 induce cellular DNA fragmentation apoptosis in HEp-2, HeLa, SK-N-SH, and Vero cells. Replicate cultures of SK-N-SH, Vero, HEp-2, and HeLa cells were infected with gD−/− (A) or gD−/+ (B) stocks as shown. The cell were harvested at 18 h after infection and processed for the DNA fragmentation assay as described in Materials and Methods.
Chloroquine blocks fragmentation of DNA induced by gD−/− virus but not by gD−/+ virus.
Subconfluent (70%) replicate cultures of SK-N-SH cells were exposed to chloroquine as described in Materials and Methods either 2 h before (−2, Fig. 2, lanes 3 to 6), concurrent with (0, Fig. 2, lanes 7 to 10), or 2 h after infection (+2, Fig. 2, lanes 11 to 14) with either gD−/− stock (Fig. 2A, 100 PFU/cell) or gD−/+ stock (Fig. 2B, 10 PFU/cell). The cells were harvested at 18 h after infection and examined for the presence of fragmented DNA.
FIG. 2.

Chloroquine blocks the fragmentation of cellular DNA in cells exposed to gD−/− (A) but not gD−/+ (B) stocks of gD null mutant. Replicate 25-cm2 flasks of subconfluent cultures of SK-N-SH cells were exposed to chloroquine for 1 h and then infected with either 100 PFU equivalents of gD−/− (A) or 10 PFU of gD−/+ (B) stock per cell. The cells were harvested at 18 h postinfection and processed as described in Materials and Methods.
As shown in Fig. 2A, 100 μM chloroquine pretreatment partly protected the cells from apoptosis induced by gD−/− stocks (lane 5). Fragmented DNA was not detectable in cells pretreated with 200 μM of chloroquine (Fig. 2A, lane 6). Moreover, DNA fragmentation was blocked by chloroquine added to the medium before or concurrent with exposure to the virus (0 h) but not at 2 h after infection (Fig. 2A, lanes 11 to 14).
As shown in Fig. 2B, chloroquine treatment of cells infected with gD−/+ stocks did not block fragmentation of cellular DNA.
Chloroquine blocked the degradation of gD−/− virus.
An earlier report has shown that gD−/− virions were internalized into cytoplasmic vesicles and degraded (29). To determine whether in addition to blocking apoptosis chloroquine also blocks degradation of the endocytosed virions, subconfluent cultures of SK-N-SH cells in a six-well plate were exposed to 100 PFU equivalents of gD−/− stock per cell for 30 min, then mock treated or overlaid with medium containing 200 μM chloroquine for 1 h. The cells were harvested 18 h after infection and stained with X-Gal as described in Materials and Methods. The X-Gal-positive cells were 0 for mock-infected cells, 30 for infected, untreated cells, and 320 for infected and chloroquine-treated cells.
Interpretation of these results hinges in part on the experimental design, which limited exposure of cells to the virus for 30 min and exposure to chloroquine for only 1 h after exposure to virus, and in part on the absence of data on the efficiency of uptake of gD−/− virus into endocytic vesicles. Notwithstanding the limitations imposed by the experimental design, the data indicate that the treatment with chloroquine significantly increased the number of cells expressing the reporter protein encoded in the gD−/− virus.
Expression of cloned mannose 6-phosphate receptor in sense and antisense orientations.
As described in Materials and Methods, cloned CI-MPR was inserted into a baculovirus in the sense and antisense orientations driven by the immediate-early cytomegalovirus promoter. The objective of the series of experiments described below was to verify the expression of the cloned sequences.
In the first series of experiments, SK-N-SH cells in replicate cultures were infected with 10 PFU of recombinant baculovirus carrying the CI-MPR gene in a sense orientation. The cells were harvested 24 h after infection, solubilized, subjected to electrophoresis in a denaturing polyacrylamide gel, transferred to a nitrocellulose sheet, and reacted with an anti-HA tag polyclonal antibody. The results (Fig. 3) indicate that the recombinant baculovirus expressed a protein that reacted with the antibody to the HA tag (Fig. 3, lane 2). The size of the protein is consistent with the reported size of CI-MPR, Mr 275,000.
FIG. 3.

HA-tagged CI-MPR encoded in a baculovirus is expressed in mammalian cells. SK-N-SH cells in 25-cm2 flasks were exposed to 10 PFU of CI-MPR recombinant baculovirus per cell. The cells were harvested at 24 h after infection, solubilized, subjected to electrophoresis in 7% denaturing polyacrylamide gels, electrically transferred onto a nitrocellulose sheet, and reacted with a polyclonal antibody to the HA tag. The heavy band migrating between Mr 97,000 and 116,000 reacted with anti-HA antibody and is probably a degradation product.
In the second series of experiments, SK-N-SH cells were infected with 10 PFU of a recombinant baculovirus carrying the CI-MPR gene in the antisense orientation. Total cell RNA was extracted from cells harvested 24 h after infection and processed by RT-PCR as described in Materials and Methods. The results (Fig. 4) were as follows. As expected, RNA antisense to CI-MPR was detected by RT-PCR initiated with the forward primer (Fig. 4, lane 3). Trace amounts of endogenous MPR RNA were detected with the reverse primer (Fig. 4, lane 4). The size of the RT-PCR product could not be differentiated from that obtained by PCR using the anti-MPR-pAc-CMV as the template (Fig. 4, lane 5). Lastly, the RT reaction was essential for the detection of RNA; in its absence, no products were detected (Fig. 4, lanes 1 and 2).
FIG. 4.
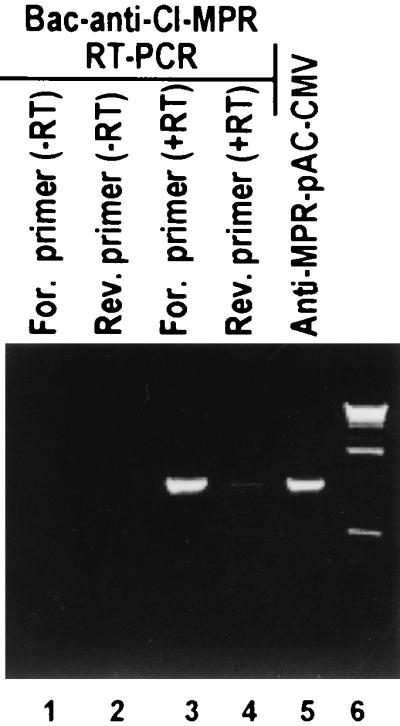
CI-MPR antisense RNA is expressed from baculoviruses encoding the gene in antisense orientation. SK-N-SH cells in 25-cm2 flasks were exposed to recombinant baculovirus encoding CI-MPR in an antisense orientation vis-a-vis the cytomegalovirus promoter (10 PFU/cell). The cells were harvested at 24 h after infection. Total RNA was extracted and subjected to RT-PCR as detailed in Materials and Methods.
We conclude from these series of experiments that the recombinant baculoviruses expressed both the sense and antisense products of the CI-MPR gene.
Expression of CI-MPR protects SK-N-SH cells from apoptosis induced by either gD−/− or gD−/+.
SK-N-SH cells were exposed to 10 PFU of recombinant baculovirus per cell for 2 h and then infected with gD−/− or gD−/+ virus. The cells were harvested 18 h after HSV-1 infection and processed as described in Materials and Methods. The results (Fig. 5) were as follows. DNA fragmentation induced by gD−/− or gD−/+ stocks was blocked by baculovirus expressing the CI-MPR protein (Fig. 5, lanes 5 and 6). The mechanism by which CI-MPR blocks the fragmentation of cellular DNA appears to be restricted inasmuch as the DNA fragmentation induced by the d120 mutant in SK-N-SH cells was not blocked by infection of the cells with the baculovirus expressing CI-MPR in the sense orientation (Fig. 5, lanes 7 and 8).
FIG. 5.

CI-MPR blocks apoptosis induced by gD−/− or gD−/+ stocks but not by the d120 mutant of HSV-1. Replicate cultures of subconfluent SK-N-SH cells in 25-cm2 flasks were infected with 10 PFU of baculovirus per cell for 2 h and then infected with 100 PFU equivalents of gD−/−, 10 PFU of gD−/+, or 10 PFU of d120 virus per cell. The cells were harvested 18 h after HSV infection and processed for the presence of fragmented cellular DNA as described in Materials and Methods.
Antisense CI-MPR induced apoptosis in SK-N-SH by itself.
In this series of experiments, SK-N-SH cells infected with 10 PFU of baculovirus expressing the CI-MPR in an antisense orientation were harvested at 18 after infection and processed for the presence of fragmented DNA as described in Materials and Methods. As shown in Fig. 6, lanes 1 and 3, the expression of sequences antisense to the CI-MPR gene induced DNA fragmentation in the absence of any other exogenous gene expression.
FIG. 6.

Expression of the sequences antisense to CI-MPR causes fragmentation of cellular DNA that is not blocked by gD. Replicate subconfluent SK-N-SH cell cultures in 25-cm2 flasks were exposed for 2 h to baculovirus encoding CI-MPR in the antisense orientation relative to the cytomegalovirus promoter (10 PFU/cell) alone or in mixtures with 10 PFU of baculovirus encoding wild-type gD (Bac-gD) and then infected with 100 PFU equivalents of gD−/− virus or 10 PFU of gD−/+ virus stock per cell. The cells were harvested 18 h after HSV infection and processed for the presence of fragmented cellular DNA as described in Materials and Methods.
To test whether gD blocks apoptosis induced by expression of sequences antisense to the CI-MPR gene, SK-N-SH cells were doubly infected with 100 PFU of gD−/−stock or 10 PFU of gD−/+ stock and 10 PFU of baculovirus expressing the CI-MPR gene in the antisense orientation or 10 PFU of baculovirus expressing wild-type gD per cell for 1 h, then incubated for an additional 18 h. The results (Fig. 6 and 7) were as follows.
FIG. 7.
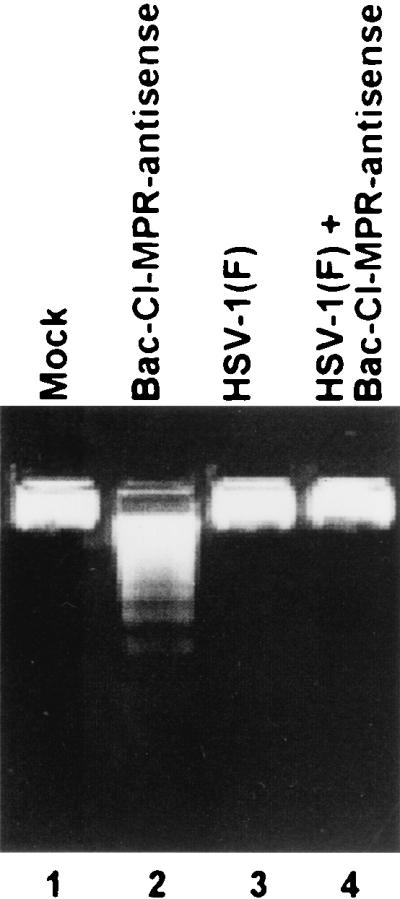
HSV-1(F) blocks apoptosis induced by the expression of sequences antisense to CI-MPR. Subconfluent SK-N-SH cells in 25-cm2 flasks were infected with 10 PFU of recombinant baculovirus expressing the sequences antisense to CI-MPR per cell for 2 h and then infected with 10 PFU of HSV-1(F) stock per cell. Cells mock infected or infected with baculovirus or HSV-1(F) alone served as controls. The cells were harvested 18 h after HSV infection and processed for the presence of fragmented DNA as described in Materials and Methods.
Neither gD−/− stocks nor gD−/+ stocks were able to block apoptosis induced by the expression of sequences antisense to the CI-MPR gene (Fig. 6, lanes 5 and 6 and 9 and 10). As expected from results published earlier, the fragmentation induced by the gD−/+ or gD−/− stocks was blocked by concurrent transduction of cells with baculoviruses expressing wild-type gD (Fig. 6, lanes 4 and 8).
The objective of the last series of experiments was to determine whether wild-type virus could block apoptosis induced by the expression of sequences antisense to the CI-MPR. Replicate cultures of SK-N-SH cells in 25-cm2 flasks were infected with 10 PFU of the baculovirus carrying CI-MPR in an antisense orientation and 10 PFU of HSV-1(F). The cells were harvested 18 h after infection and processed as described above. The results (Fig. 7) show that HSV-1(F) blocks apoptosis induced by the antisense expression of the CI-MPR gene (Fig. 7, lanes 2 and 4), As expected, HSV-1(F) did not cause fragmentation of cellular DNA (Fig. 7, lane 3).
DISCUSSION
Glycoprotein D interacts with at least three classes of cellular proteins. These are HveA, members of the nectin family, and CI-MPR (4, 27). The studies described in this report are the first step in the process of defining the role of cellular proteins that interact with gD in apoptosis induced by the mutants lacking the glycoprotein. The salient features of this report are as follows.
CI-MPR, the protein known to interact with gD, does appear to play a role in apoptosis. Specifically, the results presented in this report show that overexpression of CI-MPR blocks apoptosis induced by both gD−/− and gD−/+ viruses. It is convenient to consider the role of CI-MPR in blocking apoptosis induced by gD−/− and gD−/+ stocks separately.
As noted in this and earlier studies, the quantities of gD−/− particles required to induce apoptosis appear to be significantly higher than those required to induce apoptosis by gD−/+ stocks. The gD−/− virions are taken up in endosome-like vesicles and appear to be rapidly degraded, and ultimately the infected cells undergo programmed cell death. The hypothesis that explains these observations is that gD−/− particles are endocytosed and degraded by lysosomal enzymes and that when the concentration of endocytosed particles is very high, as would be expected from the ratio of PFU equivalents per cell required to induce apoptosis, the enzymes engaged in the degradation of virus particles initiate a process that ultimately leads to cell death. The results presented in this report are consistent with and support this hypothesis.
(i) Chloroquine, which blocks acidification of endosomal vesicles, blocks apoptosis in cells exposed to this drug before or concurrent with exposure to gD−/− mutant stocks but not in cells exposed to the drug 2 h after infection, that is, after the virus is degraded. A potentially causal link between degradation of virus particles by lysosomal enzymes and apoptosis is also apparent from the observation that even under suboptimal conditions chloroquine precluded total destruction of gD−/− virus.
(ii) As noted in the introduction, CI-MPR is involved in the transport of lysosomal enzymes to the endocytic pathway from which CI-MPR is recycled and lysosomal enzymes are diverted to lysosomes (3, 7). CI-MPR has been shown by Brunetti and colleagues (5) to interact with gD and to colocalize either with gD alone or with virus particles in endosomes. As illustrated diagrammatically in Fig. 8, a function of gD may be to retain CI-MPR in endosomes by binding the protein and preventing the release of lysosomal enzymes. Overexpression of CI-MPR may have a similar effect by affecting the transport of lysosomal enzymes.
FIG. 8.

Schematic representation of a model of the intracellular events in cells infected with gD null mutants. (A) Exposure of cells to gD−/− stocks of gD null mutants. Virions are taken up in endocytic vesicles (a) to which CI-MPR delivers lysosomal enzymes (b, yellow vesicles). CI-MPR is recycled (green arrow, white vesicles). gD−/− virions are degraded (c) and lysosomal enzymes cause nucleosomal degradation of cellular DNA. (d) Chloroquine blocks acidification of the vesicles and the fragmentation of DNA. There is reduced degradation of virions inasmuch as a small fraction expressed reporter genes encoded in the viral genome. (B) Exposure of gD−/− stocks in cells expressing gD. Glycosylated gD with mannose 6-phosphate interacts with CI-MPR and consequently apoptosis is blocked but viral infection does not ensue. gD containing the transmembrane domain could be expected to be transported to the Golgi stacks and be incorporated in the membranes of endocytic vesicles. (C) Exposure of cells to wild-type virus. Since the multiplicity is lower, fewer virions are endocytosed, and hence the induction of apoptosis at early stages of infection either does not ensue or is blocked by gD present in virion envelopes. At late stages of infection, gD made by the virus relatively early in infection saturates cellular compartments (a) and is present in the secretory pathway used by the virus to egress from the cell (b). (D) gD−/+ virus infects cells by fusion of the plasma membrane with the viral envelope, and the multiplicity of infection required to induce apoptosis is significantly lower than that required with gD−/− stocks. At lower multiplicities of infection, fewer particles are endocytosed, and most likely apoptosis is activated at late stages of infection rather than at the time of entry into cells. gD−/+ stocks produce gD−/− virus in infected cells. In the course of entry into the secretory pathway, the progeny gD−/− virus induces apoptosis (a). In cells expressing gD (b), apoptosis is blocked. Intact gD or gD ectodomain minus the transmembrane domain would also be excreted via the secretory pathway and be available to interact with CI-MPR to block apoptosis. N, nucleus.
Support for the hypothesis that gD specifically interacts with CI-MPR through its phosphorylated mannose residues also emerged from extensive studies of the function of numerous gD mutants (G. Zhou, E. Avitabile, G. Campadelli-Fiume, G. H. Cohen, R. J. Eisenberg, and B. Roizman, unpublished data). In these studies we found that gD carrying amino acid substitutions that precluded N-linked glycosylation failed to block apoptosis induced by either gD−/− or gD−/+ mutant stocks. The hypothesis that CI-MPR ligands can affect the life or death of the cell is also evident from the report that induction of apoptosis by cytotoxic T cells depends on the interaction of granzyme B with its ligand, the CI-MPR (21).
(iii) At face value, the evidence that cells transduced with constructs expressing sequences antisense to CI-MPR undergo apoptosis adds weight to the antiapoptotic role of CI-MPR. This interpretation of the data may not be correct; we cannot differentiate between interference with the synthesis or accumulation of CI-MPR and the translation of some toxic peptide from the antisense mRNA. If the function of gD is to specifically interact with CI-MPR, the failure of gD to block apoptosis in these cells would be expected. On the other hand, HSV-1(F) has proven to be very adept at blocking apoptosis by a vast variety of exogenous stimuli that include, in addition to Fas ligand, thermal and osmotic shock (9-11, 15, 16, 26).
(iv) The role of lysosomal enzymes in the degradation of nucleosomes of apoptotic cells has been established (22). Although the evidence in support of the endocytic pathway as the site of initiation of proapoptotic events is compelling, we sought to block endocytosis by transducing cells with mutant constructs of dynamin 1 and dynamin 2 (1). The results were inconclusive because the cells transduced with the mutant construct underwent apoptosis (G. Zhou and B. Roizman, unpublished studies).
Apoptosis induced by gD−/+ stocks differs from that induced by gD−/− stocks in several respects. Thus, apoptosis is induced by significantly smaller amounts of virus, chloroquine does not block apoptosis, and the ectodomain of gD is sufficient to block apoptosis by gD−/+ stocks. In contrast, in the case of gD−/− virus either the intact gD or a mixture of gene expressing ectodomain and transmembrane domain plus transmembrane domain and carboxyl-terminal cytoplasmic domain is required to block apoptosis. Either intact gD or gJ or overexpression of CI-MPR is sufficient to block apoptosis.
As illustrated diagrammatically in Fig. 8, one hypothesis that explains the events occurring in cells infected with gD−/+ mutant stocks is that apoptosis is triggered not at the time of entry of the virus into cells but rather during the egress from infected cells. This conclusion is based on three lines of evidence.
(i) Chloroquine failed to block apoptosis induced by gD−/+ mutant stocks. This is consistent with the observation that significantly less virus is required to induce apoptosis by gD−/+ stocks than by gD−/− stocks and hence the level of activated lysosomal enzyme would be below the threshold for induction of apoptosis.
(ii) Brunetti et al. reported that CI-MPR colocalized with gD or virus particles in endosomes (5). Given the time of fixation of cells postinfection, it is likely that these were newly assembled virus particles in the exocytic pathway impressed for virus egress (13) rather than virus particles internalized at the time of infection, since the latter are rapidly degraded.
(iii) The observations that the minimal gD sequences required to block apoptosis by gD−/− and gD−/+ stocks also support the conclusion that apoptosis induced by the mutant stocks initiates in different cellular compartments. The presence of the signal sequence in the construct encoding the gD ectodomain would insure that the translation product would be targeted for a secretary pathway usurped during productive infection for viral egress. The glycosylated polypeptide representing the ectodomain could therefore be expected to be present in the compartment containing CI-MPR and virions on its way out of the cell and would be available to interact with CI-MPR to block apoptosis. The ectodomain destined for the secretary pathway would not be expected to be present in vesicles containing internalized, endocytosed virus particles. In contrast, intact glycoproteins or heterodimers linked through transmembrane domain could potentially find their way to the endosomal vesicles and be available to interact with CI-MPR to preclude apoptosis.
In early studies by Johnson and coworkers, the interaction of CI-MPR with gD was sufficiently strong and compelling to suggest that CI-MPR plays a role in the entry and dissemination of HSV-1 from cell to cell (4). The more likely function of the interaction of gD with CI-MPR is to block activation of lysosomal enzymes that would be harmful to the infected cells and hence to virus replication. The results presented in this report add to the existing evidence that CI-MPR through its regulatory functions is a determinant of the life or death of the cell.
Acknowledgments
We thank Gabriella Campadelli-Fiume and David C. Johnson for useful discussions.
These studies were aided by grants from the National Cancer Institute (CA87661, CA83939, CA71933, CA78766, and CA88860).
REFERENCES
- 1.Altschuler, Y., S. M. Barbas, L. J. Terlecky, K. Tang, S. Hardy, K. E. Mostov, and S. L. Schmid. 1998. Redundant and distinct functions for dynamin-1 and dynamin-2 isoforms. J. Cell Biol. 143:1871-1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braulke, T., L. Mach, B. Hoflack, and J. Glossl. 1992. Biosynthesis and endocytosis of lysosomal enzymes in human colon carcinoma SW 1116 cells: impaired internalization of plasma membrane-associated cation-independent mannose 6-phosphate receptor. Arch. Biochem. Biophys. 298:176-181. [DOI] [PubMed] [Google Scholar]
- 3.Brown, W. J., J. Goodhouse, and M. G. Farquhar. 1986. Mannose-6-phosphate receptors for lysosomal enzymes cycle between the Golgi complex and endosomes. J. Cell Biol. 103:1235-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunetti, C. R., R. L. Brurke, S. Kornfeld, W. Gregory, F. R. Masiarz, K. S. Dingwell, and D. C. Johnson. 1994. Herpes simplex virus glycoprotein D acquires mannose 6-phosphate residues and binds to mannose 6-phosphate receptors. J. Biol. Chem. 269:17067-17074. [PubMed] [Google Scholar]
- 5.Brunetti, C. R., K. S. Dingwell, C. Wale, F. L. Graham, and D. C. Johnson. 1998. Herpes simplex virus gD and virions accumulate in endosomes by mannose 6-phosphate-dependent and -independent mechanisms. J. Virol. 72:3330-3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campadelli-Fiume, G., F. Cocchi, L. Menotti, and M. Lopez. 2000. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol. 10:305-319. [DOI] [PubMed] [Google Scholar]
- 7.Chayko, C. A., and M. C. Orgebin-Crist. 2000. Targeted disruption of the cation-dependent or cation-independent mannose 6-phosphate receptor does not decrease the content of acid glycosidases in the acrosome. J. Androl. 21:944-953. [PubMed] [Google Scholar]
- 8.DeLuca, N. A., A. McCarth, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galvan, V., R. Brandimarti, and B. Roizman. 1999. Herpes simplex virus 1 blocks caspase-3-independent and caspase-dependent pathways to cell death. J. Virol. 73:3219-3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galvan, V., R. Brandimarti, J. Munger, and B. Roizman. 2000. Bcl-2 blocks a caspase-dependent pathway of apoptosis activated by herpes simplex virus 1 infection in HEp-2 cells. J. Virol. 74:1931-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galvan, V., and B. Roizman. 1998. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell-type-dependent manner. Proc. Natl. Acad. Sci. USA 95:3931-3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geraghty, R. J., C. Krummenacher, G. M. Cohen, R. J. Eisenberg, and P. G. Spear. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618-1620. [DOI] [PubMed] [Google Scholar]
- 13.Harley, C. A., A. Dasgupta, and D. W. Wilson. 2001. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: role for organelle acidification in assembly of infectious particles. J. Virol. 75:1236-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klumperman, J., A. Hille, T. Veenendaal, V. Oorschot, W. Stoorvogel, K. von Figura, and H. J. Geuze. 1993. Differences in the endosomal distributions of the two mannose 6-phosphate receptors. J. Cell Biol. 121:997-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koyama, A. H., and Y. Miwa. 1997. Suppression of apoptotic DNA fragmentation in herpes simplex virus type 1-infected cells. J. Virol. 71:2567-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leopardi, R., and B. Roizman. 1996. The herpes simplex virus 1 major regulatory protein ICP4 blocks apoptosis induced by the virus or by hyperthermia. Proc. Natl. Acad. Sci. USA 93:9583-9587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez, M., F. Cocchi, E. Avitabile, A. Leclerc, J. Adelaide, G. Campadelli-Fiume, and P. Dubreuil. 2001. Novel, soluble isoform of the herpes simplex virus (HSV) receptor nectin1 (or PRR1-HIgR-HveC) modulates positively and negatively susceptibility to HSV infection. J. Virol. 75:5684-5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masters, S. A., T. M. Ayres, M. Skubatch, C. L. Gray, M. Rothe, and A. Ashkenazi. 1997. Herpesvirus entry mediator, a member of the tumor necrosis factor receptor (TNFR) family, interacts with members of the TNFR-associated factor family and activates the transcription factors NF-κB and AP-1. J. Biol. Chem. 272:14029-14032. [DOI] [PubMed] [Google Scholar]
- 19.Miwako, I., A. Yamamoto, T. Kitamura, K. Nagayama, and M. Ohashi. 2001. Cholesterol requirement for cation-independent mannose 6-phosphate receptor exit from multivesicular late endosomes to the Golgi. J. Cell Sci. 114:1765-1776. [DOI] [PubMed] [Google Scholar]
- 20.Montgomery, R. I., M. S. Warner, B. J. Lum, and P. G. Spear. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427-436. [DOI] [PubMed] [Google Scholar]
- 21.Motyka, B., G. Korbutt, M. J. Pinkoski, J. A. Heibein, A. Caputo, M. Hobman, M. Barry, I. Shostak, T. Sawchuk, C. F. Holmes, J. Gauldie, and R. C. Bleackley. 2000. Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell 103:491-500. [DOI] [PubMed] [Google Scholar]
- 22.Okada, C., and T. Mizuochi. 1999. Role of macrophage lysosomal enzymes in the degradation of nucleosomes of apoptotic cells. J. Immunol. 163:5346-5352. [PubMed] [Google Scholar]
- 23.Oshima, A., C. M. Nolan, J. W. Kyle, J. H. Grubb, and W. Sly. 1988. The human cation-independent mannose 6-phosphate receptor. Cloning and sequence of the full-length cDNA and expression of functional receptor in COS cells. J. Biol. Chem. 263:2553-2562. [PubMed] [Google Scholar]
- 24.Reymond, N., J. P. Borg, E. Lecocq, J. Adelaide, G. Campadelli-Fiume, P. Dubreuil, and M. Lopez. 2000. Human nectin3/PRR3: a novel member of the PVR/PRR/nectin family that interacts with afadin. Gene 255:347-355. [DOI] [PubMed] [Google Scholar]
- 25.Rooney, I. A., K. D. Butrovich, A. A. Glass, S. Borboroglu, and C. A. Benedict. 2000. The lymphotoxin-β receptor is necessary and sufficient for LIGHT-mediated apoptosis of tumor cells. J. Biol. Chem. 275:14307-14315. [DOI] [PubMed] [Google Scholar]
- 26.Sieg, S., Z. Yildirim, D. Smith, N. Kayagaki, H. Yagita, Y. Huang, and D. Kaplan. 1996. Herpes simplex virus type 2 inhibition of Fas ligand expression. J. Virol. 70:8747-8751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spear, P. G., R. J. Eisenberg, and G. H. Cohen. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1-9. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi, K., H. Nakanishi, M. Miyahara, K. Mandai, K. Satoh, A. Satoh, H. Nishioka, J. Aoki, A. Nomoto, A. Mizoguchi, and Y. Takai. 1999. Nectin/PRR: an immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with afadin, a PDZ domain-containing protein. J. Cell Biol. 145:539-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou, G., V. Galvan, G. Campadelli-Fiume, and B. Roizman. 2000. Glycoprotein D or J delivered in trans blocks apoptosis in SK-N-SH cells induced by a herpes simplex virus 1 mutant lacking intact genes expressing both glycoproteins. J. Virol. 74:11782-11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou, G., and B. Roizman. 2001. The domains of glycoprotein D required to block apoptosis depend on whether glycoprotein D is present in the virions carrying herpes simplex virus 1 genome lacking the gene encoding the glycoprotein. J. Virol. 75:6166-6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu, Y., B. Doray, A. Poussu, V. P. Lehto, and S. Kornfeld. 2001. Binding of GGA2 to the lysosomal enzyme sorting motif of the mannose 6-phosphate receptor. Science 1:1716-1718. [DOI] [PubMed] [Google Scholar]