

Abstract
Reverse transcription in hepadnaviruses is primed by the viral reverse transcriptase (RT) (protein priming) and requires the specific interaction between the RT and a viral RNA signal termed ɛ, which bears the specific template sequence for protein priming. The product of protein priming is a short oligodeoxynucleotide which represents the 5′ end of the viral minus-strand DNA and is covalently attached to the RT. We have now identified truncated RT variants from the duck hepatitis B virus that were fully active in the initial step of protein priming, i.e., the covalent attachment of the first nucleotide to the protein (RT deoxynucleotidylation), but defective in any subsequent DNA polymerization. A short sequence in the RT domain was localized that was dispensable for RT deoxynucleotidylation but essential for the subsequent DNA polymerization. These results have thus revealed two distinct stages of protein priming, i.e., the initial attachment of the first nucleotide to the RT (RT deoxynucleotidylation or initiation of protein priming) and the subsequent DNA synthesis (polymerization) to complete protein priming, with the second step entailing additional RT sequences. Two models are proposed to explain the observed differential sequence requirement for the two distinct stages of the protein priming reaction.
Reverse transcription in hepadnaviruses (hepatitis B viruses, [HBVs]) is carried out by a unique virus-encoded reverse transcriptase (RT) (26, 28). The RT is able to initiate DNA synthesis de novo, using the RT itself as a protein primer (19, 32, 34, 35; for a review, see reference 12). This protein priming reaction requires the specific interaction between the RT and a short RNA signal, termed ɛ, located at the 5′ end of the viral pregenomic RNA (pgRNA) (the template for reverse transcription) (23, 33). The unique ability of the hepadnavirus RT to carry out specific RNA recognition and protein priming is reflected in its structural organization, which is both similar to, and distinct from, that of conventional RTs (6, 12, 24). The N-terminal TP (so-called terminal protein) domain is conserved among all hepadnaviruses but absent from all other known RTs encoded by retroviruses or other retroelements. In contrast, the central RT domain and the C-terminal RNase H domain share sequence homologies with conventional RTs. A highly variable spacer or tether domain appears to link the TP and RT domains.
It is now well established that both the N-terminal TP and central RT domains are required for the RT to bind to ɛ and to carry out protein priming. Furthermore, protein priming requires additional sequences from the RT domain that are dispensable for ɛ binding (11, 23, 33). These additional amino acid sequences from the RT domain are presumably required for some aspects of viral DNA synthesis during protein priming, as follows. Using an internal bulge located on the ɛ stem-loop structure as a specific template and an invariant tyrosine residue within the TP domain as a protein primer, the RT synthesizes, de novo, a 3- to 4-nucleotide DNA oligomer. This short DNA oligomer, representing the 5′ end of the viral minus-strand DNA, is covalently attached to the RT through the primer tyrosine residue (22, 30, 31; for a review, see reference 14).
Following the synthesis of this short DNA oligomer (i.e., the completion of protein priming), viral DNA synthesis somehow pauses, and the nascent DNA-RT complex is translocated from ɛ to the 3′ end of the pgRNA (minus-strand template switch) (22, 30, 31; for a recent review, see reference 14). Only from this new (acceptor) template site does reverse transcription continue. Genetic and biochemical evidence indicates that protein priming and the subsequent DNA elongation reactions entail distinct RT activities. For example, specific point mutations in the RT that do not affect protein priming can nevertheless abolish the subsequent DNA elongation following template switching (25). In addition, certain RT inhibitors, such as the pyrophosphate analog phosphonoformic acid and some nucleoside analogs, have been shown to block viral DNA synthesis beyond protein priming but show no effect on protein priming itself (27, 32). It is generally thought that the RT must undergo some conformational change, during (or following) the template switching reaction, from a priming mode to an elongation mode, the latter being sensitive to the aforementioned inhibitors.
Due to the difference in primer usage, the protein priming reaction can be further divided into the initial step that attaches the first deoxyribonucleotide to the TP domain (RT deoxynucleotidylation or initiation of protein priming) and the subsequent DNA polymerization step that attaches the additional 2 to 3 nucleotides to the first nucleotide (DNA polymerization). It is during the initiation stage that the tyrosine residue in the TP domain is used as an unconventional (protein) primer for DNA synthesis. The ensuing DNA polymerization step resembles a conventional DNA synthesis reaction in that the primer for DNA synthesis is the preceding deoxyribonucleotide residue. Because of this mechanistic difference, it is conceivable that the two steps in protein priming may show distinct requirements, just as protein priming and the subsequent DNA elongation (following template switching) do. Here, we report the identification of RT deletion variants from the duck HBV (DHBV) that maintained normal activity for the initiation step of the protein priming reaction but were severely defective for the subsequent step of DNA polymerization. Our results thus support the notion that the two stages in the protein priming reaction do indeed exhibit differential requirements from the RT. We discuss the implications of these results for understanding the mechanism of protein-primed initiation of reverse transcription in hepadnaviruses and for protein-primed DNA synthesis in general.
MATERIALS AND METHODS
Plasmids.
pHTP, used for in vitro expression of the full-length DHBV RT, has been described before (35), as are plasmids expressing the DHBV mini-RT proteins, pcDNA-MiniRT1 and pcDNA-MiniRT2 (for in vitro and mammalian cell expression), and pGST-MiniRT1 and pGST-MiniRT2 (for bacterial expression of glutathione (GSH)-S-transferase [GST]or fusion proteins) (11). pEBG-MiniRT1 and -MiniRT2 (for expression of the GST mini-RT fusion proteins in mammalian cells) were constructed by inserting the MiniRT cassettes into a modified pEBG vector (9). All RT and mini-RT proteins were tagged with a synthetic hemagglutinin epitope inserted into the spacer domain and the mini-RT proteins harbor an additional c-myc epitope tag at their C terminus.
Antibodies.
The monoclonal antibody (MAb) against p23 (clone JJ3) was generously provided by David Toft (Mayo Clinic) (17). The MAb anti-Hsp90 (clone AC16) and anti-c-myc (9E10) were purchased from Sigma, and the anti-hemagglutinin MAb HA.11 (clone16B12) was purchased from the Berkeley Antibody Co.
In vitro transcription and translation.
RNAs used for in vitro translation were transcribed from linearized plasmids using an in vitro transcription kit (MEGAscript; Ambion) and purified as described before (11, 13). Purified RNAs were then translated using the rabbit reticulocyte lysate in vitro-translation system (Promega).
Protein expression and purification.
Two truncated minimal DHBV RT fusion proteins, GST-MiniRT1 and GST-MiniRT2, were expressed in Escherichia coli and purified by using GSH-agarose beads as described before (11). The same GST-mini-RT fusion proteins were also expressed in 293T cells following transient transfection of pEBG-MiniRT1 and pEBG-MiniRT2. The transfected cells were lysed in lysis buffer (LB) (50 mM Tris [pH 8.0], 150 mM NaCl, 1 mM EDTA, 0.1% Nonidet P-40) 2 days after transfection, and the GST-mini-RT fusion proteins were purified by using GSH beads.
Protein-protein interactions.
To identify cellular proteins associated with the RT in cultured cells, the GST-tagged mini-RT proteins were expressed in 293T cells by transient transfection of pEBG-MiniRT1 and pEBG-MiniRT2. The transfected cells were lysed in LB, and the GST-mini-RT fusion proteins were purified by using GSH beads. Unbound materials were removed by extensive wash using LB, and proteins bound to the beads were eluted by using GSH. Proteins associated with the RT were then detected by resolving the eluate by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blot analyses or Coomassie staining.
In vitro protein priming.
Five microliters of the RT in vitro translation reaction or approximately 10 ng of GST-MiniRT proteins, purified either from bacteria or 293T cells, was used in an in vitro protein priming reaction in a total volume of 10 μl, as described before (11, 13), except that the α-32P-labeled deoxynucleoside triphosphate (dNTP) used varied between different experiments as indicated in each figure. When the purified RT proteins were used, rabbit reticulocyte lysate (nuclease [Promega]-treated), supplemented with an ATP-regenerating system (5 mM ATP, 10 mM creatine phosphate, creatine phosphokinase [50 μg/ml]), was used to reconstitute the protein priming reaction as described before (11).
RESULTS
DHBV mini-RT protein active in covalent attachment of the first nucleotide to the RT but defective in subsequent DNA polymerization.
We have recently reported the successful expression and purification of two functional DHBV mini-RT proteins (as GST fusions) using a bacterial expression system and the reconstitution of their ɛ binding and protein priming activities in vitro using either the reticulocyte lysate (11) or purified chaperone proteins (15). When assayed for ɛ binding, both mini-RT proteins showed wild-type-like activity (11). However, when assayed for protein priming, the shorter MiniRT2 protein, with a more extensive C-terminal truncation into the RT domain than MiniRT1 (see Fig. 6 for a schematic diagram), showed very little activity in protein priming (less than 5% of MiniRT1 activity) (Fig. 1A, compare lanes 2 and 8) (11). In those experiments, we routinely used [α-32P]dATP as the labeled nucleotide precursor during the in vitro protein priming reaction. Surprisingly, when [α-32P]dGTP was used as the radiolabeled nucleotide, MiniRT2 showed strong protein priming activity, approaching that of MiniRT1 (Fig. 1A, lanes 4 and 10). Since dGMP is the first nucleotide of the GTAA DNA oligomer that becomes covalently attached to the RT as a result of the protein priming reaction, these findings suggested that MiniRT2 was able to carry out the first step of the priming reaction, i.e., the attachment of dGMP to the RT (RT deoxyguanylation), but was defective in the subsequent DNA polymerization to incorporate the third or fourth nucleotide (dAMP). To test if MiniRT2 was able to incorporate the second nucleotide (TMP) to the nascent DNA oligomer, we measured the incorporation of the radiolabeled [α-32P]TTP into the protein-DNA complex. As shown in Fig. 1A (lane 6), MiniRT2 was not able to incorporate even the second nucleotide into the protein primer. Interestingly, a degradation product from MiniRT1 that comigrated with MiniRT2 (and presumably had a C-terminal truncation similar to that of MiniRT2 [band 3]) behaved exactly the same as MiniRT2 in that it was able to label the RT with [α-32P]dGTP (Fig. 1A, lane 10) but only very weakly with [α-32P]dATP or [α-32P]TTP (Fig. 1A, lanes 8 and 12). In contrast, a slightly longer degradation product from MiniRT1 (band 2), migrating between MiniRT1 and MiniRT2, behaved the same way as the intact MiniRT1 (band 1) did and could incorporate all 3 nucleotides into the RT.
FIG. 6.

Summary of RT sequences required for the initiation and polymerization stages of protein priming. Shown on the top is a schematic diagram of the DHBV RT domain structure (TP, spacer, RT, and RNase H). The primer tyrosine residue and the double aspartate residues at the RT active site are indicated. The RT domain is further divided into the “finger” (F), “palm” (P), and “thumb” (T) subdomains (with the approximate boundaries marked), based on alignment with the RT structure of the human immunodeficiency virus (8, 25). Summarized below the diagram are the activities of various RT deletion/truncation variants in the initiation and polymerization steps of the protein priming reaction, which were measured by dGTP and dATP incorporation, respectively, into the RT proteins. The truncations at the C terminus are indicated by the corresponding restriction sites.
FIG. 1.

RT sequence requirement for the initiation and polymerization stage of protein priming. (A) GST-MiniRT1 (lanes 7 to 12) and GST-MiniRT2 (lanes 1 to 6) were purified from bacteria and assayed for in vitro protein priming activity, with (lanes 2, 4, 6, 8, 10, and 12) or without (lanes 1, 3, 5, 7, 9, and 11) reconstitution with the reticulocyte lysate (RL). In addition, MiniRT2 (without the GST fusion) was expressed in the RL by in vitro translation and assayed for protein priming activity (lanes 13 to 15). As indicated, different 32P-labeled nucleotide precursors (dATP, dA; dGTP, dG; TTP, T) were used; unlabeled dNTP mixtures (without the nucleotide corresponding to the labeled one) were also added to the reaction mixtures. Labeled RT proteins, as indicated by the arrows, were then detected by resolving the reactions by SDS-PAGE and autoradiography. Three distinct labeled products were detected in the reactions using GST-MiniRT1, as indicated by the numbers 1, 2, and 3. The top band (band 1) represents the intact GST-MiniRT1 protein, whereas the middle (band 2) and bottom (band 3) bands represent degradation products from GST-MiniRT1. Note that product 3 from GST-MiniRT1 migrated almost exactly as GST-MiniRT2. (B) C-terminal truncations of MiniRT1 were expressed in the RL by in vitro translation from templates linearized at the indicated restriction sites (Pml, PmlI; Nsp, NspI) (see Fig. 6 for a schematic diagram of the RT structure and the positions of these restriction sites). The truncated RT proteins were then tested for in vitro protein priming activity in the presence of either 32P-labeled dGTP (lanes 1 and 2) or dATP (lanes 3 and 4). Unlabeled dNTP mixtures (without the nucleotide corresponding to the labeled one) were also added to the reactions.
To rule out the possibility that the fusion with GST might have affected the behavior of the mini-RT proteins, we translated MiniRT2 (with no fusion) in the reticulocyte lysate and performed an assay to determine its activity in the priming reaction using different labeled nucleotide precursors. The MiniRT2 thus expressed in vitro behaved exactly the same way as the GST fusion protein purified from bacteria; it efficiently incorporated the first nucleotide (dGMP), but not the second (TTP) or third nucleotide (dATP), into the protein primer (Fig. 1A, lanes 13 to 15).
Protein priming by MiniRT2 required the ɛ RNA, the RT polymerase activity, and host factors.
To verify that the priming activity detected with MiniRT2 reflected authentic protein priming activity, we assessed whether it required the same factors as shown for the wild-type RT. As shown in Fig. 2, a protein priming reaction detected with MiniRT2 and [α-32P]dGTP, just like that with MiniRT1, required ɛ RNA binding to the RT, as omission of ɛ from the reaction (Fig. 2, lanes 2 and 5) (11, 33) or elimination of the ɛ binding activity of the RT by mutation (CA29) (25) (Fig. 2, lane 3) abolished protein priming activity. Also as expected, an RT active-site mutation (YMHA) (6) completely abolished all priming activity (Fig. 2, lane 6). Furthermore, in vitro protein priming using MiniRT2, as with MiniRT1 (11), required reconstitution with the reticulocyte lysate (as a source of host cell chaperones) (Fig. 1A, lanes 1 to 12) or with purified components of the Hsp90 chaperone complex (15; data not shown).
FIG. 2.

Protein priming by both MiniRT1 and MiniRT2 required ɛ binding and RT catalytic activity. GST-MiniRT1, its mutant derivative GST-MiniRT1/YMHA, GST-MiniRT2, and its mutant derivative GST-MiniRT2/CA29 were purified from bacteria and assayed for in vitro protein priming activity. All reactions were carried out in the presence of reticulocyte lysate and [α-32P]dGTP. The ɛ RNA was added to all reactions except those shown in lanes 2 and 5. The labeled RT proteins are indicated as in the legend to Fig. 1.
Since MiniRT2 was active not only in ɛ RNA binding (11) but also in the first step of protein priming and since it was more stable and could be expressed to much higher levels than the longer MiniRT1 or the full-length RT in bacteria (11), we attempted to express it in mammalian cells, hoping that the additional truncation and the GST fusion would stabilize the normally very unstable RT (3, 12) in mammalian cells. When the GST-tagged MiniRT2 was expressed in 293T cells, we could indeed detect and purify (by affinity purification via the GST tag) significant amounts of MiniRT2 protein. Approximately 100 ng of purified MiniRT2, as estimated by Coomassie blue staining of the mini-RT band on the SDS-polyacrylamide gel against known amounts of protein standards, could be obtained from 107 cells, although some degradation of GST-MiniRT2 (mainly to GST alone) seemed to have occurred (Fig. 3).
FIG. 3.

Association of MiniRT2 with Hsp90 and p23 in mammalian cells. Plasmid DNA expressing the GST-tagged MiniRT2 (pEBG-MiniRT2) or GST alone (pEBG vector) was transfected into 293T cells. Transfected cells were lysed, and the GST proteins were purified by using GSH affinity beads. Bound proteins were eluted with GSH, resolved by SDS-PAGE, and detected by Western blot analyses using MAbs against Hsp90 (Anti-Hsp90) or p23 (Anti-p23) or by Coomassie blue staining (top panel). Hsp90, p23, GST-MiniRT2, and GST are indicated. The star to the left of the top panel denotes a nonspecific band associated with the GSH beads.
With significant amounts of MiniRT2 purified from mammalian cells at hand, we wished to test if this purified RT was active in protein priming, with or without in vitro reconstitution. By using labeled dGTP as the nucleotide precursor, we could show that the purified MiniRT2 was active in protein priming, in an ɛ-dependent fashion, but again only after reconstitution with the reticulocyte lysate (Fig. 4A, lane 6; >60-fold stimulation compared to the control reaction shown in lane 2) or with purified Hsp90 chaperone components (15; data not shown). As the mini-RT was purified from mammalian cells under nondenaturing conditions, we thought at least a portion of the purified RT might be associated with the endogenous Hsp90 chaperone components. Indeed, we could detect both Hsp90 and p23, an Hsp90 cochaperone, in the purified GST-MiniRT2 preparation, but not in the GST preparation purified under the same conditions (Fig. 3). These results suggested that although some of the purified MiniRT2 was still associated with some components of the Hsp90 machinery, other essential factors required for RT activity (such as Hsp70, Hsp40, or p60 [15]) may have been lost following purification. On the other hand, we noticed that addition of an ATP-regenerating system to the purified MiniRT2 could weakly, but consistently, stimulate protein priming (Fig. 4A, lane 4; approximately fourfold stimulation compared to the control reaction shown in lane 2), suggesting that the chaperone components that remained associated with the RT (including Hsp90 and p23), though not fully functional by themselves, could still activate the RT weakly in an ATP-dependent fashion.
FIG. 4.

GST-MiniRT2 purified from mammalian cells was active in the initiation of protein priming but defective in DNA polymerization. GST-MiniRT2 was expressed in 293T cells following transient transfection of pEBG-MiniRT2 and purified using GSH affinity resin. (A) Purified GST-MiniRT2 was assayed for in vitro protein priming activity, supplemented with buffer alone (lanes 1 and 2), with an ATP regenerating system (ATP RS [lanes 3 and 4), or with reticulocyte lysate (RL [lanes 5 and 6]). The ɛ RNA was added to the indicated reaction mixtures only (lanes 2, 4, and 6). [α-32P]dGTP was used as the nucleotide precursor. (B) Purified GST-MiniRT2 was assayed for in vitro protein priming, all reactions being supplemented with reticulocyte lysate. Either [α-32P]dGTP (lanes 1 and 2) or [α-32P]dATP (plus unlabeled dGTP and TTP) (lanes 3 and 4) was used as the labeled nucleotide precursor. The ɛ RNA was added to the mixtures for reactions 1 and 3 only.
We then wanted to test if the MiniRT2 protein purified from mammalian cells was able to carry out the full protein priming reaction (synthesis of the 4-nucleotide oligomer attached to the RT) or still only the first step (i.e., the attachment of the first nucleotide to the RT), like the same protein purified from bacteria or expressed in vitro (Fig. 1). As shown in Fig. 4B, MiniRT2 purified from mammalian cells was still only able to attach the first nucleotide (dGMP) to the RT and unable to elongate the primer (very little dATP incorporation) (Fig. 4B, lanes 1 and 3).
In summary, the protein priming activity of MiniRT2, as detected by the attachment of the first nucleotide to the RT, displayed the same requirement previously shown for the longer MiniRT1 or the full-length RT, in that it required the ɛ RNA, the RT active site, and specific host cell factors. Furthermore, MiniRT2 purified from mammalian cells showed the same priming activity and displayed the same requirements as that purified from bacteria.
Failure of DNA polymerization by MiniRT2 was not a result of low affinity for nucleotide substrates.
The above results strongly suggested that MiniRT2 was able to attach the first nucleotide of the viral minus-strand DNA to the RT but was defective in any subsequent DNA polymerization. In essence, MiniRT2 was able to carry out an abbreviated protein priming reaction, attaching only 1 nucleotide (instead of the normal, 4-nucleotide DNA oligomer) to the RT protein primer. However, since the labeled nucleotides were present in the priming reaction at much lower concentrations compared to the unlabeled nucleotides, it was formally possible that MiniRT2 may have had a drastically decreased affinity for dATP or TTP (but normal affinity for dGTP) and thus could not efficiently utilize the radiolabeled dATP or TTP. To test this possibility, we used an ɛ RNA variant with a single-nucleotide substitution (C to U) at the first position (3′ end) of the template sequence (the internal bulge of the ɛ RNA), so that the RNA template sequence for protein priming was changed from UUAC to UUAU. With this UUAU ɛ variant, the first deoxyribonucleotide attached to the RT following protein priming is predicted to be dAMP (instead of dGMP as it is normally) and the nascent DNA oligomer attached to the RT will have the sequence ATAA (instead of GTAA). If the reason for the inability of MiniRT2 to incorporate dATP to the RT primer was the decreased affinity of MiniRT2 for dATP, then it should still be unable to attach dAMP to the protein, even with the variant ɛ RNA. If, on the other hand, MiniRT2 was only able to attach the first nucleotide (independent of the nucleotide identity) to the RT and was defective in any subsequent polymerization, it should then efficiently attach dAMP to the protein when the UUAU ɛ variant is used.
Indeed, the second prediction was exactly what we observed. Thus, when the variant ɛ RNA was used, MiniRT2 now efficiently attached dAMP, but not dGMP, to the protein (Fig. 5, lanes 4 to 6). Since dAMP was the first nucleotide attached to the RT when the UUAU ɛ variant was used, no other nucleotides were required for dAMP to be incorporated into the protein (Fig. 5, lanes 5 and 6). In contrast, the very inefficient incorporation of the labeled dAMP to MiniRT2 using the wild-type ɛ RNA template (UUAC), i.e., the elongation of the nascent DNA strand to position 3 on the ɛ RNA template (the synthesis of the dGTA oligonucleotide), required the presence of the other nucleotides (dGTP and TTP) as expected (Fig. 5, lanes 2 and 3; data not shown). These results thus confirmed that the ɛ variant RNA template behaved as predicted, and more importantly, they indicated that MiniRT2 could attach any nucleotide to the RT (depending on the template sequence) but was unable to extend beyond this first nucleotide.
FIG. 5.

Protein priming in the presence of variant ɛ RNA template. GST-MiniRT2 purified from bacteria was assayed for in vitro protein priming activity reconstituted by reticulocyte lysate, using as template either the wild-type ɛ RNA (with the bulge sequence UUAC as the template residues [lanes 1 to 3]) or a variant ɛ with the 3′ nucleotide of the template sequence changed from C to U (UUAU [lanes 4 to 6]). As indicated, either [α-32P]dGTP (dG [lanes 1 and 4]) or [α-32P]dATP (dA) was used as the labeled nucleotide precursor (lanes 2, 3, 5, and 6). Unlabeled dNTP mixture (without dATP) was also added to the reactions shown in lanes 2 and 5.
Mapping of RT domain sequences required for DNA polymerization.
Since a degradation product from MiniRT1 (band 2 in Fig. 1A and Fig. 2) was still able to complete protein priming, it suggested that MiniRT1 could be truncated further while still maintaining RT activity in both steps of protein priming. Therefore, we constructed additional C-terminal truncations from the RT domain between the C termini of MiniRT1 (amino acid 734) and MiniRT2 (amino acid 575) in order to narrow down the sequences that are important for the DNA polymerization step of the protein priming reaction (but dispensable for the initiation step). As shown in Fig. 1B and summarized in Fig. 6, truncation up to the PmlI site (amino acid 661) still allowed equivalent levels of both dGTP and dATP incorporation, indicating that deletion of the C-terminal 125 amino acids from the RT did not affect either stage of the protein priming reaction. Thus, the 86-amino-acid segment between position 661 and 575 (or an even shorter segment thereof) presumably bears the sequence requirement that permits the transition from the initiation to the polymerization stage of protein priming.
To rule out the possibility that the N-terminal and spacer deletions in MiniRT2 (in addition to the C-terminal truncation) may have affected its ability to carry out dATP (but not dGTP) incorporation to the RT, we made an RT variant with a C-terminal truncation similar to that in MiniRT2 in the context of the full-length RT (without any N-terminal or spacer deletions) (Fig. 4). This RT variant behaved just like MiniRT2 in that it was able to carry out dGTP but not dATP incorporation. We noted that a similarly truncated RT mutant was also reported previously to be competent for dGTP incorporation, but its ability to incorporate dATP was not tested (29).
DISCUSSION
Protein-primed initiation of reverse transcription in hepadnaviruses is a complex process involving the viral RT, the pgRNA, and specific host cell factors. To initiate DNA synthesis, the primer tyrosine residue located within the N-terminal TP domain, the RT active site in the central RT domain, and the RNA template sequence (the internal bulge of the ɛ stem-loop) must be brought into precise spatial arrangement. We have demonstrated here that a substantial portion of the RT domain can be removed without any detrimental effect on the initiation of protein priming, i.e., the covalent attachment of the first nucleotide of the viral minus-strand DNA to the RT. However, a short segment (86 amino acids or less) from the C-terminal portion of the RT domain appears to be specifically required for DNA synthesis subsequent to this initial reaction and, thus, for the completion of protein priming (the synthesis of the 3- to 4-nucleotide minus-strand DNA oligonucleotide). Both stages of the protein priming reaction required specific RT-ɛ interaction and the same RT active site. These results have thus revealed that protein priming in hepadnaviruses can be divided into two distinct, enzymatic steps: the initial attachment of the first nucleotide of viral minus-strand DNA to the RT (i.e., RT deoxynucleotidylation) and the subsequent DNA synthesis to complete protein priming, generating the nascent DNA oligonucleotide. We choose here to call the first step “initiation” and the latter step “polymerization” (instead of “elongation” to differentiate it from the extensive DNA synthesis following minus-strand template switching).
One interpretation for the distinct requirements during these two stages of protein priming suggests that the requirement for the additional RT domain sequences, specifically for polymerization but not for initiation, reflects the need to properly position the nascent DNA primer (but not the protein primer) that is fulfilled by the additional C-terminal RT domain sequences. Evidently, during the initiation stage, the protein primer (the tyrosine residue in the TP domain) can be positioned properly relative to the RT active site and the ɛ template, solely through the specific protein-protein interactions between the TP domain and the N-terminal part of the RT domain as well as the protein-RNA interactions between these domains and the ɛ RNA. Indeed, the sequence requirement from the RT domain for the initiation stage of protein priming was remarkably short; only 50 amino acids C-terminal to the double aspartic acid residues in the RT active site were needed (Fig. 6). Following the initiation step, the tyrosine residue in the TP domain has to exit the RT active site and the newly attached deoxyribonucleotide (dGMP in DHBV) has to be positioned properly in the RT active site for the subsequent DNA polymerization. Our results suggest that the proper positioning of this 1-nucleotide DNA primer may then require the additional sequences from the C-terminal portion of the RT domain (specifically from amino acid 575 to 661), probably through protein-DNA interactions. Interestingly, recent model-building efforts have suggested that this same region of the RT domain forms the major part of the “thumb” subdomain that is thought to be responsible for positioning the nucleic acid template and primer in the hepadnavirus RT active site (1, 8, 20, 25). Our results therefore suggest that the thumb subdomain is entirely dispensable for positioning the protein primer during the initiation of protein priming and that the mechanisms of positioning the protein primer and those of positioning the nucleotide primer are fundamentally different.
In addition to this structural requirement for nucleotide (as opposed to protein) primer positioning, the C-terminal sequence of the RT domain may also be required, either directly or indirectly, to facilitate a conformational change in the RT itself that may be necessary for the transition from initiation (RT deoxynucleotidylation) to DNA polymerization during protein priming. In this regard, it is informative to compare protein-primed DNA synthesis in hepadnaviruses to that in other viruses (such as the adenovirus and the phage φ29) that also employ protein priming to initiate DNA synthesis. Due to the special nature of the protein primer, all protein-primed DNA syntheses studied so far require the polymerase to undergo some structural changes as it switches from an initiation mode, when the primer for DNA synthesis is a protein, to an elongation mode, when the primer for DNA synthesis is the newly formed DNA oligomer (18, 21). However, the structural transition from the protein-primed initiation mode to the DNA-primed elongation mode has been shown to occur only after the synthesis of a DNA oligomer of at least several (from 3 to 9) nucleotides in the case of adenoviruses and the bacteriophages (5, 18, 21). In hepadnaviruses, the corresponding transition is likely the RT conformational change accompanying minus-strand template switch, also occurring after the synthesis of the 3- to 4-nucleotide DNA oligomer (i.e., the completion of protein priming) (27, 32). The results presented here are the first to suggest that the incorporation of even the second nucleotide into the nascent DNA strand can have a different structural requirement compared to the initial attachment of the first nucleotide to the protein primer during protein-primed DNA synthesis.
As the protein primer in hepadnaviruses is a domain of the polymerase that is covalently attached to the RT catalytic domain, the interactions between the TP and the RT domains likely exhibit important differences compared to the other viruses in which the protein primer and DNA polymerase reside on separate proteins. In addition, the specific interactions between the TP and the RT domains and the ɛ RNA are also essential for pgRNA packaging in hepadnaviruses, which further requires the C-terminal RNase H domain (2, 4, 7, 10, 23). The protein-protein and protein-RNA interactions required for pgRNA packaging may impose special constraints for TP-RT-ɛ RNA interactions that permit the initiation (i.e., RT deoxynucleotidylation), but not the completion (i.e., the “polymerization” step), of protein priming. These properties may contribute to the possibly unique requirement for the hepadnavirus polymerase to undergo some conformation change immediately after the first nucleotide is attached to the TP domain in order to accommodate the transition from TP priming to nucleotide priming. Therefore, these results, together with previous observations, support a highly dynamic, multistage model for the protein-primed initiation of reverse transcription in hepadnaviruses entailing multiple RT structural changes, as outlined in Fig. 7.
FIG. 7.
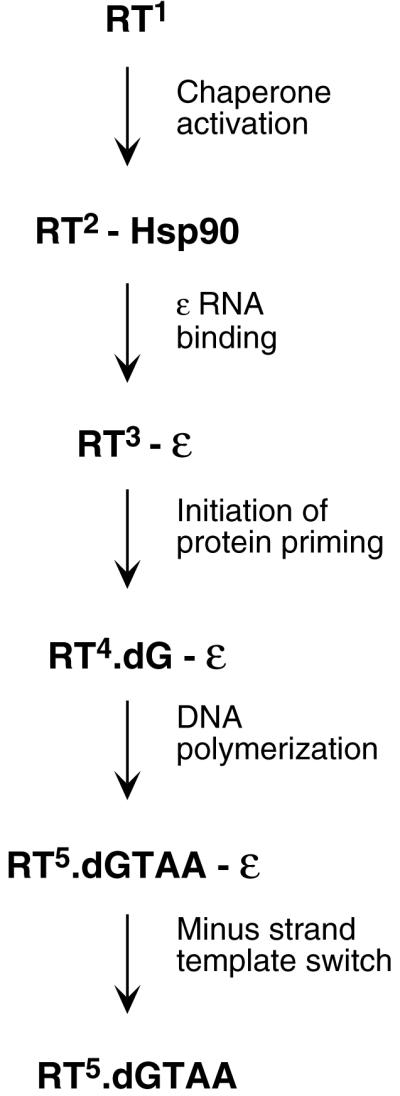
Multistage, protein-primed initiation of reverse transcription in hepadnaviruses. Multiple functional and conformational states of the viral RT. The viral RT, without assistance of host cell factors, is in an inactive state (RT1) unable to bind to ɛ RNA or carry out protein priming. Upon association with the cellular Hsp90 chaperone complex, the RT undergoes a conformational maturation and adopts an ɛ binding-competent state (RT2) (11, 13, 15, 16). Binding of ɛ then induces another conformational change in the RT (RT3) that may be required for the RT to gain its enzymatic activity (29). The initiation of protein priming then leads to the covalent attachment of the first nucleotide of the viral minus-strand DNA (a dGMP residue in the case of DHBV) to the RT (RT deoxynucleotidylation), using ɛ as the template. Following this initiation reaction, a conformational change of the RT (RT4) may be required for further DNA synthesis to produce the 4-nucleotide nascent minus-strand DNA oligomer (dGTAA in DHBV), still templated by ɛ (30, 31). Clearly, the putative RT thumb subdomain is dispensable for the initiation of protein priming (step 3) but required for its completion (step 4). Following the completion of protein priming, the RT undergoes another major conformational change (RT5), dissociates from the ɛ RNA and the RT-nascent minus-strand DNA complex, and then translocates to the 3′ end of the pgRNA (minus-strand template switch) to continue DNA synthesis (elongation mode) (30, 31).
Acknowledgments
We thank Dana Anselmo and Joslynn Jordan for excellent technical assistance; C. Seeger, R. Corley, and G. Viglianti for critical comments on the manuscript; and David Toft for the anti-p23 MAb.
J. Hu was a Harcourt General Researcher and the recipient of an American Liver Foundation Liver Scholar Award. This work was supported by Public Health Service grant R01 AI43453 (to J.H.) from the National Institutes of Health, by a New Investigator Award of The Medical Foundation from the Harcourt General Charitable Foundation (to J.H), and by the American Liver Foundation (to J.H).
REFERENCES
- 1.Allen, M. I., M. Deslauriers, C. W. Andrews, G. A. Tipples, K. A. Walters, D. L. Tyrrell, N. Brown, L. D. Condreay, et al. 1998. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Hepatology 27:1670-1677. [DOI] [PubMed] [Google Scholar]
- 2.Bartenschlager, R., M. Junker-Niepmann, and H. Schaller. 1990. The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J. Virol. 64:5324-5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartenschlager, R., C. Kuhn, and H. Schaller. 1992. Expression of the P-protein of the human hepatitis B virus in a vaccinia virus system and detection of the nucleocapsid-associated P-gene product by radiolabelling at newly introduced phosphorylation sites. Nucleic Acids Res. 20:195-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartenschlager, R., and H. Schaller. 1992. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 11:3413-3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brenkman, A. B., M. R. Heideman, V. Truniger, M. Salas, and P. C. van der Vliet. 2001. The (I/Y)XGG motif of adenovirus DNA polymerase affects template DNA binding and the transition from initiation to elongation. J. Biol. Chem. 276:29846-29853. [DOI] [PubMed] [Google Scholar]
- 6.Chang, L. J., R. C. Hirsch, D. Ganem, and H. E. Varmus. 1990. Effects of insertional and point mutations on the functions of the duck hepatitis B virus polymerase. J. Virol. 64:5553-5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, Y., W. S. Robinson, and P. L. Marion. 1994. Selected mutations of the duck hepatitis B virus P gene RNase H domain affect both RNA packaging and priming of minus-strand DNA synthesis. J. Virol. 68:5232-5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Das, K., X. Xiong, H. Yang, C. E. Westland, C. S. Gibbs, S. G. Sarafianos, and E. Arnold. 2001. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine and emtricitabine. J. Virol. 75:4771-4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grammatikakis, N., J.-H. Lin, A. Grammatikakis, P. N. Tsichlis, and B. H. Cochran. 1999. p50cdc37 acting in concert with Hsp90 is required for Raf-1 function. Mol. Cell. Biol. 19:1661-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirsch, R. C., J. E. Lavine, L. J. Chang, H. E. Varmus, and D. Ganem. 1990. Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as well as for reverse transcription. Nature 344:552-555. [DOI] [PubMed] [Google Scholar]
- 11.Hu, J., and D. Anselmo. 2000. In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J. Virol. 74:11447-11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu, J., and C. Seeger. 1996. Expression and characterization of hepadnavirus reverse transcriptases. Methods Enzymol. 275:195-208. [DOI] [PubMed] [Google Scholar]
- 13.Hu, J., and C. Seeger. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. USA 93:1060-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu, J., and C. Seeger. 1997. RNA signals that control DNA replication in hepadnaviruses. Semin. Virol. 8:205-211. [Google Scholar]
- 15.Hu, J., D. Toft, D. Anselmo, and X. Wang. 2002. In vitro reconstitution of functional hepadnavirus reverse transcriptase with cellular chaperone proteins. J. Virol. 76:269-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu, J., D. O. Toft, and C. Seeger. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 16:59-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson, J. L., T. G. Beito, C. J. Krco, and D. O. Toft. 1994. Characterization of a novel 23-kilodalton protein of unactive progesterone receptor complexes. Mol. Cell. Biol. 14:1956-1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King, A. J., W. R. Teertstra, and P. C. van der Vliet. 1997. Dissociation of the protein primer and DNA polymerase after initiation of adenovirus DNA replication. J. Biol. Chem. 272:24617-24623. [DOI] [PubMed] [Google Scholar]
- 19.Lanford, R. E., L. Notvall, and B. Beames. 1995. Nucleotide priming and reverse transcriptase activity of hepatitis B virus polymerase expressed in insect cells. J. Virol. 69:4431-4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin, X., Z. H. Yuan, L. Wu, J. P. Ding, and Y. M. Wen. 2001. A single amino acid in the reverse transcriptase domain of hepatitis B virus affects virus replication efficiency. J. Virol. 75:11827-11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mendez, J., L. Blanco, and M. Salas. 1997. Protein-primed DNA replication: a transition between two modes of priming by a unique DNA polymerase. EMBO J. 16:2519-2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nassal, M., and A. Rieger. 1996. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J. Virol. 70:2764-2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pollack, J. R., and D. Ganem. 1994. Site-specific RNA binding by a hepatitis B virus reverse transcriptase initiates two distinct reactions: RNA packaging and DNA synthesis. J. Virol. 68:5579-5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radziwill, G., W. Tucker, and H. Schaller. 1990. Mutational analysis of the hepatitis B virus P gene product: domain structure and RNase H activity. J. Virol. 64:613-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seeger, C., E. H. Leber, L. K. Wiens, and J. Hu. 1996. Mutagenesis of a hepatitis B virus reverse transcriptase yields temperature-sensitive virus. Virology 222:430-439. [DOI] [PubMed] [Google Scholar]
- 26.Seeger, C., and W. S. Mason. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Staschke, K. A., and J. M. Colacino. 1994. Priming of duck hepatitis B virus reverse transcription in vitro: premature termination of primer DNA induced by the 5′-triphosphate of fialuridine. J. Virol. 68:8265-8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Summers, J., and W. S. Mason. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403-415. [DOI] [PubMed] [Google Scholar]
- 29.Tavis, J. E., B. Massey, and Y. Gong. 1998. The duck hepatitis B virus polymerase is activated by its RNA packaging signal, epsilon. J. Virol. 72:5789-5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tavis, J. E., S. Perri, and D. Ganem. 1994. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J. Virol. 68:3536-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang, G. H., and C. Seeger. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 67:6507-6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang, G. H., and C. Seeger. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663-670. [DOI] [PubMed] [Google Scholar]
- 33.Wang, G. H., F. Zoulim, E. H. Leber, J. Kitson, and C. Seeger. 1994. Role of RNA in enzymatic activity of the reverse transcriptase of hepatitis B viruses. J. Virol. 68:8437-8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber, M., V. Bronsema, H. Bartos, A. Bosserhoff, R. Bartenschlager, and H. Schaller. 1994. Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J. Virol. 68:2994-2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zoulim, F., and C. Seeger. 1994. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J. Virol. 68:6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]