

Abstract
Human immunodeficiency virus type 1 (HIV-1) entry requires conformational changes in the transmembrane subunit (gp41) of the envelope glycoprotein (Env) involving transient fusion intermediates that contain exposed coiled-coil (prehairpin) and six-helix bundle structures. We investigated the HIV-1 entry mechanism and the potential of antibodies targeting fusion intermediates to block Env-mediated membrane fusion. Suboptimal temperature (31.5°C) was used to prolong fusion intermediates as monitored by confocal microscopy. After transfer to 37°C, these fusion intermediates progressed to syncytium formation with enhanced kinetics compared with effector-target (E/T) cell mixtures that were incubated only at 37°C. gp41 peptides DP-178, DP-107, and IQN17 blocked fusion more efficiently (5- to 10-fold-lower 50% inhibitory dose values) when added to E/T cells at the suboptimal temperature prior to transfer to 37°C. Rabbit antibodies against peptides modeling the N-heptad repeat or the six-helix bundle of gp41 blocked fusion and viral infection at 37°C only if preincubated with E/T cells at the suboptimal temperature. Similar fusion inhibition was observed with human six-helix bundle-specific monoclonal antibodies. Our data demonstrate that antibodies targeting gp41 fusion intermediates are able to bind to gp41 and arrest fusion. They also indicate that six-helix bundles can form prior to fusion and that the lag time before fusion occurs may include the time needed to accumulate preformed six-helix bundles at the fusion site.
The human immunodeficiency virus type 1 (HIV-1) envelope glycoprotein (Env) forms trimers on the virion surface, with each monomer consisting of two subunits, gp120 and gp41 (25, 31). gp120 binds to CD4 molecules on target cells and undergoes conformational changes that allow gp120 to interact with certain chemokine receptors on the same target membranes (1, 20). Env-receptor binding triggers a series of conformational changes in gp41 that facilitate membrane fusion. The gp41 ectodomain contains two 4,3 hydrophobic repeat regions, N-HR and C-HR, that can self-assemble into a trimer of antiparallel dimers (hairpins) (21). Crystallographic studies confirmed that this gp41 core structure is a six-helix bundle in which the N-HR forms three central helices arranged in a trimeric coiled coil. The C-HR forms three outer helices that pack in an antiparallel manner into highly conserved, hydrophobic grooves on the surface of this coiled coil (3, 26, 28).
The six-helix bundle likely represents a fusion-active conformation of gp41 that forms after receptor binding. Support for this model includes the demonstration that synthetic peptides derived from the C-HR (DP178 and C34) inhibit HIV infection and cell-cell fusion at nanomolar concentrations (3, 16, 29, 30) and that a C-HR peptide binds gp41 after receptor activation (11). Peptides derived from the N-HR (DP-107 and N-36) and a short peptide representing a prominent “pocket” on the surface of the central coiled-coil (IQN17) also block fusion (9, 10). Both C-HR and N-HR peptides are believed to bind to the gp41 fusion intermediates prior to formation of the six-helix bundle complex (29). Once this gp41 core is assembled, it is extremely stable (with a melting temperature in excess of 90°C) and is unlikely to be disrupted by exogenous peptides (4).
Previously, we generated rabbit antisera against peptides derived from the N-HR and C-HR as well as to mixtures of N-HR and C-HR that self-assemble into six-helix bundles. These sera were used to investigate fusion-inducing conformational changes in Env. Several of these sera were shown to immunoprecipitate receptor-activated forms of gp41 (7), but these antibodies were not neutralizing under conventional infectivity conditions at 37°C. Similarly, monoclonal antibodies specific for the six-helix bundle have also been found to be nonneutralizing (5, 14, 17). It was postulated that antibody molecules might be too large to access the fusion intermediates at the interface of effector-target (E/T) or virus-target cell membranes (steric problem), or that fusion may occur too quickly once fusion intermediates form (kinetics problem) (7, 23).
In the present studies, we slowed the fusion process by using suboptimal temperature (31.5°C) to dissect steps in HIV entry and to reevaluate the potential of antibodies targeting fusion intermediates to block HIV-1 entry. Under these conditions, antibodies targeting the N-HR and the six-helix bundle blocked E/T cell fusion and viral entry. Confocal microscopy demonstrated binding of antibundle antibodies to effector cells interacting with target cells (E/T conjugates) prior to fusion. These data indicate that fusion intermediates are accessible to antibodies and that the lack of neutralization at 37°C is probably related to the kinetics of conformational changes and membrane fusion. Our data further suggest that six-helix bundle formation can precede fusion and that the lag period before fusion occurs may include the time needed to accumulate preformed six-helix bundles at the fusion site.
MATERIALS AND METHODS
Recombinant vaccinia viruses and fusion inhibition assay.
Recombinant vaccinia viruses were described previously (2). vCB28 (JR-FL envelope) was from Christopher Broder (U.S. Uniformed Health Services University, Bethesda, Md.); v89.6 and a control recombinant vaccinia virus expressing the bacterial β-galactosidase gene were from Bernard Moss (National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Md.). Syncytium formation was measured at different times after cocultures (1:1 ratio, 105 cells each, in triplicate) of target cells and effector cells (CD4− 12E1 cells infected overnight with 10 PFU of recombinant vaccinia viruses/cell expressing HIV-1 envelopes). For measurements of X4 Env-mediated fusion, we used the human lymphoid cell line TF228.1.16, which stably expresses HIV-1 IIIB/BH10 envelope (18). Where indicated, preimmune rabbit immunoglobulin G (IgG), rabbit anti-N-HR, rabbit anti-C-HR, or rabbit anti-six-helix bundle or monoclonal antibodies against gp120 (2G12; IGb12) (National Institutes of Health AIDS Reagent Repository, McKesson BioServices Corp., Rockville, Md.), 48d and 17b (from J. Robinson, Tulane University, New Orleans, La.) (27), or human monoclonal antibody against gp41, 2F5 (24) (National Institutes of Health AIDS Reagent Repository) and 181-D, 98-6, 1281, and 167-D (12, 14, 32) were added to the Env-expressing effector cells for 1 h at 37°C, at 30 or 10 μg/ml, before the addition of target cells. Alternatively, antibodies were added to mixtures of effector and target cells at threefold serial dilutions and incubated for 60 to 120 min at 31.5°C. The plates were then transferred to 37°C, and syncytia were scored during the next 5 h.
gp41-derived inhibitory peptides DP178 (T20) and DP 107 (T21) were produced at the Core Facility, CBER. IQN17 peptide was a gift from Debra M. Eckert and Peter Kim (currently at Merck & Co., West Point, Pa.). IQN17 is a soluble trimeric coiled-coil (GCN4-plql) fused to the gp41 hydrophobic pocket (amino acids 565 to 581 of HXB2 gp160). All inhibitors were serially diluted in phosphate-buffered saline (PBS) and added to mixtures of effector and target cells either at 37°C or for 60 min at 31.5°C and then transferred to 37°C.
Animal immunization.
Synthetic peptides modeling the N-HR and C-HR of HIV-1 gp41 prepared by using standard solid-phase techniques (7) were used as immunogens. The N- and C-HR peptides were administered alone or as mixtures (N34/C36 and DP107/DP178). New Zealand White rabbits were immunized subcutaneously with 200 μg of peptide with complete Freund's adjuvant, followed by two boosts of peptides mixed with incomplete Freund's adjuvant at approximately 4-week intervals. Following the third immunization, sera were collected for characterization as previously described (7). In most experiments, total IgG and IgG specific for N-HR were purified by using protein A- and N-HR peptide-conjugated Sepharose beads. Sera were enriched for binding to the six-helix bundle by sequential adsorption to biotinylated N-HR and C-HR peptides bound to avidin-agarose. The flowthrough fractions were purified on protein A-agarose columns and verified for specificity to the six-helix bundle by enzyme-linked immunosorbent assay (ELISA).
HIV-1 neutralization.
HIV-1 LAI (X4 strain) was obtained from Keith Peden (CBER, Food and Drug Administration, Bethesda, Md.). Viral stock was produced in phytohemagglutinin (PHA)-activated peripheral blood mononuclear cells (PBMC), and titers were counted on PM1 cells. For viral neutralization by different rabbit anti-N-HR or anti-six-helix bundle antibodies, twofold-diluted preimmune and immune rabbit sera were added to mixtures of target cells (PM1) and LAI virus containing 100 50% tissue culture infectious doses (TCID50). These cell-virus-antibody mixtures were incubated for 4 h at 31.5°C and then transferred to 37°C for an additional 14 h. Unbound virus and antibodies were washed away, and the plates were cultured for 2 weeks (5 × 104 cells/well, five replicates per group). Every second day, supernatants were removed, and the cultures were supplemented with fresh medium. Viral production was determined by p24 ELISA of culture supernatants (NEN Life Science Products Inc., Boston, Mass.). Viral neutralization is expressed as percent inhibition of p24 production at a given day of culture (usually at the peak infection, days 9 to 11).
Mathematical model.
Due to the limited number of data points, syncytium formation was viewed as a two-stage process: binding of effector cells expressing Env to target cells expressing receptor molecules and formation of intermediates during time τ (A, number of intermediates), and formation of syncytia for t ⩾ τ (B, number of syncytia). Without information about the dynamics of A in the interval 0 ⩽ t < τ, it was assumed that at t = τ there are A(τ) = Aτ intermediates. It was also assumed that syncytia do not form during this interval [B(τ) = 0]. For t ⩾ τ, syncytium formation is proportional to the concentration of fusion intermediates with an effective proportionality constant k:
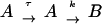 |
So, for the time interval 0 ⩽ t < τ, B(t) = 0 and A(t) = unknown function, and for t ⩾ τ,
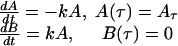 |
The solution of this system of equations is
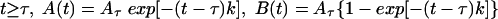 |
The lag periods (τ) and the fusion exponential rate constant k derived from the kinetic curves for E/T mixtures preincubated at 31.5°C (suboptimal temperature) and then transferred to 37°C were compared with the kinetic curves of E/T mixtures that were incubated at 37°C from time zero by using standard statistical methods (t test).
Confocal microscopy of E/T cell conjugates and rabbit antibodies binding to fusion intermediates.
Effector cells (TF228) and target cells (CEM) were labeled with membrane dyes (PKH26 and PKH67, respectively), or TF228 cells were labeled with the cytoplasmic dye calcein AM (Molecular Probes, Eugene, Oreg.) and CEM cells were labeled with the membrane dye PKH26. After 2 h of incubation at different temperatures (4, 31.5, and 37°C), cells were gently harvested and centrifuged for 4 min at 800 rpm. Cells were fixed in 4% paraformaldehyde for 10 min on ice, washed, and resuspended in PBS. Fixed cells were added to poly-l-lysine-treated glass slides (0.1% [wt/vol], 100 μl/slide) and allowed to sediment for 45 min at room temperature. The remaining buffer was removed; cell monolayers were mounted with ProLong Antifade kit (Molecular Probes). Slides were inspected under confocal microscope LSM 5 Pascal, and cell images were acquired by using AxioVision software 2.05 (Zeiss, Munich-Hallbergmoos, Germany).
To detect binding of antibundle antibodies to E/T conjugates, CEM target cells were labeled with PKH67 (membrane green dye), and TF228 cells were labeled with calcein AM (diffuse cytoplasmic green dye). Labeled effector and target cells were incubated for 2 h at 31.5°C in complete medium, and rabbit antibodies specific for the six-helix bundle were added for the last hour of incubation (at 30 μg/ml). Cells were harvested, washed once, and incubated with Alexa Fluor 546 (red)-goat anti-rabbit IgG (3 μg/ml) (Molecular Probes). Cells were washed, fixed, applied to poly-l-lysine-coated glass slides, mounted, and observed under the confocal microscope.
RESULTS
Kinetics of syncytium formation after incubation at suboptimal temperature.
Interaction of HIV-1 Env with target cell receptors triggers a series of temperature-dependent conformational changes in gp120 and gp41 that eventually lead to fusion between viral and cell membranes. To better characterize the fusion intermediates, assays were set up at suboptimal temperatures in order to slow down the fusion process. HIV-1 envelope-expressing effector (E) cells were mixed (1:1 ratio) with target (T) cells expressing appropriate receptor and coreceptors for HIV-1 and incubated for 60 min at 31.5 or 23°C. The cocultures were then transferred to 37°C, and syncytia were scored immediately and every 15 to 30 min. The kinetics of syncytium formation under these conditions was compared with that in E/T cell mixtures that were placed at 37°C without preincubation at the suboptimal temperature (Fig. 1A and B). With effector cells expressing diverse HIV-1 envelopes (IIIB, X4; JR-FL, R5; and 89.6, X4R5), no syncytia were visible at time zero.
FIG. 1.



Enhanced kinetics of syncytium formation at 37°C after preincubation of E/T cells at 31.5°C. Effectors were 12E1 cells infected for 18 h with recombinant vaccinia virus expressing 89.6 (dual tropic) Env (A) or JR-FL (R5) Env (B). Effector and target (PM1) cells were mixed at a 1:1 ratio and either incubated at 37°C or preincubated at 31.5°C (A and B) or at 23°C (B, broken line) for 60 min before transfer to 37°C. (C) TF228 (IIIB Env) effector cells were mixed with target (PM1) cells and preincubated at 31.5°C for the indicated times before transfer to 37°C. No syncytia were scored at the time of transfer. The data were fitted according to the mathematical model described in Materials and Methods. Statistically significant differences in lag time and fusion rate constant were seen after 30 and 60 min of preincubation at 31.5°C (see Table 1).
To compare the kinetics of syncytium formation under the different preincubation protocols and to conduct statistical analyses, we developed a semiempirical mathematical model to fit the experimental data (see Materials and Methods). According to this model, the dynamics of syncytium formation is reflected in the lag time, τ, and the rate constant k. These constants approximately account for all events leading to prefusion state and the rate of its conversion to fusion, respectively. However, because the dynamics of syncytium formation is complex, these parameters should be considered effective constants, reflecting an average of these key aspects of cell fusion. Figure 1 depicts fitted data for syncytium formation with 89.6 and JR-FL Env-expressing cells cocultured with PM1 target cells. In both cases, the lag time (τ) from time zero at 37°C to the first appearance of syncytia was shorter for E/T cells that were preincubated at 31.5°C for 60 min than for E/T cells that were placed immediately at 37°C (Fig. 1A and B) or preincubated at 23°C for 60 min (Fig. 1B).
The mathematical model allowed us to determine the lag times (τ) and the rate constants (k) characterizing the kinetic curves. Statistically significant differences between the τ and k values of E/T cells preincubated at 31.5°C and E/T cells placed directly at 37°C were seen for all E/T combinations (Table 1 and data not shown). Enhanced fusion kinetics was dependent on the preincubation time. When TF228(IIIB env)/PM1 mixtures were placed at 31.5°C for different times before transfer to 37°C, statistically significant differences for τ and k were observed after 30 min and were more prominent after 1 h at 31.5°C (Fig. 1C).
TABLE 1.
Comparison of kinetics of syncytium formation by E/T mixtures with and without preincubation for 60 min at a suboptimal temperature (31°C)a
| Cells | τ (min)
|
P |
k (nM)
|
P | ||
|---|---|---|---|---|---|---|
| 0 min | 60 min | 0 min | 60 min | |||
| TF228 (IIIB Env) + PM1 | 75.38 ± 1.37 | 15.88 ± 2.55 | 0.000 | 0.013 ± 0.003 | 0.008 ± 0.002 | 0.038 |
| 12E/v CB28 (JR-FL Env) + PM1 | 42.94 ± 1.90 | 23.00 ± 0.54 | 0.046 | 0.015 ± 0.0003 | 0.043 ± 0.002 | 0.054 |
| 12E/v 89.6 + PM1 | 57.47 ± 0.44 | 21.05 ± 0.57 | 0.000 | 0.010 ± 0.003 | 0.029 ± 0.001 | 0.000 |
The lag periods (τ) and the fusion exponential rate constant k derived from the kinetic curves for E/T mixtures preincubated at 31.5°C (suboptimal temperature) and then transferred to 37°C were compared with the kinetic curves of E/T mixtures that were incubated at 37°C from time zero by using standard statistical methods (t test).
Monitoring E/T cell interactions by confocal microscopy.
Confocal microscopy was used to monitor receptor-mediated E/T cell interactions. PKH26-Red-labeled TF228 cells and PKH67-Green-labeled CEM cells were mixed at a 1:1 E/T ratio and either kept at 4°C or incubated for 1 to 3 h at either 31.5 or 37°C before analysis by confocal microscopy (Fig. 2). Confocal microscopy demonstrated that a significant percentage of the double-stained cell conjugates were involved in active fusion at 37°C, as determined by E/T membrane mixing (yellow patches) (Fig. 2C) as well as transfer of the cytoplasmic dye calcein AM from effector to target cells (Fig. 2F). In contrast, neither membrane dye nor cytoplasmic dye transfer was observed between effector and target cells that were incubated for 2 h at 31.5°C (Fig. 2B and E). They looked indistinguishable from E/T cells remaining at 4°C for the entire incubation period (Fig. 2A and D).
FIG. 2.

Confocal microscopy of effector/target cell conjugates. In panels A to C, TF228 (IIIB Env) and CEM cells were labeled with the membrane dyes PKH26 (red) and PKH67 (green), respectively. In panels D to F, TF228 cells were labeled with the cytoplasmic dye calcein AM (diffused green) and CEM cells were labeled with the membrane dye PKH26 (red). Cell mixtures were incubated for 2 h at 4°C (A and D), 31.5°C (B and E), or 37°C (C and F). Conjugates were analyzed by confocal microscopy. Three-dimensional (A and B) or two-dimensional (C to F) images are presented at either ×100 (A, B, D, and E) or ×40 (C and F) magnification. Insets in panels C and F show ×100 images of syncytia formed at 37°C, as indicated by yellow patches (C) or by arrowheads (F). Data represent several experiments.
After longer incubations at 31.5°C (4 to 5 h), some mixing of membrane and cytoplasmic dyes was observed even at this suboptimal temperature, in agreement with previous studies using other E/T cell combinations (especially with adherent cells) (15; C. Weiss and D. Dimitrov, unpublished data). Thus, the enhanced kinetics of syncytium formation after transfer to 37°C could not be explained by initiation of membrane fusion pore formation during the 2-h preincubation at 31.5°C. However, it was possible that under these conditions, accumulation of receptor-triggered gp41 fusion intermediates took place.
Enhanced potency of peptide fusion inhibitors by preincubation at 31.5°C.
To test the hypothesis that preincubation at 31.5°C leads to accumulation of gp41 fusion intermediates, several gp41-derived peptides previously shown to inhibit HIV-1 fusion and viral cell entry at nanomolar concentrations were evaluated for potency after preincubation at 31.5°C. We used T20 and T21, corresponding to the C-HR (DP178) and the N-HR (DP107), respectively, as well as a trimeric coiled-coil peptide (IQN17) containing the “pocket” sequence exposed on the outer face of the N-HR coiled-coil (9, 10, 29, 30). Serial dilutions of T20, T21, and IQN17 were added to E/T cell mixtures that were either placed directly at 37°C or incubated at 31.5°C for 60 min prior to transfer to 37°C (Fig. 3). In all cases, the dose-response curves showed a 4- to 10-fold reduction in 50% inhibitory dose (ID50) when mixed with E/T cells during the preincubation at 31.5°C for 60 min. Similar reductions in ID50 values were seen with effector cells expressing X4 (IIIB), R5 (JR-FL), or dual-tropic (89.6) envelopes (Fig. 3 and data not shown). These data suggested that gp41 fusion intermediates, characterized by the exposed N-HR and C-HR (prehairpin conformation), form at 31.5°C and may persist longer at this temperature prior to fusion.
FIG. 3.

Increased potency of fusion inhibition mediated by gp41 peptides when added to E/T cells at 31.5°C. Serial dilutions of T20 and T21 (A) or IQN17 (B) were added to E/T cells that were either incubated at 37°C (▪) or preincubated with the inhibitors for 60 min at 31.5°C and then transferred to 37°C (♦). Syncytia were scored 3 to 4 h after transfer to 37°C. The data are plotted as percent inhibition of syncytia in control cultures (medium control) as a function of the inhibitor dose. Three wells per group were set up in all experiments, and the standard deviations did not exceed 15% of the means. The ID50 values calculated from the curves were as follows: T20, 37°C (65 ng/ml), 31.5°C (5 ng/ml); T21, 37°C (540 ng/ml), 31.5°C (65 ng/ml); IQN17 (89.6 Env), 37°C (400 nM), 31.5°C (50 nM); and IQN17 (IIIB Env), 37°C (120 nM), 31.5°C (28 nM).
Inhibition of fusion by rabbit anti-N-HR and anti-N+C-HR antibodies after preincubation at 31.5oC.
We previously generated a panel of rabbit polyclonal antibodies against the gp41 N-HR and C-HR peptides, as well as against mixtures of N-HR and C-HR that self-assemble into stable six-helix bundles, and showed that the anti-N-HR and anti-six-helix bundle (antibundle) antibodies can bind to receptor-activated conformations of gp41 (7). Although these antibodies were not neutralizing in conventional assays (37°C), they were reevaluated in the present fusion assay involving preincubation of E/T cells at 31.5°C for 1 h prior to transfer to 37°C (Fig. 4). Purified IgG fractions from the anti-N-HR and antibundle sera had no inhibitory activity when added to E/T cells at 37°C (at 10 or 30 μg/ml) (Fig. 4, dark bars). However, the same polyclonal IgG inhibited fusion when added to E/T cells that were preincubated at 31.5°C for 1 h before being transferred to 37°C (Fig. 4, light bars). The anti-C-HR sera, which had low titers by ELISA, did not show significant inhibition of fusion under either condition (data not shown).
FIG. 4.

Inhibition of fusion with rabbit anti-N-HR and anti-N+C-HR-peptide antibodies when added to E/T cells at 31.5°C before transfer to 37°C. TF228 (IIIB Env)/PM1 cell mixtures were incubated with various human monoclonal antibodies (or human IgG [HuIgG] control) or with IgG from rabbits immunized with N-HR peptide (R288) or with N+C-HR peptide mixture (antibundle, R948) (or with preimmune rabbit IgG) at 10 μg/ml. The antibodies were preincubated with the E/T cell mixtures at 31.5°C for 1 h before transfer to 37°C (light columns) or only at 37°C (dark columns). In the case of human monoclonal antibodies, the monoclonal antibodies were incubated with the Env-expressing cells for 1 h at 37°C before addition of the target cells (dark columns).
In parallel, we assessed other antibodies recognizing various neutralizing epitopes on gp120 (2G12, IG1b12, 48d, and 17b) or gp41 (2F5). No significant difference in their inhibitory activities was observed when these monoclonal antibodies were preincubated with E/T cells at 31.5°C compared with 37°C (Fig. 4). Two other gp41-specific monoclonal antibodies (T9 and D61) that do not bind to N- or C-HR peptides derived from mice immunized with oligomeric gp160 (by Patricia Earl and Christopher Broder, National Institutes of Health) (8) were tested and found to lack inhibitory activity under both 37°C and 31.5→37°C conditions (data not shown).
The inhibitory activity was also evaluated in viral neutralization assays, in which virus-cell mixtures were incubated with the rabbit anti-N-HR, anti-C-HR, and antibundle antibodies (or preimmune sera) for 4 h at 31.5°C, before transfer to 37°C for an additional 14 h of incubation (Table 2). Significant viral neutralization (on days 9 to 13) was observed in cultures treated with either anti-N-HR or antibundle serum (but not preimmune or anti-C-HR serum), in agreement with the cell-cell fusion inhibition assays. Similar results were obtained with purified IgG from multiple rabbits immunized against slightly different versions of N-HR, C-HR, and N+C-HR (six-helix bundle) peptides (7). Immune sera derived from three of four and four of four rabbits immunized with N-HR or N+C-HR peptides, respectively, inhibited both fusion and viral infection when added to E/T cells or to virus-T-cell mixtures at 31.5°C (but not at 37°C) (data not shown).
TABLE 2.
Inhibition of HIV-1 infection and syncytia by rabbit antibodies against gp41a
| Inhibitor | HIV-1 infection
|
Syncytium formation
|
||
|---|---|---|---|---|
| p24 (pg/ml) | % Inhibition | No. of syncytia | % Inhibition | |
| Rabbit 979 (preimmune) | 184,648 ± 67,544 | 0 | 666 ± 34 | 0 |
| Rabbit 979 (α-N-peptide) | 671 ± 105 | 99 | 287 ± 18 | 57 |
| Rabbit 948 (α-N+C-peptides) | 873 ± 246 | 99 | 297 ± 14 | 55 |
| Rabbit 980 (α-C-peptide) | 140,730 ± 9,585 | 24 | 631 ± 86 | 5 |
To test neutralization of HIV-1, LAI virus was incubated with PM1 cells (100 TCID50/well, five replicates) in the absence or presence of rabbit antibodies (1:50 final dilution) at 31.5°C for 4 h, and the plates were then transferred to 37°C for 14 h. All wells were washed to remove unbound virus and antibodies and cultured for 16 days. To test fusion inhibition, TF228 (IIIB Env) and PM1 cells were cocultured for 1 h at 31.5°C (three wells/group) for 60 min in the absence or presence of rabbit antibodies (1:10 dilution) and then transferred to 37°C. Syncytia were scored after 4 to 5 h.
These results suggested that rabbit anti-N-HR and antibundle antibodies may bind to receptor-activated gp41 fusion intermediate structures in spite of the close proximity of the E/T and virus-T-cell membranes.
Binding of rabbit antibundle antibodies to E/T cells incubated at 31.5°C.
Confocal microscopy was performed to confirm that rabbit antibundle antibodies bind to cell surface gp41 after interaction with target cells. TF228 (IIIB Env) cells were labeled with the cytoplasmic dye calcein AM (diffuse green staining) and CEM cells with the membrane dye PKH67-Green (Fig. 5). Mixed labeled effector and target cells (or effector cells alone) were incubated for 1 h at 31.5°C and then treated with rabbit antibundle IgG (Fig. 5B) or preimmune IgG (Fig. 5A) for an additional 1 h at 31.5°C, followed by Alexa Fluor 546 (red)-conjugated goat anti-rabbit IgG at 4°C. Confocal microscopy demonstrated binding of antibundle antibodies at the E/T cell-membrane interfaces (Fig. 5B). The same antibodies did not bind to effector cells that were incubated without target cells (data not shown).
FIG. 5.

Binding of the antibundle antibodies to fusion intermediates at E/T cell contacts. TF228 (IIIB Env) cells were labeled with calcein AM and cocultured with CEM cells labeled with PKH67 for 2 h at 31.5°C. IgG from a nonimmunized rabbit (A) or from the rabbit immunized with gp41-derived peptides (N36+C34) (B) was added to the cell cultures for the last hour of incubation. Cells were harvested, incubated with Alexa Fluor 546 goat anti-rabbit IgG, and mounted on slides. Digital images of the cell cultures are presented as two-dimensional images scanned at ×100 magnification. CEM cells are marked with arrowheads, TF228 cells are marked with asterisks, and E/T cell junctions are indicated with arrows. The presence of red color in the E/T cell junction areas indicates binding of antibundle antibodies.
Kinetics of formation of gp41 fusion intermediates.
In order to further dissect the kinetics of formation of gp41 fusion intermediates, we performed a series of experiments where antibodies were added to E/T cells at various times during a 90-min preincubation at 31.5°C, prior to transfer to 37°C. Adding anti-N-HR antibodies to E/T cells after a 60-min delay resulted in a significant reduction in their inhibitory activity, as judged by 4- to 30-fold increase in the ID50 values for different antibodies (Table 3). In contrast, the efficiency of fusion inhibition by antibundle IgG increased slightly when the antibodies were added to E/T cells after 60 min at 31.5°C (Table 3). These data suggested that during the incubation at 31.5°C, both types of intermediates are formed and can be recognized by specific antibodies, resulting in fusion arrest. Furthermore, our data indicate that formation of the prehairpin intermediates precedes the transition to six-helix bundles.
TABLE 3.
Decreased potency of fusion inhibition by anti-N-HR antibodies after preincubation of E/T cells at 31.5°C for 60 mina
| Rabbit antibody | ID50 (μg/ml)
|
|
|---|---|---|
| A | B | |
| R288 (α-DP107 IgG) | 5.1 | 60 |
| R979 (α-N36 IgG) | 2.9 | 50 |
| R3303 (α-N36 IgG) | 7.8 | >100 |
| R3249 (α-DP107 IgG) | 4.2 | 17 |
| R948 (α-N36+C34) | 21 | 4.8 |
| R3250 (α-N36+C34) | 16 | 5.2 |
Total IgG antibodies were purified by adsorption and elution from protein A-conjugated agarose beads. Experiment shown is representative of three independent experiments. Bundle-specific antibodies were isolated after multiple adsorptions of R948 and R3250 IgG antibodies to immobilized N-peptide and C-peptide columns (see Materials and Methods). The nonadsorbed antibodies were shown to bind to six-helix bundles (N+C peptides) in ELISA. Rabbit antibodies (in multiple doses) were added to mixtures (1:1) of effector (TF228, IIIB Env) and target (PM1) cells either at time zero (A) or after 60 min of incubation at 31.5°C (B). The total incubation time of the E/T cells at 31.5°C was 90 min, after which the cell-antibody mixtures were transferred to 37°C. Syncytia were scored 4 h later. ID50 values were calculated from the dose-response curves for each antibody preparation added according to the two regimens.
To confirm the transition from prehairpin intermediate to six-helix bundle after 1 to 2 h of incubation at 31.5°C, the same timing experiments were repeated by using peptides targeting the prehairpin structures (i.e., T20 and T21). When T20 was added to E/T cells at the beginning of the preincubation at 31.5°C, the dose-response curve reflected an increased potency of inhibition corresponding to 7- and 13-fold reductions in ID50 values for 89.6 and IIIB envelope-expressing cells, respectively, compared with T20 added to E/T cells at 37°C (Fig. 6), in agreement with earlier experiments (Fig. 3). In contrast, if T20 was added to E/T cells after incubation for 1 h at 31.5°C, just prior to their transfer to 37°C, a significant reduction in inhibition efficiency was observed, as judged by 22- and 36-fold increases in the ID50 values for 89.6 and IIIB envelope-expressing cells, respectively (Fig. 6). These data suggest that most of the fusion intermediates present after 60 min at 31.5°C are at the six-helix bundle conformation and therefore are less susceptible to T20.
FIG. 6.

Preincubation of E/T cells at 31.5°C before transfer to 37°C results in reduced susceptibility to inhibition by T20 peptide. Serial dilutions of T20 were added to E/T cells that were incubated at 37°C (▪) or preincubated with the inhibitor for 60 min at 31.5°C and then transferred to 37°C (⧫) or preincubated without the inhibitor at 31.5°C for 60 min. T20 was added to these cells at the time of transfer to 37°C (▴). Syncytia were scored 3 h (89.6 Env) or 4 h (IIIB Env) after transfer to 37°C. The data are plotted as percent inhibition of syncytia in control cultures (medium control) as a function of the inhibitor dose. Three wells per group were set up in all experiments, and the standard deviations did not exceed 15% of the means. The ID50 values calculated from the curves were as follows: 89.6 Env, 11 ng/ml (▪), 1.5 ng/ml (⧫), and 250 ng/ml (▴); IIIB Env, 5.2 ng/ml (▪), 0.4 ng/ml (⧫), and 190 ng/ml (▴).
Additional timing experiments were performed by using affinity-purified anti N-HR and antibundle antibodies and an antibody that binds CD4 and prevents interaction with Env (13) (Table 4). As expected, if antibodies were added to E/T cell mixtures at the beginning of the 31.5°C preincubation (Table 4, protocol A), the anti-N and antibundle antibodies blocked 70 and 64% of subsequent syncytium formation, respectively. On the other hand, if antibodies were added to E/T cells after 60 min of incubation at 31.5°C, just prior to transfer to 37°C (Table 4, protocol B), the anti-N-HR antibodies did not inhibit syncytium formation, while under the same conditions, the antibundle antibodies showed partial inhibition of subsequent syncytium formation (35 to 45% inhibition in three experiments with two different envelopes). In this protocol, the anti-CD4 monoclonal antibody gave only partial inhibition, suggesting the presence of a significant number of postbinding fusion-active structures that are resistant to blocking by anti-CD4 monoclonal antibody.
TABLE 4.
Antibundle but not anti-N-HR partially blocks syncytium formation after incubation of E/T at 31.5°C for 60 mina
| Protocol | Antibody (dose, μg/ml) | No. of syncytia | % Inhibition |
|---|---|---|---|
| A | None | 553 ± 21 | |
| R3249 (α-N-peptide) (30) | 168 ± 37 | 70 | |
| R3250 (α-bundle) (30) | 198 ± 38 | 64 | |
| α-CD4 (3) | 5 ± 2 | 99 | |
| B | None | 496 ± 12 | |
| R3249 (α-N-peptide) (30) | 545 ± 26 | 0 | |
| R3250 (α-bundle) (30) | 272 ± 8 | 45 | |
| α-CD4 (3) | 197 ± 29 | 60 |
Rabbit antibodies were added to mixtures (1:1) of E/T cells according to two different temperature transition protocols (protocol A, E/T + Ab [31.5°C, 1 h] → 37°C; protocol B, E/T [31.5°C, 1 h] → add Ab → 37°C). Under both protocols, incubation time of the cells at 31.5°C was 60 min, after which the cells were transferred to 37°C. Syncytia were scored at 4 h. Data are representative of three experiments (different Envs) with similar results.
Together these findings suggest that prehairpin fusion intermediates are precursors to the six-helix bundles and that during the prefusion lag time the prehairpin intermediates may be shorter lived than the six-helix bundles.
Fusion inhibition with human monoclonal antibodies specific for gp41 conformational epitopes.
To confirm the conclusions derived from experiments with polyclonal IgG, we tested a panel of human monoclonal antibodies previously shown to recognize conformational gp41 epitopes (12, 32). When added to effector cells at 37°C for 1 h prior to the addition of target cells, minimal or no inhibition was observed with these monoclonal antibodies, while the 2F5 monoclonal antibody effectively blocked fusion, as expected (ID50, 2.5 μg/ml) (Table 5). Monoclonal antibody 98-6 was mapped to a C-HR epitope (5′ to the 2F5 epitope [32]). It blocked fusion when added to E/T cells at the beginning of the 2-h incubation at 31.5°C. However, if added to E/T cells after 1 h at the suboptimal temperature, its inhibitory activity dropped dramatically (ID50 of 15 and 90 μg/ml, respectively), similar to what was observed with rabbit anti-N-HR IgG (above).
TABLE 5.
Fusion inhibition with human monoclonal antibodies specific for gp41 conformational epitopes
| Protocola | Antibodyb (dose, μg/ml) | Specificity | % Fusion inhibition at 37°C (ID50, μg/ml) |
|---|---|---|---|
| A | MAb 240-D (30) | Cluster I | 0 |
| MAb 181-D (30) | Cluster I | 0 | |
| MAb 98-6 (30) | α-C-HR | 18 | |
| MAb 1281 (30) | α-Bundle | 0 | |
| MAb 167-D (30) | α-Bundle | 0 | |
| MAb 2F5 (30) | ELDKW | 78 (2.5) | |
| B | MAb 240-D (30) | Cluster I | 0 |
| MAb 181-D (30) | Cluster I | 1 | |
| MAb 98-6 (30) | α-C-HR | 75 (15) | |
| MAb 1281 (30) | α-Bundle | 52 (25) | |
| MAb 167-D (30) | α-Bundle | 64 (10) | |
| MAb 2F5 (30) | ELDKW | 65 (4.2) | |
| C | MAb 240-D (30) | Cluster I | 0 |
| MAb 181-D (30) | Cluster I | 0 | |
| MAb 98-6 (30) | α-C-HR | 38 (90) | |
| MAb 1281 (30) | α-Bundle | 78 (4) | |
| MAb 167-D (30) | α-Bundle | 74 (3.5) | |
| MAb 2F5 (30) | ELDKW | 60 (6.2) |
Protocol A, E + Ab (37°C, 1 h) → add T cells; protocol B, E/T + Ab (31.5°C, 1 h) → 37°C; protocol C, E/T (31.5°C, 1 h) → add Ab (31.5°C, 1 h) → 37°C.
MAb, monoclonal antibody.
Importantly, two monoclonal antibodies that were found to be specific for the six-helix bundles, 167-D and 1281 (12), exhibited an opposite pattern of inhibition. They inhibited fusion more efficiently (lower ID50 values) when added to E/T cells after 1 h of incubation at 31.5°C (Table 5). These findings were reproduced with effector cells expressing 89.6 envelope and confirm our conclusions from the data with the rabbit antibundle polyclonal antibodies.
Transition from six-helix bundles to syncytium formation.
In order to track the lag time between six-helix bundle formation and cell-cell fusion after transfer to 37°C, antibundle IgG was added in the presence of anti-CD4 antibody to block new E/T cell interactions that may happen at 37°C. In these experiments, anti-CD4 monoclonal antibody (Q4120) was added after 2 h of preincubation at 31.5°C prior to transfer to 37°C (time zero), and purified antibundle IgG (from R948) was added either together with the anti-CD4 monoclonal antibody (time zero) or at different times after transfer of the cultures to 37°C (Fig. 7). The anti-CD4 monoclonal antibody only partially blocked subsequent syncytium formation at 37°C (45 to 55% inhibition) (Table 4, Fig. 7). When added with anti-CD4 at time zero, the antibundle antibodies blocked 72% of syncytium formation. Delayed addition of the antibundle antibodies resulted in gradual loss of inhibition during the first 30 min at 37°C, with the preformed six-helix bundles having an apparent half-life of 9 to 10 min. In an identical experiment with 12E1 effector cells expressing 89.6 Env, the half-life of the preformed bundles was only 3.5 min after transfer to 37°C (data not shown).
FIG. 7.

Dynamics of the transition from six-helix bundles to membrane fusion: loss of sensitivity to neutralization by antibundle antibodies after transfer of E/T cells from 31.5°C (2 h) to 37°C in the presence of anti-CD4 monoclonal antibody. TF228 (IIIB Env)/PM1 cells were incubated for 2 h at 31.5°C in the absence of antibodies (Abs). Immediately prior to transfer to 37°C (time zero), anti-CD4 monoclonal antibody Q4120 was added at 3 μg/ml. Six-helix bundle-specific IgG from R948 was added at 30 μg/ml at the indicated times. Syncytia were scored 4 h after transfer to 37°C.
DISCUSSION
Our findings shed new light on the sequence of conformational changes preceding membrane fusion and the accessibility of gp41 fusion intermediates to antibodies and inhibitors. Earlier studies concluded that the thermodynamically stable six-helix bundles form coincidently with membrane fusion (22) and that antibodies are not likely to bind fusion intermediates due to close proximity of E/T membranes (23). In contrast, our data suggest not only that six-helix bundles can form prior to membrane fusion, but that antibodies targeting the six-helix bundle and prehairpin structures bind fusion intermediates and arrest fusion under certain conditions. The different conclusions are likely explained by differences in the experimental approaches we used compared to those used in the study by Melikyan et al. (22) as well as the use in our study of both polyclonal and monoclonal antibodies targeting the prehairpin and the six-helix fusion intermediates.
Our strategy involved preincubations of E/T cells at 31.5oC prior to transfer to 37oC, compared to the 23 to 25oC used by Melikyan et al., in both fusion and infectivity assays. Determinations of the kinetics and ID50s for a variety of gp41 peptide inhibitors and antibodies targeting gp41 fusion intermediates were conducted. Our kinetics studies showed statistically significant shortening of the fusion lag period and increased kinetic rate constants of syncytium formation (at 37oC) after incubations at 31.5oC but not at 23oC compared to continuous cultures at 37oC (Fig. 1). Preincubation at either suboptimal temperature may be permissive for progression to the prehairpin structures, as judged by increased susceptibility to fusion inhibition by peptides targeting the N-HR and C-HR (T21 and T20) (Fig. 4) (22). However, the enhanced fusion kinetics with preincubations at 31.5oC further suggested that this temperature allowed accumulation of more advanced gp41 fusion intermediates. Importantly, under these conditions (i.e., 31.5oC for 1 to 2 h), we did not observe transfer of either membrane dye or cytoplasmic dye between effector and target cells (Fig. 2), confirming fusion arrest in our system.
To determine whether progression from prehairpin to six-helix bundle fusion intermediates occurs at 31.5°C, we tested the ability of the gp41 peptides and anti-N-HR and -bundle antibodies to inhibit fusion when added to E/T cells at various times after preincubation at 31.5°C. When the peptides were added to E/T cells at the beginning of 31.5°C incubation, the calculated ID50 values were 5- to 10-fold lower than the ID50 values for E/T cells that were incubated with the same inhibitors at 37°C (Fig. 3). However, when these peptides were added to E/T cells after 1 h of preincubation at 31.5°C (at the time of transfer to 37°C), there was a >30-fold increase in the ID50 values (Fig. 6). Furthermore, if an anti-CD4 monoclonal antibody was added at the end of the 2-h incubation at 31.5°C to prevent new conjugate formation at 37°C, the T20 peptide lost inhibitory activity (data not shown), indicating that conformational changes had proceeded beyond the prehairpin intermediate.
Antibody experiments also confirmed the stepwise progression of fusion intermediates from the prehairpin to six-helix bundle structure at 31.5°C prior to fusion. IgG recognizing either the N-HR or six-helix bundles inhibited fusion when added to E/T cells and incubated at 31.5°C for 1 h before transfer to 37°C, but no fusion inhibition was seen in assays that were maintained only at 37°C. However, when added to E/T cultures after 60 min of preincubation at 31.5°C, the anti-N-HR antibodies had reduced inhibitory activity (5- to >50-fold increase in ID50 values), while the antibundle antibodies maintained potency, which appeared even to be slightly enhanced. Identical results were obtained with several human monoclonal antibodies that are specific for either the six-helix bundle or the C-HR (12; unpublished data), while other anti-gp41 monoclonal antibodies specific for the disulfide loop between the HR regions were not inhibitory under any conditions tested.
The patterns of inhibition observed and the correlation between the efficiency of blocking and time of addition to the temperature-arrested intermediates strongly indicate that they recognize structures that are relevant for fusion and are formed at a stepwise manner after effector/target mixing at the suboptimal temperature used in our studies. Furthermore, since the monoclonal antibodies were isolated from HIV-1-infected individuals, our data suggest that during infection, some exposure of gp41 prefusion conformational structures takes place.
Our data indicate that after 1 h at 31.5°C, a significant portion of the prehairpin fusion intermediates have undergone transition to six-helix bundles, which are resistant to inhibition by the anti-N-HR or anti-C-HR antibodies and gp41-derived peptides (T20) but remain sensitive to antibundle antibodies.
Our data further demonstrate that the inability of the anti-N-HR and antibundle antibodies to block fusion or to neutralize virus in previous studies is not due to steric interference of antibody binding due to close proximity of E/T or virus-cell membranes. Rather, the lack of inhibition at 37°C is more likely related to unfavorable competition between the avidity and/or kinetics of antibody binding and the kinetics of the E/T membrane fusion process.
The increased efficiency of inhibition with antibundle antibodies when added after 60 min at 31.5oC rather than at the beginning of the preincubation is not understood. Conceivably, it reflects an increase in the density of the six-helix bundles in localized membrane patches. Under such a scenario, the overall avidity of antibody binding may increase due to cross-linking of adjacent bundles.
Additional studies demonstrated that after transfer to 37°C, preformed six-helix bundles transitioned to syncytium formation with a half-life of 3.5 to 9.5 min (for different Env-expressing cells). Together, these findings lead us to speculate that a rate-limiting step in fusion at 31.5°C may reflect the time needed to accumulate critical numbers of six-helix bundles into fusion pores rather than the time needed to form six-helix bundles (Fig. 8). Our findings support the model proposed by Chernomordik et al. (6), who postulated that the six-helix bundles help bring the cellular and viral membranes in close proximity and that subsequent higher-order clustering of the six-helix bundles facilitates membrane fusion.
FIG. 8.

Proposed model of gp41 conformational changes leading to fusion. Arrows indicate potential binding of peptides and antibodies (Ab) targeting the prehairpin and six-helix bundle fusion intermediates.
We conclude that six-helix bundles can form prior to membrane fusion and that both the prehairpin fusion intermediates and the six-helix bundles can be accessible to antibodies. Our data support further investigations of fusion intermediates as targets in the rational design of novel HIV-1 inhibitors and vaccines. The high degree of conservation in the gp41 N- and C-HR sequences among HIV-1 strains of different clades makes these structures desirable targets for inhibition. The broadly neutralizing antibodies generated in mice vaccinated with formaldehyde-fixed fusion-competent E/T conjugates may have included antibodies specific for gp41 fusion active intermediates similar to those used in our studies (19).
Acknowledgments
We are grateful to Debra Eckert and Peter Kim for providing us with IQN17 peptide. We thank Keith Peden and Judy Beeler for critical review of the manuscript.
The studies described in this manuscript were partly funded by the NIH Intramural AIDS Targeted Antiviral Program (H. Golding and C. Weiss).
REFERENCES
- 1.Berger, E. A., P. M. Murphy, and J. M. Farber. 1999. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism and disease. Annu. Rev. Immunol. 17:657-700. [DOI] [PubMed] [Google Scholar]
- 2.Broder, C. C., and E. A. Berger. 1995. Fusogenic selectivity of the envelope glycoprotein is a major determinant of human immunodeficiency virus type 1 tropism for CD4+ T-cell lines vs. primary macrophages. Proc. Natl. Acad. Sci. USA 92:9004-9008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan, D. C., D. Fass, J. M. Berger, and P. S. Kim. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263-273. [DOI] [PubMed] [Google Scholar]
- 4.Chan, D. C., and P. S. Kim. 1998. HIV entry and its inhibition. Cell 93:681-684. [DOI] [PubMed] [Google Scholar]
- 5.Chen, C. H., M. L. Greenberg, D. P. Bolognesi, and T. J. Matthews. 1995. Monoclonal antibodies that bind to the core of fusion-active glycoprotein 41. J. Virol. 69:1462-1472. [DOI] [PubMed] [Google Scholar]
- 6.Chernomordik, L. V., E. Leikina, V. Frolov, P. Bronk, and J. Zimmerberg. 1997. An early stage of membrane fusion mediated by the low pH conformation of influenza hemagglutinin depends upon membrane lipids. J. Cell Biol. 136:81-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Rosny, E., V. Vassell, P. T. Wingfield, C. T. Wild, and C. D. Weiss. 2001. Peptides corresponding to the heptad repeat motifs in the transmembrane protein (gp41) of human immunodeficiency virus type 1 elicit antibodies to receptor glycoprotein. J. Virol. 75:8859-8863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Earl, P. L., C. C. Broder, R. W. Doms, and B. Moss. 1997. Epitope map of human immunodeficiency virus type 1 gp41 derived from 47 monoclonal antibodies produced by immunization with oligomeric envelope protein. J. Virol. 71:2674-2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eckert, D. M., V. N. Malashkevich, and P. S. Kim. 1998. Crystal structure of GCN4-pIqI, a trimeric coiled coil with buried polar residues. J. Mol. Biol. 284:859-865. [DOI] [PubMed] [Google Scholar]
- 10.Eckert, D. M., V. N. Malashkevich, L. H. Hong, P. A. Carr, and P. Kim. 1999. Inhibiting HIV-1 entry: discovery of d-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 99:103-115. [DOI] [PubMed] [Google Scholar]
- 11.Furuta, R. A., C. T. Wild, Y. Weng, and C. D. Weiss. 1998. Capture of an early HIV-1 gp41 six-helix bundle formation occurs rapidly after the engagement of gp120 by CXCR4 in the HIV-1 Env-mediated fusion process. Biochemistry 40:12231-12236. [DOI] [PubMed] [Google Scholar]
- 12.Gorny, M. K., and S. Zolla-Pazner. 2000. Recognition by human monoclonal antibodies of free and complexed peptides representing the prefusogenic and fusogenic forms of human immunodeficiency virus type 1 gp41. J. Virol. 74:6186-6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Healey, D., L. Dianda, J. P. Moore, J. S. McDougal, M. J. Moore, P. Estess, D. Buck, P. D. Kwong, P. C. Beverley, and Q. J. Sattentau. 1990. Novel anti-CD4 monoclonal antibodies separate human immunodeficiency virus infection and fusion of CD4+ cells from virus binding. J. Exp. Med. 172:1233-1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hioe, C. E., S. Xu, P. Chigurupati, S. Burda, C. Williams, M. K. Gorny, and S. Zolla-Pazner. 1997. Neutralization of HIV-1 primary isolates by polyclonal and monoclonal human antibodies. Int. Immunol. 9:1281-1290. [DOI] [PubMed] [Google Scholar]
- 15.Jernigan, K. M., R. Blumenthal, and A. Puri. 2000. Varying effects of temperature, Ca2+ and cytochalasin on fusion activity mediated by human immunodeficiency virus type 1 and type 2 glycoproteins. FEBS Lett. 474: 246-251. [DOI] [PubMed] [Google Scholar]
- 16.Jiang, S., K. Lin, N. Strick, and A. R. Neurath. 1993. HIV-1 inhibition by a peptide. Nature 365:113-114. [DOI] [PubMed] [Google Scholar]
- 17.Jiang, S., K. Lin, and M. Lu. 1998. A conformation-specific monoclonal antibody reacting with fusion-active gp41 from the human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 72:10213-10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jonak, Z. L., R. K. Clark, D. Matour, S. Trulli, R. Craig, E. Henri, E. V. Lee, R. Greig, and C. Debouck. 1993. A human lymphoid recombinant cell line with functional human immunodeficiency virus type 1 envelope. AIDS Res. Hum. Retroviruses 9:23-32. [DOI] [PubMed] [Google Scholar]
- 19.LaCasse, R. A., K. E. Follis, et al. 1999. Fusion-competent vaccines: broad neutralization of primary isolates of HIV. Science 283:357-362. [DOI] [PubMed] [Google Scholar]
- 20.Lapham, C. K., J. Ouyang, B. Chandrasekhar, N. Y. Nguyen, D. S. Dimitrov, and H. Golding. 1996. Evidence for cell-surface association between fusin and the CD4-gp120 complex in human cell lines. Science 274:602-605. [DOI] [PubMed] [Google Scholar]
- 21.Lu, M., and P. S. Kim. 1997. A trimeric structural subdomain of the HIV-1 transmembrane glycoprotein. J. Biol. Struct. Dyn. 15:465-471. [DOI] [PubMed] [Google Scholar]
- 22.Melikyan, G. B., R. M. Markosyan, H. Hemmati, M. K. Delmedico, D. M. Lambert, and F. S. Cohen. 2000. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 151:413-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore, J. P., P. W. H. I. Parren, and D. R. Burton. 2001. Genetic subtypes, humoral immunity, and human immunodeficiency virus type 1 vaccine development. J. Virol. 75:5721-5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muster, T., F. Steindl, M. Purtscher, A. Trkola, A. Klima, G. Himmler, F. Ruker, and H. Katinger. 1993. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 67:6642-6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skehel, J. J., and D. C. Wiley. 1998. Coiled coils in both intracellular vesicle and viral membrane fusion. Cell 95:871-874. [DOI] [PubMed] [Google Scholar]
- 26.Tan, K., J. Liu, J.-H. Wang, S. Shen, and M. Lu. 1997. Atomic structure of thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. USA 94:12303-12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thali, M., J. P. Moore, C. Furman, M. Charles, D. D. Ho, J. Robinson, and J. Sodroski. 1992. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J. Virol. 67:3978-3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weissenhorn, W., A. Dessen, S. C. Harrison, J. J. Skehel, and D. C. Wiley. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426-430. [DOI] [PubMed] [Google Scholar]
- 29.Wild, C. T., D. C. Shugars, T. K. Greenwell, C. McDanal, and T. J. Mathews. 1994. Propensity for leucine zipper-like domain of human immunodeficiency virus type 1 gp41 to form oligomers correlates with a role in virus-induced fusion rather than assembly of the glycoprotein complex. Proc. Natl. Acad. Sci. USA 91:12676-12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wild, C. T., T. Greenwell, D. Shugars, L. Rimsky-Clarke, and T. J. Mathews. 1995. The inhibitory activity of an HIV type 1 peptide correlates with its ability to interact with leucine zipper structure. AIDS Res. Hum. Retroviruses 11:323-325. [DOI] [PubMed] [Google Scholar]
- 31.Wyatt, R., and J. Sodroski. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280:1884-1888. [DOI] [PubMed] [Google Scholar]
- 32.Xu, J.-Y., M. K. Gorny, T. Palker, S. Karwowska, and S. Zolla-Pazner. 1991. Epitope mapping of ten human monoclonal antibodies to gp41, the transmembrane protein of human immunodeficiency virus type 1. J. Virol. 65:4832-4838. [DOI] [PMC free article] [PubMed] [Google Scholar]