

Abstract
Responses in three chimpanzees were compared following challenge with a clonal hepatitis C virus (HCV) contained in plasma from an animal that had received infectious RNA transcripts. Two of the chimpanzees (Ch1552 and ChX0186) had recovered from a previous infection with HCV, while the third (Ch1605) was a naïve animal. All animals were challenged by reverse titration with decreasing dilutions of plasma and became serum RNA positive following challenge. Ch1605 displayed a typical disease profile for a chimpanzee. We observed increasing levels of serum RNA from week 1 postinoculation (p.i.), reaching a peak of 106 copies/ml at week 9 p.i., and alanine aminotransferase (ALT) elevations and seroconversion to HCV antibodies at week 10 p.i. In contrast, both Ch1552 and ChX0186 exhibited much shorter periods of viremia (4 weeks), low serum RNA levels (peak, 103 copies/ml), and minimal ALT elevations. A comparison of intrahepatic cytokine levels in Ch1552 and Ch1605 showed greater and earlier gamma interferon (IFN-γ) and tumor necrosis factor alpha responses in the previously infected animal, responses that were 30-fold greater than baseline responses at week 4 p.i. for IFN-γ in Ch1552 compared to 12-fold in Ch1605 at week 10 p.i. These data indicate (i) that clonal HCV generated from an infectious RNA transcript will lead to a typical HCV infection in naïve chimpanzees, (ii) that there are memory immune responses in recovered chimpanzees that control HCV infection upon rechallenge, and (iii) that these responses seem to be T-cell mediated, as none of the animals had detectable antibody against the HCV envelope glycoproteins. These observations have encouraging implications for the development of a vaccine for HCV.
Infection with hepatitis C virus (HCV) poses a serious public health problem. It frequently leads to chronic hepatitis and cirrhosis in humans and is associated with the development of hepatocellular carcinoma (26). Persistent infection occurs in up to 85% of patients (1) and at least 30% of chimpanzees (2). Persistently infected patients are the major source for new infections. Drug therapy consists of alpha interferon (IFN-α) or IFN plus ribavirin; however, only a minority of those infected respond to treatment, making vaccine development of major importance.
HCV has a genomic organization similar to those of the pestiviruses and flaviviruses and has been classified as a separate genus, Hepacivirus, within the family Flaviviridae (25). The viral particle consists of a nucleocapsid containing a positive-sense, single-stranded RNA genome of approximately 9,500 nucleotides (nt) (5) surrounded by an envelope derived from host membranes into which are inserted the virally encoded glycoproteins (E1 and E2). The genome contains highly conserved untranslated regions at both the 5′ and 3′ termini (13, 16, 28). The viral products (core, E1, E2, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) are processed from a ∼3,000-amino-acid polyprotein expressed from a single open reading frame (11, 14).
It has been noted that antibody to the surface proteins of HCV (E1 and E2) occurs more frequently in persistent infections than in those that are self-limiting (24). There is some evidence that antibody to surface proteins can neutralize virus in vitro (9, 18), and vaccine studies have been carried out with recombinant E1E2 antigens to induce neutralizing antibody (4). Although protection in chimpanzees against low-level challenge (10 50% chimpanzee infectious doses [CID50]) was observed following vaccination, this was only during periods of high anti-E1E2 antibody titer (4). It has therefore been suggested that clearance following infection is not mediated through neutralizing antibody (24) and that in fact strong T-cell responses, often to nonstructural proteins, in humans and chimpanzees are a better correlate of viral clearance and recovery (6, 7, 12, 21, 27).
The chimpanzee is currently the only animal model for HCV infection. These animals seem to clear HCV infection more frequently than humans and develop only mild hepatitis upon progression to chronicity. However, this model represents well the features of HCV infection in humans, such as the kinetics of viremia and persistent infection despite the presence of humoral and cellular immune responses (29). Of the chimpanzees that clear HCV, it has long been known that reinfection can occur with homologous and heterologous strains of the virus (8, 23). The suggestion has been made that reinfection can occur due to the variability of the HCV genome and the fact that HCV exists in clinical isolates as a population of quasispecies that may include viral variants which can escape the preexisting neutralizing or cellular immune responses (19, 30). These observations have led to the belief that immunity following HCV infections is weak and that the development of an effective vaccine against HCV poses a serious challenge.
We wished to revisit the question of reinfection of chimpanzees that have cleared HCV to determine whether there is some control of viral replication upon rechallenge. One naïve chimpanzee and two previously infected animals were compared in their responses to infection with a clonal virus stock that lacks quasispecies variability. We examined the clinical outcome of infection of these three animals by comparing the levels and durations of viremia, HCV-specific antibody responses, alanine aminotransferase (ALT) elevation, and intrahepatic cytokine levels. In this study, we saw a clear difference in the course of infection between the animals rechallenged following clearance and the naïve animal. These results indicate that a memory immune response is present in animals that clear infections and that this response can significantly control virus upon rechallenge, suggesting that a vaccine against HCV is a feasible prospect.
MATERIALS AND METHODS
Challenge inoculum.
The chimpanzees in this study were challenged intravenously with 1 ml of plasma from Ch1536 serially diluted in Hanks buffer containing 0.5% human serum albumin. Ch1536 was inoculated with RNA transcripts from an HCV cDNA clone by direct intrahepatic injection (15). At week 2 postinoculation (p.i.), serum from the animal became positive for HCV RNA and the animal developed an apparently normal HCV infection, with elevated liver enzymes and seroconversion to HCV antibodies (15, 22). The sample taken at week 4 p.i. was found to have a real-time PCR titer for HCV RNA of 4 × 105 copies/ml (22) and was free of detectable HCV antibodies, and the viral population sequence was indistinguishable from that of the parental cDNA clone. This plasma sample was serially diluted 10-fold and used to challenge chimpanzees ChX0186, Ch1552, and Ch1605 by reverse titration.
Chimpanzees.
The serologic status of the animals used in this study is shown in Table 1. Both ChX0186 and Ch1552 had been previously infected with HCV and had cleared the virus from their sera. ChX0186 had received H77 plasma in 1994, and Ch1552 had received infectious RNA in 1998, approximately 48 and 15 months prior to the initiation of this experiment, respectively. At the time of challenge, these animals were HCV antibody positive by enzyme immunoassay (EIA) 3.0 (Ortho-Clinical Diagnostics, Rochester, N.Y.) but negative for anti-E1E2 antibody and negative in a T-cell proliferation assay to several HCV antigens. Ch1605 was a naïve animal and had no signs of prior exposure to HCV. Sera from these animals were collected at weekly intervals following challenge with diluted virus and tested by nested reverse transcription (RT)-PCR for HCV RNA as described below. If the serum sample was negative at 3 weeks postchallenge, the animal received the next highest dilution at week 4. This continued until a positive serum sample was detected. Serum samples were tested for ALT elevations, anti-HCV antibodies, and serum RNA for up to 51 weeks postchallenge with the infecting dilution. Liver biopsies were obtained biweekly and tested for HCV RNA, IFN-γ, tumor necrosis factor alpha (TNF-α), and interleukin-4 (IL-4) mRNA.
TABLE 1.
Serological status of chimpanzees prior to initiation of this study
| Chim- panzee no. | Previous infection (date, type) | Status regarding:
|
|||
|---|---|---|---|---|---|
| Serum RNA | T-cell pro- liferation assay | EIA 3.0 | Anti- E1E2 | ||
| X0186 | 1994, H plasma | Negative | Negative | Positive | Negative |
| 1552 | 1998, infectious RNA transcript | Negative | Negative | Positive | Negative |
| 1605 | Naïve | Negative | Negative | Negative | Negative |
RNA extractions.
Total RNA was prepared from 100 μl of serum or plasma or approximately 1 mm3 of liver biopsy by using TRIzol reagent (Life Technologies, Gaithersburg, Md.) according to the manufacturer's instructions. Twenty micrograms of glycogen (Boehringer Mannheim, Mannheim, Germany) was added at the precipitation step to facilitate the recovery of RNA. The RNA pellet was resuspended in 10 μl of RNasin-dithiothreitol water (0.2 U of RNasin/μl, 10 mM dithiothreitol) (Promega, Madison, Wis.) and stored at −80°C until use.
Nested PCR.
HCV RNA was analyzed by nested PCR with forward and reverse primers located in the 5′ noncoding region. The primers used were as follows: outer sense primer 47F1, GTGAGGAACTACTGTCTTCACG (nt 47 to 68); outer antisense primer 263R1, CACTACTCGGCTAGCAGTCTT (nt 263 to 243); inner sense primer 86F2, TGGCGTTAGTATGAGTGTCGTG (nt 86 to 107); and inner antisense primer 227R2, AATCTCCAGGCATTGAGCG (nt 227 to 209). The RT-PCR was carried out with 10 μl of RNA in a 50-μl reaction mixture by using a GeneAmp EZ rTth RNA PCR kit (Perkin-Elmer Applied Biosystems, Foster City, Calif.) according to the manufacturer's instructions. Reactions were run on a Perkin-Elmer GeneAmp 2400 PCR system machine. The reaction mixture contained 300 μM (each) dATP, dCTP, dGTP, and dTTP and primers 47F1 and 263R1. Cycle conditions consisted of a 30-min RT step at 60°C, 40 cycles of 15 s at 94°C and 30 s at 60°C, and then 7 min at 60°C. The nested reaction mixture, with Taq gold polymerase (Perkin-Elmer Applied Biosystems) used according to the manufacturer's instructions, contained 5 μl from the first reaction mixture, primers 86F2 and 227R2 at 300 nM, and dATP, dCTP, dGTP, and dTTP at 300 μM. Cycle conditions consisted of 10 min at 94°C, 40 cycles of 15 s at 94°C and 30 s at 60°C, and then 7 min at 60°C. Products were visualized on 1% agarose gels with ethidium bromide staining.
HCV RNA quantification.
RNA levels in plasma and serum samples were quantified by real-time PCR with the PRISM 7700 sequence detection system (Perkin-Elmer Applied Biosystems) as previously described (22).
T-cell proliferation assays.
Peripheral blood mononuclear cells (PBMCs) were examined for proliferative responses to specific HCV proteins. PBMCs (105 per well) were cultured in 200 μl of RPMI 1640 (GIBCO Laboratories, Grand Island, N.Y.) supplemented with l-glutamine (2 mM), penicillin (100 U/ml), streptomycin (100 μg/ml), HEPES (10 mM), and 10% heat-inactivated human AB serum in a 96-well flat-bottom plate (Costar, Cambridge, Mass.). Cells were stimulated with 1 μg of HCV proteins/ml in four replicate cultures per stimulation condition. Cultures were labeled with 1 μCi of [3H]thymidine (Amersham, Little Chalfont, England) on day 5 and harvested 16 h later. Responses were expressed as a stimulation index (SI), which was calculated as the incorporation of [3H]thymidine in stimulated cultures divided by the incorporation in unstimulated cultures.
Relative quantification of cytokine mRNA.
Relative mRNA levels for IFN-γ, TNF-α, and IL-4 in chimpanzee liver biopsies were determined by using predeveloped TaqMan assay reagent kits for human targets (Applied Biosystems). Primer-probe sets for the endogenous controls human β-actin and human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were also obtained from Applied Biosystems and used according to the manufacturer's instructions.
RT of RNA isolated from chimpanzee liver biopsies was carried out by using a First-Strand cDNA synthesis kit (Pharmacia) according to the manufacturer's instructions with 10 pg of random hexamers in a 33-μl reaction mixture. The volume of the RT reaction mixture was increased to 132 μl with distilled water. Five microliters was used to test for IFN-γ, TNF-α, IL-4, β-actin, and GAPDH in separate tubes. Relative mRNA quantification was calculated by the comparative cycle threshold (CT) method by performing the arithmetic operation
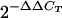 |
. This calculated the amount of target normalized to an endogenous reference (β-actin or GAPDH) and relative to a calibrator (a liver biopsy taken prior to challenge), as described in Perkin-Elmer Applied Biosystems user bulletin no. 2. In brief, ΔCT is the difference between the threshold cycles for target (cytokine) mRNA and endogenous reference (β-actin or GAPDH) mRNA. ΔΔCT is the difference between the ΔCT of the target mRNA postchallenge and the ΔCT of the target mRNA prechallenge.
Enzyme-linked immunosorbent assay (ELISA) for E1E2- or NS3-specific antibodies.
Antibodies to the HCV envelope E1E2 and NS3 were determined by using recombinant antigens derived from the Sindbis virus expression system as previously described (22).
RESULTS
Virological responses to infection.
Chimpanzees ChX0186, Ch1552, and Ch1605 were challenged with increasing amounts of RNA-positive HCV plasma until RNA positivity was detected in the serum by nested PCR. Following infection, serum RNA was analyzed both qualitatively and quantitatively by nested and real-time PCR, respectively. The sensitivity of the real-time PCR system with a specific fluorogenic probe was calculated to be approximately 400 RNA copies/ml of serum. The nested PCR system has a sensitivity of <40 RNA copies/ml; therefore, some serum samples could not be quantitated, even though they were shown to contain HCV RNA. However, in these cases, the serum RNA titer in the animal was known to be low (<400 copies/ml).
In this study, the challenge inoculum was plasma derived from a chimpanzee (Ch1536) infected with in vitro transcribed RNA. Although Ch1536 developed a classical response to infection, with increasing HCV RNA titers in the serum and elevated liver enzymes, the infectious qualities of the virus produced in this animal had not been determined. The RNA titer was calculated to be 4 × 105 RNA copies/ml by real-time PCR, but the relationship of this to actual infectious particles was also unknown. Therefore, the animals were challenged by reverse titration in order to assess the infectious titer of this plasma sample and determine whether the virus produced from the infectious RNA transcripts induced a normal infection in naïve and previously infected chimpanzees.
Ch1605.
Ch1605 was a naïve animal with no detectable serologic evidence of prior exposure to HCV. Viral RNA was not detected following challenge with plasma diluted to 10−6 or 10−5. However, Ch1605 became serum RNA positive 1 week after challenge with the 10−4 dilution of plasma from Ch1536. The clinical profile closely resembled those observed for Ch1535 and Ch1536 (15, 22), with rapidly increasing viral titers, elevated liver enzymes, and seroconversion to HCV-specific antibodies (Fig. 1). Much like those observed for Ch1535 and Ch1536, the viral RNA level began at about 104 RNA copies/ml at week 1 p.i. and reached a peak of 106 RNA copies/ml at week 7 p.i. Similarly, an increase in ALT levels correlated with a decrease in serum RNA. ALT levels reached a peak at week 9 p.i. and returned to normal by week 13 p.i. In contrast to those observed for Ch1535 and Ch1536, for which serum RNA levels remained around 104 copies/ml, the serum RNA titer for Ch1605 dropped below the level of detection for real-time PCR (<400 copies/ml), although it could still be detected intermittently by nested PCR (top of Fig. 1). These data suggested that Ch1605 was clearing the infection, which occurs in approximately 50% of chimpanzees. During further monitoring of this animal up to week 91 p.i., we detected only one positive serum sample at week 59 p.i. All serum and liver biopsies tested subsequently to this were negative for HCV RNA. In our studies, we considered a chimpanzee to have cleared an infection if three consecutive monthly liver biopsies and all serum samples obtained during this period (taken weekly) were negative for HCV RNA. Therefore, Ch1605 was classified as having cleared the infection by week 91 p.i.
FIG. 1.

Clinical response of Ch1605 to challenge with increasing doses of plasma from Ch1536. Arrowheads indicate the dates of challenge with increasing doses. Weeks postinoculation are counted as postchallenge with the 10−4 dilution of plasma. Results from nested PCR are indicated at the top of the graph. Vertical open bars represent negative PCR signals, and vertical filled bars represent positive PCRs. Serum positivity in Ortho EIA 3.0 is indicated by a horizontal shaded bar.
Ch1605 became infected with the 10−4 dilution of Ch1536 plasma, but no apparent infection occurred with the 10−5 dilution. Therefore, the infectious titer for the plasma derived from Ch1536 is approximately 104.5 CID50/ml.
Ch1552.
Ch1552 had been previously infected with in vitro transcribed RNA. This resulted in a classic productive infection, with rapidly increasing HCV RNA titers and elevated ALT levels (17) similar to those described above for Ch1605. Ch1552 cleared the infection and remained serum and liver biopsy negative for HCV RNA for 34 weeks, at which point the rechallenge experiments with the diluted Ch1536 plasma were started. The RNA used to infect Ch1552 was derived from the same cDNA clone used to transcribe the RNA inoculated into Ch1536. Therefore, the viruses produced in these two animals were as genetically similar as any two HCV isolates can be.
As observed for Ch1605, Ch1552 became serum RNA positive 1 week after challenge with the 10−4 diluted plasma. There were no indications of infection following challenge with the lower dilution, 10−5. However, in contrast to those of naïve animals, the clinical response of Ch1552 was substantially reduced in terms of viral titer, longevity of viremia, and ALT elevations (Fig. 2). Ch1552 was only briefly positive on three dates following challenge with the 10−4 diluted plasma, and only on one of those dates (week 2 post-10−4 dilution challenge) were there quantifiable levels of RNA (4 × 103 copies/ml). Slightly elevated ALT was also observed at this same sample date, increasing from a baseline of 17 mIU/liter to a value of 31 mIU/liter. Again, this is substantially lower than the ALT elevation normally observed after the infection of naïve animals. In the previous infection with in vitro transcribed RNA, Ch1552 had an ALT peak of 249 mIU/liter at week 10 p.i. and a viremic peak of 107 RNA copies/ml at week 9 p.i. (17). Four weeks after rechallenge with the 10−4 dilution of plasma, Ch1552 also received an inoculation of the 10−3 dilution of Ch1536 plasma, but there was no evidence of reinfection. These data suggest that Ch1552 had a preexisting immune response that allowed it to control infection.
FIG. 2.

Clinical response of Ch1552 to challenge with increasing doses of plasma from Ch1536. Arrowheads indicate the dates of challenge with increasing doses. Weeks postinoculation are counted as postchallenge with the 10−4 dilution of plasma. Results from nested PCR are indicated at the top of the graph. Vertical open bars represent negative PCR signals, and vertical filled bars represent positive PCRs. Serum positivity in Ortho EIA 3.0 is indicated by a horizontal shaded bar.
Following its recovery from infection, Ch1552 was moved from the Food and Drug Administration facility to the Coulston Foundation, Alamogordo, N.Mex. Three weeks after this move, Ch1552 became serum RNA positive. Ch1552 was again able to control the infection, as demonstrated by the occurrence of intermittent and low-level viremia, the fact that the RNA concentrations were not always high enough to be quantifiable, and clearance. This second bout of viremia led to some elevation of ALT levels, which were higher than those observed following challenge with 10−4 diluted plasma but lower than those observed in acute infections of naïve animals. Attempts to amplify the viral populations in the sera from these sample dates for sequence analysis were unsuccessful due to the low viral titers. Therefore, the possibility of escape mutations causing this reappearance of viremia could not be examined for this animal.
ChX0186.
Chimpanzee X0186 had been previously infected with H plasma in 1994 and recovered from infection. The animal was challenged with diluted plasma from Ch1536, starting with a 10−7 dilution. There was no indication of infection in this animal following either the 10−4 dilution or the 10−3 dilution. Two weeks after receiving the 10−2 dilution of plasma, ChX0186 became serum RNA positive (Fig. 3). Much like that observed for Ch1552, the infection in ChX0186 was significantly reduced and was characterized by intermittent viremia, RNA titers below the level of quantification, and minimal ALT elevations. These observations served as a further indication that previously infected animals have long-term memory immune responses to HCV that can control infection.
FIG. 3.

Clinical response of ChX0186 to challenge with increasing doses of plasma from Ch1536. Arrowheads indicate the dates of challenge with increasing doses. Weeks postinoculation are counted as postchallenge with the 10−4 dilution of plasma. Results from nested PCR are indicated at the top of the graph. Vertical open bars represent negative PCR signals, and vertical filled bars represent positive PCRs. Serum positivity in Ortho EIA 3.0 is indicated by a horizontal shaded bar.
Antibody responses.
Serum samples from all chimpanzees were tested for antibodies to HCV by using the commercial tests Ortho EIA 3.0 (Ortho-Clinical Diagnostics) and RIBA 2.0 (Chiron Corp., Emeryville, Calif.) and in-house ELISAs with the recombinant antigens NS3 and E1E2.
Ch1605 seroconverted to HCV-specific antibodies 9 weeks after challenge with plasma containing clonal HCV (Fig. 1). This coincided with the peak in ALT elevation. The resolved chimpanzees 1552 and X0186 were positive by EIA 3.0 before rechallenge; hence, any boosting of antibody levels following reinfection could not be determined with the qualitative commercial EIA. Analyses of the antibody profiles to individual antigens for all three chimpanzees with RIBA 2.0 and an in-house NS3-specific ELISA are shown in Fig. 4 (RIBA 2.0 contains antigens from the HCV core [c22], NS2 and NS3 [c33], and NS4 [5-1-1/c100]). Ch1605 (Fig. 4A, top panel) seroconverted by RIBA at week 9 p.i., at the same time as the EIA seroconversion. The strongest response was against c33, the NS3 antigen in the assay. A weaker response developed later against the 5-1-1/c100 peptides located in the NS4 region of the HCV polyprotein. Ch1552 had weak preexisting antibody to the same two RIBA antigens (Fig. 4B, top panel). Three weeks after challenge with the 10−4 dilution of plasma, antibody titers to these antigens rapidly increased to levels comparable to those seen for Ch1605. Chimpanzee X0186 also had preexisting antibodies to the NS3 and NS4 antigens (Fig. 4C, top panel), and these antibody levels were also boosted following infection with HCV. Testing of sera against recombinant NS3 antigen in an in-house ELISA showed similar increases in specific antibody following infection with HCV, although this decreased rapidly following clearance of the virus (Fig. 4, lower panels). No antibody to the HCV envelope was detected in any of the animals at any time prior to or following the HCV challenge (data not shown).
FIG. 4.

RIBA and NS3 ELISA results obtained for sera from Ch1605 (A), Ch1552 (B), and ChX0186 (C) following challenge with plasma from Ch1536. In the top panels, filled bars represent 5-1-1/c100 and open bars represent C33c; the filled bar at the extreme right in panel C represents C22. In the bottom panels, shaded bars represent NS3 (derived from Sindbis virus). OD, optical density.
Proliferative T-cell responses.
PBMCs from all chimpanzees were examined prior to challenge with plasma and then again during the acute phase of infection. PBMCs collected on consecutive dates were tested, and the strongest responses observed for each animal are shown in Fig. 5. No significant proliferative responses were observed in any of the animals prior to initiation of this study, even in animals that had been previously infected with HCV. Following infection with HCV, Ch1605 had significant proliferative responses at week 10 p.i. to the NS3 and NS4 antigens, reaching an SI of >15. This time point was after the peaks of viremia and ALT, which occurred at weeks 7 and 9 p.i., respectively. The strong T-cell response in Ch1605 may have contributed to the animal's eventual clearance of the HCV infection. The response in Ch1552 was lower than expected given the rapid clearance of the infection. Ch1552 had a detectable response only to the helicase portion of NS3 at 3 weeks p.i., and the magnitude of this response was reduced compared to that of Ch1605. By week 3 p.i., the viremia and ALT elevations in Ch1552 had peaked and it had already begun to clear the infection. In contrast to Ch1552, ChX0186 had a vigorous response to HCV antigens, particularly to the NS4 antigen (SI of almost 70). This sample date was 3 weeks after inoculation with the 10−2 plasma dilution and 12 weeks after challenge with the 10−4 dilution, which was infectious in Ch1605 and Ch1552 but did not lead to a detectable infection of ChX0186.
FIG. 5.

T-cell proliferation assay with PBMCs from Ch1605, Ch1552, and ChX0186 prechallenge (top panels) and postchallenge (lower panels) with infecting plasma. The cutoff for positive responses was an SI of 3, indicated by a dashed line. pos., positive; Hel., helicase portion of NS3.
Intrahepatic cytokine levels.
To examine the possible correlation between intrahepatic cytokine levels and virus clearance after rechallenge, we analyzed mRNA levels of IFN-γ, TNF-α, and IL-4 in snap-frozen liver biopsies from Ch1605 and Ch1552. No liver biopsies were available from ChX0186 to perform this assay. The levels of mRNA were determined by using real-time PCR and normalized relative to the GAPDH and β-actin housekeeping genes. Both endogenous controls yielded similar results. For clarity, the data shown are normalized to GAPDH and are expressed as values relative to the first prechallenge sample (Fig. 6). For Ch1605, we observed a brief 12-fold increase in intrahepatic IFN-γ that coincided with the peak in ALT at week 10 p.i. No significant elevations in TNF-α or IL-4 were observed for this animal (Fig. 6A). For Ch1552, a very different profile was observed. The increase in intrahepatic IFN-γ mRNA began earlier, at week 2 p.i., and persisted until at least week 6 p.i. An almost 30-fold increase over baseline was observed at week 4 p.i. In addition, an increase in TNF-α mRNA was observed for Ch1552, peaking at week 2 p.i. at approximately ninefold above baseline (Fig. 6B). No increase in IL-4 was detected. Only minimal ALT elevations above baseline were observed for Ch1552, but these coincided with the increase in cytokine levels observed at week 2 p.i.
FIG. 6.

Relative cytokine levels (measured in fold increase over baseline) observed in liver biopsies from Ch1605 (A) and Ch1552 (B).
Rechallenge of Ch1605.
Following clearance of the virus by Ch1605, we rechallenged this animal to determine whether the response profiles observed for Ch1552 and ChX0186 were reproducible in Ch1605 by using a higher challenge dose. This animal was rechallenged with 100 CID50 of Ch1536 plasma.
A response similar to those of Ch1552 and ChX0186 was observed for Ch1605 following rechallenge (Fig. 7). The animal became serum RNA positive at week 1 p.i. but had short, low-level viremia and reduced ALT elevations. Serum samples were quantifiable for viral RNA only at week 1 p.i., with a titer of 5 × 104 RNA copies/ml. ALT levels increased above normal at week 2 p.i. for only 1 week, to 70 mIU/liter. This contrasts with a peak of 176 mIU/liter during the primary infection and a total elevation time above normal of 9 weeks (Fig. 1). The intrahepatic cytokine pattern was also similar to that seen in Ch1552 following rechallenge. The peak in IFN-γ was ninefold above baseline and coincided with the ALT peak at week 2 p.i.; it returned to normal levels by week 9 p.i. (Fig. 7).
FIG. 7.

Clinical profile and intrahepatic cytokine levels of Ch1605 following rechallenge with 100 CID50 of Ch1536 plasma. Relative cytokine levels are measured in fold increase over baseline. Results from nested PCR are indicated at the top of the graph. Vertical open bars represent negative PCR signals, and vertical filled bars represent positive PCRs.
DISCUSSION
Studies of HCV-infected humans (20) and experimentally infected chimpanzees (8, 23) have suggested that natural HCV infection does not induce protective immunity at the humoral or cellular levels. Farci et al. (8) studied chimpanzees sequentially inoculated with different HCV strains. Each rechallenge with either a homologous or a heterologous strain resulted in the recurrence of viremia and often the reappearance of HCV-specific antibodies and changes in liver pathology. Prince et al. (23) made similar observations, finding no significant difference in the responses to homologous and heterologous challenges. The viral titers in the sera of the rechallenged chimpanzees were not determined in either of these studies. However, in most cases, the viremia was shorter following rechallenge. These observations have led to questions regarding the feasibility of a vaccine for this virus, as it appears that natural infection does not confer any protection from reinfection.
One of the major questions regarding reinfection is whether it could be due to the quasispecies nature of HCV isolates and therefore be the result of preexisting escape mutants. In our study, a virus stock was used for rechallenge that had no detectable sequence heterogeneity, greatly reducing the quasispecies problem one usually faces with natural isolates. Our observations correlate with those previously made for chimpanzees and suggest that HCV infection does not elicit a sterilizing immune response. However, it does induce a response that curtails the infection in terms of its duration, viral replication, and degree of hepatitis and leads to rapid clearance and recovery. This response appears to be cell mediated, as no antibodies to surface antigens were detected in these animals, and it is active at the primary site of HCV replication, the liver. Our observations are also consistent with those of recent reports examining the reinfection of chimpanzees (3, 31). Bassett et al. (3) examined PBMCs in rechallenged animals and found similarly rapid clearance that coincided with higher proliferative T-cell responses; intrahepatic responses were not examined. Weiner et al. (31) used intrahepatic inoculation of RNA transcribed from cDNA clones and found reduced infection levels in rechallenged animals. Control of infection correlated with HCV-specific T-cell responses in the PBMCs and liver, with a possible contribution of anti-E1E2 or -HVR1 antibodies, indicating that the role of HCV-specific antibodies in clearing virus in a rechallenged chimpanzee cannot be eliminated.
The previously infected animals in this study, ChX0186 and Ch1552, did not have antibody to the surface envelope proteins of HCV, as would be expected for most acute viral infections (24). Nor did the chimpanzees produce any detectable antibodies to these antigens upon reinfection. Therefore, there is presumably no first line of defense in the form of neutralizing antibody to prevent reinfection. Virus replication, once initiated, appears to be contained through cellular responses directed towards the liver, responses that would first require some viral replication for the activation of memory cells.
Inoculation of the naïve chimpanzee, Ch1605, with 1 ml of the 10−4 dilution of serum from Ch1536 elicited a classic infection, with a rapid onset of viremia and an exponential increase in viral titer over several weeks. This viral replication resulted in acute hepatitis, as determined by elevated liver enzymes and seroconversion to HCV-specific antibody at week 9 p.i. (Fig. 1). In this animal, the immune responses led to a decrease in serum RNA below the level of quantitation by week 12 p.i. Ch1605 remained intermittently serum RNA positive at a low level for almost a year but eventually cleared the infection.
The data obtained from Ch1605 indicate that the virus derived from the infectious RNA transcripts used to inoculate Ch1536 leads to a normal infection in naïve animals. This provides further evidence that the sequence encoded by the cDNA clone constructed by Kolykhalov et al. (15) is a faithful representation of an infectious HCV genome. Since Ch1605 was not infected with the 10−5 dilution challenge but developed a normal infection following the 10−4 dilution challenge, the infectious titer of this plasma sample can be estimated to be 104.5 CID50/ml (3.16 × 104 CID50/ml). This infectious titer compares with an RNA titer of 4 × 105 copies/ml. Therefore, the particle-to-infectivity ratio in this system was approximately 12:1. This is similar to that observed for the original H77 strain, which had an infectious titer in chimpanzees of 106.5 (10) and an RNA copy number of ∼108/ml. This difference could reflect the fact that not all virus particles in serum can result in infection upon inoculation into a suitable host.
In the two previously infected animals, Ch1552 and ChX0186, a clinical profile very different from that seen in Ch1605 was observed upon challenge with virus. Although both animals became infected, the infection resulted in very low levels of serum RNA, and ALT elevations were significantly diminished (Fig. 2 and 3). However, there was a 2-log difference in the amount of virus required to infect these animals. Ch1552 became RNA positive following challenge with the 10−4 dilution of Ch1536 plasma, the same as that required to infect Ch1605. Challenge of ChX0186 with either this dilution or the 10−3 dilution elicited no apparent infection, as determined by nested PCR of serum or liver biopsies. In addition, there was no increase in anti-HCV antibody levels until challenge with the 10−2 dilution (Fig. 4C), which led to a productive infection. The reason for this difference in infectivity is unclear. ChX0186 had been previously infected with the H strain of HCV, whereas Ch1552 had received infectious RNA transcribed from the same clone as that used for the infection of Ch1536. Therefore, Ch1552 was reinfected with a virtually identical virus. ChX0186, having received the H strain isolate, was effectively challenged with only a single variant from the full repertoire of viruses present in its original infection. It may therefore have had a more diverse immune response and may have been protected from reinfection following low-dose challenge with the 10−4 (3.2 CID50) and 10−3 (32 CID50) dilutions. The animal had no detectable anti-envelope antibody prior to challenge with the diluted plasma or after infection, although the titers may have been below the detection levels of the assays used. However, these data suggest that the protection and the rapid clearance observed for both of the previously infected chimpanzees were due to cellular responses.
T-cell proliferation assays on peripheral blood do not seem to entirely support this conclusion, as Ch1552 had lower T-cell responses than Ch1605 (Fig. 5). However, analysis of liver biopsies from Ch1605 and Ch1552 for cytokine mRNA levels indicated a robust response in the liver, suggesting that responses in the peripheral blood may not be good indicators of the mechanisms controlling the viral infection at the site of replication. In the naïve Ch1605, intrahepatic IFN-γ increased to approximately 12-fold above baseline during the acute phase, but no changes in TNF-α were observed. This increase coincided with the ALT peak, occurring 10 weeks after challenge. An increase in IFN-γ was also seen in Ch1552. However, this occurred within 2 weeks of the challenge and was more pronounced, reaching levels almost 30-fold above baseline (Fig. 6B). An increase in TNF-α levels was also observed within the first 2 weeks of infection. No increases in IL-4 were observed for either animal. Thus, Ch1552 exhibited a more rapid and robust response than that seen in the primary infection of Ch1605. This response appeared to control replication and the spread of the virus in the liver, preventing liver damage and hepatitis, as indicated by only minimal and transient increases in liver enzymes. Rechallenge of Ch1605 resulted in a clinical profile similar to those seen for Ch1552 and ChX0186, with reduced viremia and ALT elevations (Fig. 7). The level of intrahepatic IFN-γ in this animal (eightfold above baseline) was not significantly greater than that seen in the primary infection, but its increase was more rapid, occurring at 2 weeks p.i.
It is widely believed that HCV is not a cytopathic virus and that hepatitis observed during infection is mediated by the host immune response. If this is true, the reduced pathology observed by us for previously infected animals could be due to the primed immune response, limiting the viral replication to a few infected cells and possibly destroying those cells before the exponential increase in viral load that is seen in naïve animals could take place (Fig. 1). As the infection was restricted to only a few cells, manifestations of hepatitis such as ALT elevations were also limited to normal or near-normal levels. In the naïve animal, Ch1605, several weeks of viral replication were necessary to induce a primary immune response. At this stage, many liver cells were infected. Therefore, more cells were damaged by the immune system, leading to control of the infection but also to high ALT levels.
The cells responsible for the intrahepatic cytokine response remain to be determined and will require analysis and characterization of a specific T-cell effector function in the liver and T-cell kinetics in the blood. Therefore, it is not yet possible to determine the specific correlates of immunity in these animals. These studies are currently being addressed and will be reported elsewhere. However, the cytokine data do indicate that the immune system in chimpanzees can be primed to elicit an immune response upon infection that controls virus replication and prevents chronic infection. In addition, this memory response was long lasting in ChX0186. Four years had elapsed between the first infection and the rechallenge in this study. In other reports, animals had not been exposed to HCV for up to 16 years but still controlled infection (3). This has promising implications for vaccine development against HCV. Thus far, vaccines to HCV have concentrated on the generation of antibodies to the surface glycoproteins (4). This approach has had limited success, in that protection could be achieved only against low-dose challenge and only when antibody titers to the envelope were high. A vaccine that primes the system to mimic the responses observed for ChX0186 and Ch1552 upon primary challenge with HCV would be an enormous advance. Although low-level, short-lived infection may still occur, it is the development of chronic infections leading to chronic liver disease and cirrhosis that needs to be prevented. The concept of T-cell vaccines is not new and has been suggested for HCV as well as other pathogens. The diversity of the envelope protein among HCV isolates has frequently led to the suggestion that the inclusion of more conserved T-cell epitopes will be a necessary requirement for an effective vaccine. The data presented in this study support this notion and extend the theory for a T-cell vaccine that can at least protect against chronic HCV infection. The coupling of a vaccine designed to induce strong T-cell responses with one that also elicits neutralizing antibody may provide the ideal means of protection from HCV infection.
Acknowledgments
We thank Estella Jones and Ray Olsen for expert care and handling of chimpanzees and samples within the FDA facility. We also thank Kris Murthy, Southwest Foundation, San Antonio, Tex., for his assistance with some of these animal studies.
This work was supported in part by NIH grant CA 85883.
REFERENCES
- 1.Alter, M. J., H. S. Margolis, K. Krawczynski, F. N. Judson, A. Mares, W. J. Alexander, P. Y. Hu, J. K. Miller, M. A. Gerber, R. E. Sampliner, E. L. Meeks, and M. J. Beach. 1992. The natural history of community-acquired hepatitis C in the United States. N. Engl. J. Med. 327:1899-1905. [DOI] [PubMed] [Google Scholar]
- 2.Bassett, S. E., K. M. Brasky, and R. E. Lanford. 1998. Analysis of hepatitis C virus-inoculated chimpanzees reveals unexpected clinical profiles. J. Virol. 72:2589-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bassett, S. E., B. Guerra, K. Brasky, E. Miskovsky, M. Houghton, G. R. Klimpel, and R. E. Lanford. 2001. Protective immune response to hepatitis C virus in chimpanzees rechallenged following clearance of primary infection. Hepatology 33:1479-1487. [DOI] [PubMed] [Google Scholar]
- 4.Choo, Q. L., G. Kuo, R. Ralston, A. Weiner, D. Chien, G. Van Nest, J. Han, K. Berger, K. Thudium, C. Kuo, J. Kansopon, J. McFarland, A. Tabrizi, K. Ching, B. Moss, L. B. Cummins, M. Houghton, and E. Muchmore. 1994. Vaccination of chimpanzees against infection by the hepatitis C virus. Proc. Natl. Acad. Sci. USA 91:1294-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choo, Q. L., G. Kuo, A. J. Weiner, L. R. Overby, D. W. Bradley, and M. Houghton. 1989. Isolation of a cDNA clone derived from a blood-born non-A, non-B viral hepatitis genome. Science 244:359-362. [DOI] [PubMed] [Google Scholar]
- 6.Cooper, S., A. L. Erickson, E. J. Adams, J. Kansopon, A. J. Weiner, D. Y. Chien, M. Houghton, P. Parham, and C. M. Walker. 1999. Analysis of a successful immune response against hepatitis C virus. Immunity 10:439-449. [DOI] [PubMed] [Google Scholar]
- 7.Diepolder, H. M., J. T. Gerlach, R. Zachoval, R. M. Hoffmann, M. C. Jung, E. A. Wierenga, S. Scholtz, T. Santantonio, M. Houghton, S. Southwood, A. Sette, and G. R. Pape. 1997. Immunodominant CD4+ T-cell epitope within nonstructural protein 3 in acute hepatitis C virus infection. J. Virol. 71:6011-6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farci, P., H. J. Alter, S. Govindarajan, D. C. Wong, R. Engle, R. Lesniewski, I. K. Mushahwar, S. M. Desai, R. H. Miller, N. Ogata, and R. H. Purcell. 1992. Lack of protective immunity against reinfection with hepatitis C virus. Science 258:135-140. [DOI] [PubMed] [Google Scholar]
- 9.Farci, P., H. J. Alter, D. C. Wong, R. H. Miller, S. Govindarajan, R. Engle, M. Shapiro, and R. H. Purcell. 1994. Prevention of hepatitis C virus infection in chimpanzees after antibody-mediated in vitro neutralization. Proc. Natl. Acad. Sci. USA 91:7792-7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feinstone, S. M., H. J. Alter, H. P. Dienes, Y. Shimizu, H. Popper, D. Blackmore, D. Sly, W. T. London, and R. H. Purcell. 1981. Non-A, non-B hepatitis in chimpanzees and marmosets. J. Infect. Dis. 144:588-598. [DOI] [PubMed] [Google Scholar]
- 11.Grakoui, A., C. Wychowski, C. Lin, S. M. Feinstone, and C. M. Rice. 1993. Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 67:1385-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grüner, N. H., T. J. Gerlach, M. C. Jung, H. M. Diepolder, C. A. Schirren, W. W. Schraut, R. Hoffmann, R. Zachoval, T. Santantonio, M. Cucchiarini, A. Cerny, and G. R. Pape. 2000. Association of hepatitis C virus-specific CD8+ T cells with viral clearance in acute hepatitis C. J. Infect. Dis. 181:1528-1536. [DOI] [PubMed] [Google Scholar]
- 13.Han, J. H., V. Shyamala, K. H. Richman, M. J. Brauer, B. Irvine, M. S. Urdea, P. Tekamp-Olson, G. Kuo, Q. L. Choo, and M. Houghton. 1991. Characterization of the terminal regions of hepatitis C viral RNA: identification of conserved sequences in the 5′ untranslated region and poly(A) tails at the 3′ end. Proc. Natl. Acad. Sci. USA 88:1711-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hijikata, M., N. Kato, Y. Ootsuyama, M. Nakagawa, and K. Shimotohno. 1991. Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc. Natl. Acad. Sci. USA 88:5547-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolykhalov, A. A., E. V. Agapov, K. Blight, K. Mihalik, S. M. Feinstone, and C. M. Rice. 1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277:570-574. [DOI] [PubMed] [Google Scholar]
- 16.Kolykhalov, A. A., S. M. Feinstone, and C. M. Rice. 1996. Identification of a highly conserved sequence element at the 3′ terminus of hepatitis C virus genome RNA. J. Virol. 70:3363-3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolykhalov, A. A., K. Mihalik, S. M. Feinstone, and C. M. Rice. 2000. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 74:2046-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krawczynski, K., M. J. Alter, D. L. Tankersley, M. Beach, B. H. Robertson, S. Lambert, G. Kuo, J. E. Spelbring, E. Meeks, S. Sinha, and D. A. Carson. 1996. Effect of immune globulin on the prevention of experimental hepatitis C virus infection. J. Infect. Dis. 173:822-828. [DOI] [PubMed] [Google Scholar]
- 19.Kumar, U., J. Brown, J. Monjardino, and H. C. Thomas. 1993. Sequence variation in the large envelope glycoprotein (E2/NS1) of hepatitis C virus during chronic infection. J. Infect. Dis. 167:726-730. [DOI] [PubMed] [Google Scholar]
- 20.Lai, M. E., A. P. Mazzoleni, F. Argiolu, S. De Virgilis, A. Balestrieri, R. H. Purcell, A. Cao, and P. Farci. 1994. Hepatitis C virus in multiple episodes of acute hepatitis in polytransfused thalassaemic children. Lancet 343:388-390. [DOI] [PubMed] [Google Scholar]
- 21.Lechner, F., D. K. Wong, P. R. Dunbar, R. Chapman, R. T. Chung, P. Dohrenwend, G. Robbins, R. Phillips, P. Klenerman, and B. D. Walker. 2000. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 191:1499-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Major, M. E., K. Mihalik, J. Fernandez, J. Seidman, D. Kleiner, A. A. Kolykhalov, C. M. Rice, and S. M. Feinstone. 1999. Long-term follow-up of chimpanzees inoculated with the first infectious clone for hepatitis C virus. J. Virol. 73:3317-3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prince, A. M., B. Brotman, T. Huima, D. Pascual, M. Jaffery, and G. Inchauspe. 1992. Immunity in hepatitis C infection. J. Infect. Dis. 165:438-443. [DOI] [PubMed] [Google Scholar]
- 24.Prince, A. M., B. Brotman, D. H. Lee, L. Ren, B. S. Moore, and J. W. Scheffel. 1999. Significance of the anti-E2 response in self-limited and chronic hepatitis C virus infections in chimpanzees and in humans. J. Infect. Dis. 180: 987-991. [DOI] [PubMed] [Google Scholar]
- 25.Robertson, B., G. Myers, C. Howard, T. Brettin, J. Bukh, B. Gaschen, T. Gojobori, G. Maertens, M. Mizokami, O. Nainan, S. Netesov, K. Nishioka, T. Shin, P. Simmonds, D. Smith, L. Stuyver, and A. Weiner. 1998. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. Arch. Virol. 143:2493-2503. [DOI] [PubMed] [Google Scholar]
- 26.Saito, I., T. Miyamura, A. Ohbayashi, H. Harada, T. Katayama, S. Kikuchi, Y. Watanabe, S. Koi, M. Onji, Y. Ohta, Q. L. Choo, M. Houghton, and G. Kuo. 1990. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 87:6547-6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takaki, A., M. Wiese, G. Maertens, E. Depla, U. Seifert, A. Liebetrau, J. L. Miller, M. P. Manns, and B. Rehermann. 2000. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat. Med. 6:578-582. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka, T., N. Kato, M. J. Cho, K. Sugiyama, and K. Shimotohno. 1996. Structure of the 3′ terminus of the hepatitis C virus genome. J. Virol. 70:3307-3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walker, C. M. 1997. Comparative features of hepatitis C virus infection in humans and chimpanzees. Springer Semin. Immunopathol. 19:85-98. [DOI] [PubMed] [Google Scholar]
- 30.Weiner, A., A. L. Erickson, J. Kansopon, K. Crawford, E. Muchmore, A. L. Hughes, M. Houghton, and C. M. Walker. 1995. Persistent hepatitis C virus infection in a chimpanzee is associated with emergence of a cytotoxic T lymphocyte escape variant. Proc. Natl. Acad. Sci. USA 92:2755-2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiner, A. J., X. Paliard, M. J. Selby, A. Medina-Selby, D. Coit, S. Nguyen, J. Kansopon, C. L. Arian, P. Ng, J. Tucker, C. T. Lee, N. K. Polakos, J. Han, S. Wong, H. H. Lu, S. Rosenberg, K. M. Brasky, D. Chien, G. Kuo, and M. Houghton. 2001. Intrahepatic genetic inoculation of hepatitis C virus RNA confers cross-protective immunity. J. Virol. 75:7142-7148. [DOI] [PMC free article] [PubMed] [Google Scholar]