

Abstract
Rheumatoid arthritis (RA) is a common autoimmune disease with a complex genetic etiology. Herein we identify a single-nucleotide polymorphism (SNP) in the promoter region of FcRH3, a member of the Fc receptor homolog family, that is associated with RA susceptibility (OR=2.15, P=0.00000085). This polymorphism alters the binding affinity of nuclear factor-κB and regulates FcRH3 expression. High FcRH3 expression on B-cells and augmented autoantibody production were observed in individuals with the disease-susceptible genotype. Associations were also found between the SNP and susceptibility to autoimmune thyroid disease and systemic lupus erythematosus. FcRH3 may thus play a pivotal role in autoimmunity.
Rheumatoid arthritis represents one of the most common autoimmune diseases, and is characterized by inflammation of synovial tissue and joint destruction. Although the disease is believed to result from a combination of genetic and environmental factors, the complete etiology of RA has not yet been clarified1. While specific haplotypes of human leukocyte antigen (HLA)-DRB1, usually referred to as shared-epitope (SE) sequences2, have been repeatedly reported as conferring RA-susceptibility3,4, other genetic components are also involved in the pathogenesis of RA5. This combination of HLA haplotypes and non-HLA genes accounting for disease susceptibility is also seen in other autoimmune diseases6–8. In autoimmune thyroid disease (AITD), for instance, studies have consistently shown that the HLA-DR3 haplotype is associated with disease risk, in addition to a functional haplotype of a non-HLA gene, CTLA4, that has recently been associated with AITD susceptibility9.
Identification of non-HLA genes associated with RA susceptibility and other autoimmunities seems difficult, due to the low relative risk of disease resulting from these non-HLA genes compared with the strong relative risk from disease-associated HLA haplotypes. In a search for non-HLA determinants of disease susceptibility, whole genome studies have been conducted for both human autoimmune diseases and experimental animal models. These studies have revealed non-random clustering of susceptibility loci for clinically distinct diseases8,10. This overlapping of susceptibility loci for multiple autoimmunities suggests the existence of common susceptibility genes in those regions. Intense studies of loci-clustering regions has revealed genes commonly associated with multiple autoimmune diseases, such as CTLA4 on 2q33 (ref. 9), SLC22A4/A5 on 5q31 (ref.11) and PTPN22 on 1p13 (ref. 12).
Cytoband 1q21-23 is one of the regions implicated in susceptibility to multiple autoimmune diseases. The Fcγ receptor (FcγR) II/III genes are located in 1q23 and a new family of genes, Fc receptor homologs (FcRHs)13,14 (also known as IRTAs15,16 or SPAPs17) clusters nearby in 1q21 (Fig. 1a). FcRHs have high structural homology with classical FcγRs, although ligands and function remain unclear. These receptors are strong candidates for involvement in autoimmunity, as they are believed to play important roles in the pathogenesis of RA and other autoimmune diseases18. Region 1q23 represents a candidate locus for susceptibility to systemic lupus erythematosus (SLE), and variants in the classical FcγR II/III genes would partially account for disease susceptibility 6,19. The FcRH gene cluster in 1q21 is also reportedly associated with SLE (Gibson, AW. et al. American College of Rheumatology 66th Annual Scientific Meeting, 2003). Region 1q21 represents a candidate locus for psoriasis (PSORS4 (ref. 7,20)) and multiple sclerosis21. The mouse syntenic region of human 1q21, on chromosome 3, also displays susceptibility loci for multiple autoimmune disease models8, including collagen-induced arthritis (Mcia2 (ref. 22), Eae3 (ref. 23), Tmevd2 (ref. 24), Idd10 and Idd17 (ref. 25)). Although 1q21-23 is a strong candidate region for RA susceptible genes, as above mentioned, the association of classical FcγRs with disease susceptibility remains controvertial26,27. The present study focused on the 1q21-23 region to identify RA-associated genes in Japanese subjects using linkage disequilibrium (LD) mapping.
Figure 1.
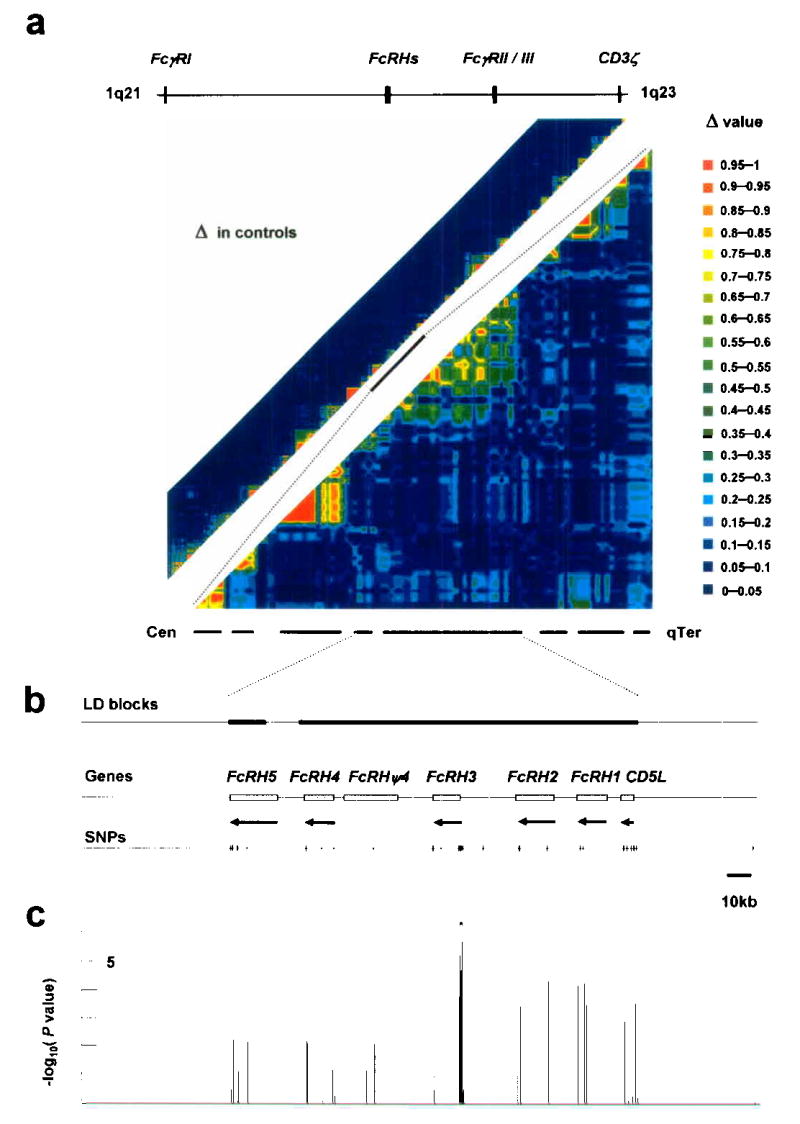
LD and association of the FcRH gene cluster. (a) Pairwise LD between SNPs, as measured by Δ in 658 controls. The 16-Mb region in 1q21-23 (upper left) and the 2-Mb region around the FcRH gene cluster (lower right) were evaluated. (b) Location of LD blocks, genes and 41 SNPs in the FcRH gene cluster. (c) Case-control association test with 41 SNPs in the FcRH gene cluster using 830 patients and 658 controls. *Peak association.
RESULTS
Case-control study by SNP-based LD-mapping in 1q21-23
To evaluate the extent of association, we analyzed linkage disequilibrium (LD) with SNPs distributed in a 16-Mb region on 1q21-23, including the FcRH gene cluster and the classical FcγRs (Fig. 1a). A total of 658 control subjects were genotyped for 742 SNPs from the JSNP database, and 491 SNPs were selected for evaluation of LD based on the following criteria: allele frequency >0.1; successful genotyping rate >0.95; and P>0.01 with Hardy-Weinberg Equilibrium (HWE) testing. The pairwise LD index Δ28 was calculated for each pair of SNPs, identifying 110 LD blocks11 at a threshold of Δ >0.5 (Fig. 1a).
For association testing, the Japanese set of 830 cases and 658 controls used for LD block evaluation was examined. Initially, 94 RA cases were genotyped for 491 SNPs and their allele frequencies were preliminarily compared with 658 control subjects. A total of 9 SNPs were identified as displaying allele frequencies differing by >0.1 between 658 controls and 94 cases with P<0.01. The remaining cases were genotyped for these 9 SNPs, and allele frequencies were tested for case-control association. The smallest P value was identified between an intronic SNP in the FcRH3 gene and RA (fcrh3_6, P=1.8 ×10−5; association was statistically significant in both RA subgroups (94 and 736)). This SNP was located in an LD block containing 4 of the 5 FcRH genes, with the fifth in the adjacent block. We thus used these 2 LD blocks to further evaluate the origin of this association (Fig. 1b), although our results do not exclude the presence of RA and/or other autoimmunity-associated variants in other LD blocks in 1q21-23.
In addition to the 25 SNPs among 491 used SNPs for LD block evaluation in the 2 LD blocks, 16 additional SNPs were identified in exons and 5′- and 3′-flanking regions of 5 FcRH genes and 1 pseudogene (FcRHψ4 or LOC343265) by searching the public database and sequencing genomic DNA from Japanese RA-patients. These additional 16 SNPs were genotyped for the identical case and control samples (830 cases, 658 controls) to increase density of variants in the targeted region. A peak of association was observed in a short segment consisting of 4 SNPs in FcRH3 (P<1.0 ×10−4) (Fig. 1c, Supplementary Table 1 online). These comprised fcrh3_3, fcrh3_4, fcrh3_5 and fcrh3_6, located at nt −169, −110, +358 (5′-untranslated region of exon 2) and +1381 (intron 3; 204 and 859bp from the 3′- and 5′-end of the flanking exons) relative to the transcription initiation site, respectively.
The smallest P value without correction was observed in recessive-trait genotype comparison of fcrh3_3 in FcRH3 (P=8.5 ×10−7; odds ratio, 2.15; 95% confidence interval, 1.58–2.93) (Table 1). This P value was still significant when the most conservative Bonferroni correction was applied (comparisons for 507 SNPs; Pcorr=0.00043). The 4 strongly associated SNPs were in LD with each other and 3 common haplotypes were inferred (Table 2; Fcrh3_3, fcrh3_5 and fcrh3_6 showed strong LD with each other, with Δ >0.99, whereas fcrh3_4 showed relatively weak LD with the other 3 SNPs (mean Δ = 0.68)).
Table 1.
Case-control analysis of FcRH3
| SNPsa | Allele | Allele1 frequency | Genotype 11 versus 12+22 | ||||
|---|---|---|---|---|---|---|---|
| ID | Location | 1/2 | Patients | Controls | OR (95% CI) | χ2 | P |
| fcrh3_3 | −169 | C/T | 0.42 | 0.35 | 2.15 (1.58–2.93) | 24.3 | 0.00000085 |
| fcrh3_4 | −110 | A/G | 0.25 | 0.18 | 3.01 (1.71–5.29) | 16.1 | 0.000060 |
| fcrh3_5 | Exon2 | C/G | 0.42 | 0.35 | 2.05 (1.51–2.78) | 21.6 | 0.0000033 |
| fcrh3_6 | Intron3 | A/G | 0.42 | 0.34 | 2.02 (1.49–2.75) | 20.8 | 0.0000052 |
SNPs with P<0.0001 in allele frequency comparison test
Table 2.
Haplotype structure and frequency in FcRH3
| Haplotype frequency
|
|||
|---|---|---|---|
| Haplotypea | fcrh3_3/4/5/6 | Patients | Controls |
| Haplotype1 | TGGG | 0.58 | 0.65 |
| Haplotype2 | CACA | 0.25 | 0.19 |
| Haplotype3 | CGCA | 0.17 | 0.14 |
Haplotypes with frequency >0.01
To identify causal variants in this segment based on genotype data, a forward stepwise-regression procedure was performed with cut-off P value to proceed next step being 0.01 (ref. 29). No SNP in FcRH genes other than FcRH3 improved the model. None of the 4 SNPs in FcRH3 were preferred over the others in these data (data not shown), which implicated that one of the SNPs in FcRH3may cause the disease, but there still remained the possibility that variants in other genes were truly associated with the disease.
To validate the case-control association test, samples (830 cases, 658 controls) were evaluated for the impact of population stratification on the case-control study. We selected 2069 SNPs, each of which was identified as a tagging SNP30 in 2069 distinct LD segments that were previously identified by genotyping 74,842 SNPs distributed in autosomal chromosomes31. Analysis of population structure32, and the χ2 sum33, were used for the evaluation of stratification. No significant evidence of population stratification was detected (Supplementary Fig. 1 online), suggesting no or negligible stratification of our samples and supporting the validity of the case-control association results by removing this confounder from further consideration.
Regulatory effect of SNP -169 C/T on FcRH3 expression
Since none of the 4 SNPs in FcRH3 (fcrh3_3, fcrh3_4, fcrh3_5, and fcrh3_6) produces amino-acid substitutions, potential effects of the SNPs on transcription factor binding were assessed using TRANSFAC software. Nuclear factor-κB (NF-κB) was predicted to bind the sequence containing RA-susceptible allele fcrh3_3 (-169C) with a high score (core match 1.000, matrix match 0.957) and substitution to a non-susceptible allele T decreasing the score of NF-κB binding substantially (core match 0.760, matrix match 0.824). The other 3 SNPs were not predicted to bind to any transcriptional factor with high score and nucleotide substitution was not predicted to affect binding at any regulatory factor. We therefore focused on the 5′-flanking region of fcrh3_3 to explore the regulatory effects on expression of FcRH3.
Reporter gene analysis was performed using the genomic sequence of FcRH3 from nt −523 to +203. Constructs were made corresponding to the 3 haplotypes using SNPs at nt -169 (C/T; fcrh3_3) and -110 (G/A; fcrh3_4) (Fig. 2a). These constructs were transfected into Raji cells, a Burkitt’s lymphoma cell line that expresses FcRH3 (ref. 13) and is derived from germinal center B-cells. Luciferase activity was significantly higher in cells transfected with -169C/-110G or -169C/-110A than in cells transfected with -169T/-110G. This suggests that -169C/T is critical for regulation of FcRH3 expression. To clarify, we cloned single or 4 tandem copies of 30-bp oligonucleotides surrounding -169T/C and control oligonucleotides into a vector with the SV40 promoter. Cells transfected with a single copy of the C allele produced significantly greater luciferase activity than in cells transfected with a single copy of the T allele. More convincingly, transfection with 4 tandem copies of the C allele enhanced luciferase activity 20-fold over those cells transfected with 4 tandem copies of the T allele (Fig. 2a).
Figure 2.
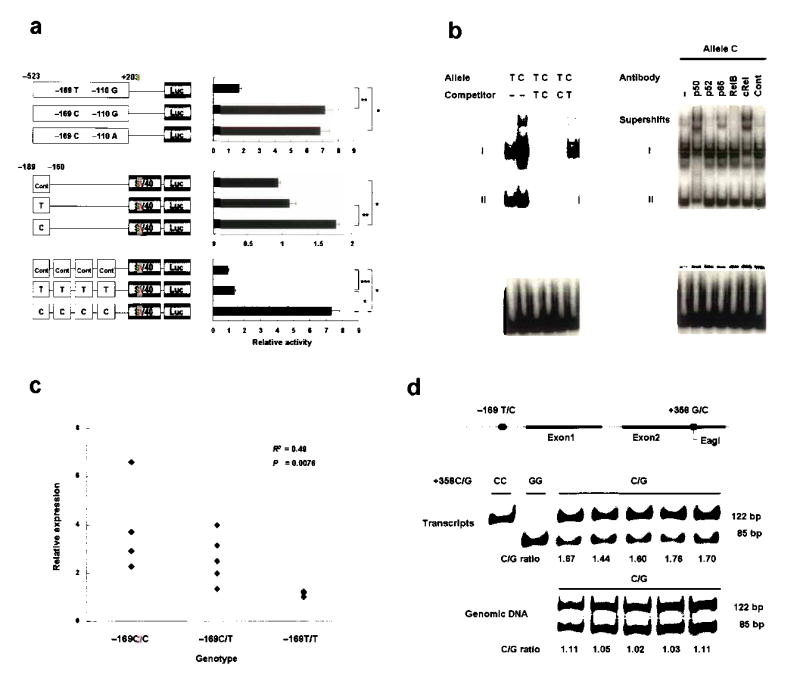
Correlation of FcRH3 expression with allele and genotype. (a) Promoter activity of haplotypes in FcRH3 (top) and enhancing activity of the 30-bp promoter region around -169 C/T (middle and bottom), as evaluated by luciferase assay. Data represent mean ±SEM. Representative data from three experiments performed in quadruplicate. *P<0.0001, **P<0.001, and ***P<0.01 by Student’s t-test. (b) Binding affinity of nuclear factors to the 30-bp promoter region around -169C/T, evaluated by EMSA. Allelic difference and competition experiment (left) and supershift experiment using antibodies for NF-κB components (right). (c) Expression of FcRH3 measured by quantitative Taqman PCR of RNA purified from CD19-positive B-cells obtained from 13 healthy volunteers (C/C, n=4; C/T, n=5; T/T, n=4). (d) Allele-specific transcript quantification (ASTQ). FcRH3 transcripts in B cells and genomic DNA from individuals (n=5) with heterozygous genotype (−169C+358C/−169T+358G) were amplified and quantified using an Eag I restriction fragment length polymorphism located at position +358. The 122-bp and 85-bp bands represent transcripts of the +358C allele and +358G allele, respectively. Transcripts from homozygous individuals (+358C/C and +358G/G) are shown as controls for digestion.
To elucidate specific nuclear factors that bind the disease-susceptible allele, we analyzed the sequence around -169C/T. These sequences were predicted by TRANSFAC software to display binding affinity for nuclear factor (NF)-κB, which regulates a wide variety of genes in the immune system. The disease-susceptible sequence GGGAAGTCCC (underlined nucleotide represents SNP at -169) displayed higher matrix similarity to the consensus NF-κB binding motif than the non-susceptible sequence GGGAAGTCCT. Electrophoretic mobility shift assay (EMSA) was then performed to examine whether differences between the susceptible -169C allele and the non-susceptible -169T allele affected binding of nuclear proteins from Raji cells. The 30-bp labeled oligonucleotides used in the luciferase assay were used again in this study. These sequences contain the predicted NF-κB binding site. Two major bands, I and II, were observed in the presence of nuclear extracts, and intensity of band I was higher for the susceptible -169C allele than for the non-susceptible -169T allele (Fig. 2b). Competition assays with unlabelled oligonucleotides revealed that these complexes were specific for the probes. In addition, competition assays with unlabelled probes of the C allele for T and the T allele for C demonstrated that the C allele was better able to compete for binding, a result consistent with the higher binding affinity exhibited by labeled C allele probes alone. A supershift experiment was also performed with antibodies specific for NF-κB components (p50, p52, p65, RelB, cRel). Supershifts were observed in some lanes with specific antibodies for p50, p65 and cRel (Fig. 2b). Among these, only anti-p50 antibody shifted band II, suggesting the presence of a p50-p50 homodimer. Band I had the highest intensity and a significant allelic difference, and was supershifted by anti-p50, anti-p65 and anti-cRel antibodies. Although these findings indicate that band I comprises a mixture of heterodimers, the greater shifts caused by anti-p50 and anti-cRel antibodies suggest that the main component is a p50-cRel heterodimer.
The two in vitro assays indicated the potent transcriptional activity of the disease-susceptible haplotype regulated by NF-κB, suggesting expression of FcRH3 from a chromosome with the disease-susceptible -169C allele be more than from a chromosome with non-susceptible -169T allele. To extend these findings, we quantified expression of FcRH3 in peripheral blood B-cells from healthy donors using quantitative Taqman methods, and analyzed the effect of the number of susceptible copies on the transcript level by regression model. Regression analysis revealed a significant positive correlation between number of susceptible chromosomes and transcription level (R2=0.49, P=0.0076) (Fig. 2c).
Allele-specific transcript quantification (ASTQ)9,34 was also performed to confirm the effect of the SNP on transcription.. Using an Eag I restriction fragment length polymorphism (RFLP) located at position +358 in exon 2 of FcRH3 (fcrh3_5, +358C/G), the relative contribution of each haplotype to transcript production in heterozygous individuals could be measured (Fig. 2d). The transcripts of 5 individuals with the -169C+358C/-169T+358G genotype were evaluated, and mean ratio (susceptible vs. non-susceptible haplotype) was 1.63, significantly higher than that of DNA amplicons (ratio=1.06, P<1 ×10−5) from the same individuals. (The quantity of template DNA from the 2 haplotypes was equal.) These results show that the expression of FcRH3 is higher in individuals with the disease-susceptible haplotype, and suggest that higher expression of FcRH3 is a potential cause and component of the pathological mechanism(s) leading to RA.
Expression of FcRH3 mRNA
FcRH3 expression in multiple tissues was then quantified using TaqMan methods. Expression of FcRH3 transcripts was high in the spleen and tonsils (Fig. 3a), which are secondary lymphoid organs. Lower expression was observed in thymus and bone marrow. Analysis of human blood fractions revealed that CD19-positive cells, which represent the B-cell population, displayed the highest level of FcRH3 expression among peripheral blood mononuclear cells. Lower expression was observed in CD4- and CD8-positive cells (Fig. 3b). Next, the effect of B-cell stimulation on FcRH3 expression was examined. Peripheral blood B-cells from a healthy donor were cultured for 4 h using known B-cell stimulants, and mRNA of FcRH3 was quantified (Fig. 3c). Expression of FcRH3 was increased using stimuli comprising anti-CD40 antibody and lipopolysaccharide (LPS).
Figure 3.
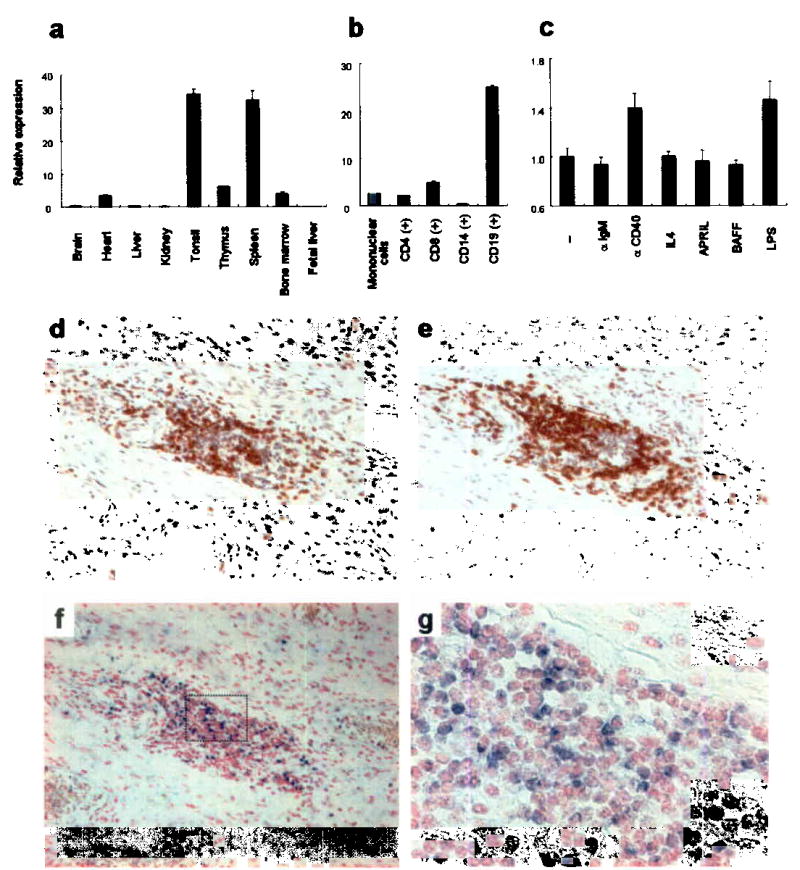
Expression patterns of FcRH3 in human tissues and cells. (a) Relative expression of FcRH3 in various tissues. (b) Relative expression of FcRH3 in fractionated leukocytes using MTC panel (Clontech). (c) Relative expression of FcRH3 in response to stimuli (anti-CD40 Ab, 1 μg/ml; anti-IgM Ab, 1 μg/ml; IL-4, 10 ng/ml; APRIL, 10 ng/ml; BAFF, 10 ng/ml; LPS 100ng/ml for 4 h). Representative data from 3 experiments performed in triplicate. (d, e) Lymphocyte aggregates in RA synovium. T-cells and B-cells in serial sections were immunostained using anti-CD3 (d) and anti-CD20 (e) antibodies respectively. (f, g) FcRH3 mRNA expression (blue stain) in RA synovium as analyzed by in situ hybridization. Higher magnification views of synovium (g) are denoted by the box in f (magnifications: d, e, f, ×100; g, ×400). Counterstaining: d, e, hematoxylin; f, g, nuclear fast red.
Expression of FcRH3 transcripts in synovial tissue was then investigated using in situ hybridization methods. T and B cells are known as the key players with regard to inflammation in synovial tissue, producing pro-inflammatory cytokines and autoantibodies that might be pathogenic1. These cells are known to show three distinct histological patterns: diffuse infiltration; clustering in aggregates; and follicles with germinal center reaction35,36. Aggregations of T and B cells were observed in paraffinized synovial sections from patients with RA, using immunostaining with anti-CD3 and -CD20 antibodies, respectively (Fig. 3d, e). In situ hybridization assay with serial sections detected FcRH3 mRNA in aggregated lymphocytes (Fig. 3f, g). Although strict differentiation between B and T cells was difficult, at least some aggregated B-cells were positive, with strong expression of FcRH3 mRNA. RA synovium from two other patients displayed similar images of lymphocyte aggregates and FcRH3 expression (Supplementary Fig. 2 online).
SNP association with autoantibody and HLA-DRB1 status
Higher FcRH3 expression was suspected as leading to B-cell abnormalities in RA, so associations were examined in RA patients between genotype and two RA-related autoantibodies: rheumatoid factor (RF); and anti-cyclic citrullinated peptide (CCP) antibody. RF is a well-known autoantibody for the Fc region of IgG, and titers correlate with RA disease activity37. Anti-CCP antibody recognizes peptides containing citrulline, and is detected in RA with extremely high specificity38,39. RF titer in RA patients was positively correlated with the number of susceptible alleles with statistical significance (R2=0.049, P=0.0065) (Table 3). The positive ratio of anti-CCP antibody in RA patients also differed significantly among genotypes (P<0.05), and correlated with numbers of susceptible alleles.
Table 3.
Genotype and autoantibodies in patients
| Rheumatoid factor
|
Anti-CCP antibody
|
|||
|---|---|---|---|---|
| Genotype | n (N=148) | Serum level ±SEM (IU/ml) | n (N=71) | Positivity (%) |
| −169 C/C | 29 | 479.9 ±91.3a | 17 | 100.0b |
| −169 C/T | 75 | 323.7 ±47.3a | 35 | 94.3b |
| −169 T/T | 44 | 216.4 ±44.0a | 19 | 73.7b |
R2=0.049, P=0.0065 by regression analysis.
P=0.029 by Fisher’s exact test.
As genetic interactions between HLA and non-HLA loci have been described in susceptibility for RA and other autoimmune diseases26,40, genotype distributions for SNP -169C/T were compared among three RA-case subgroups stratified by number of HLA-DRB1 shared-epitope (SE)-encoding alleles. We had previously genotyped HLA-DRB1 in our population and observed significant associations between RA-susceptibility and SE-encoding alleles4. Allele frequency of RA-susceptible allele, SNP -169C, in the subgroup with two copies of SE alleles was significantly higher than in the subgroup with no SE alleles (SNP -169C allele frequency: SE +/+, 0.49 (n=113); SE +/−, 0.43 (n=376); SE −/−, 0.39 (n=215)) (P<0.05).
Replication study of association in RA, ATID and SLE
To confirm associations between the FcRH3 variant and RA susceptibility, a replication study was conducted (540 RA patients, 636 control subjects). Allele-frequency comparison revealed a significant association between fcrh3_3 (-169C/T) and RA susceptibility (P=0.041, allele frequency: RA patients, 0.40; control subjects, 0.36) (Supplementary Table 2 online). No significant differences were noted between two cohorts that consisted of the replication samples. These results further confirmed the association of the fcrh3_3 -169C SNP allele with RA susceptibility in Japanese.
Since this region was known to display associations with multiple autoimmune diseases, and because several variants were known to be involved in multiple autoimmunities, associations were investigated between SNP -169C/T and two other autoimmune diseases, AITD and SLE. A total of 509 Japanese patients with AITD (Graves’ disease, n=351; Hashimoto’s thyroiditis, n=158 patients) and 564 Japanese patients with SLE were recruited and compared with 2037 Japanese controls. In addition, AITD, SLE and RA cases were combined as subjects with an autoimmune phenotype and tested for associations with the SNP. Individual diseases as well as combination of two AITDs and combination of AITD, SLE and RA were significantly associated with the SNP (OR=1.52 and P=0.00000084 in Japanese for a recessive model between all 4 autoimmunities considered in aggregate and controls; Table 4). As RA-specific autoantibodies were correlated with the number of susceptible alleles, anti-DNA antibody titer was higher in SLE patients with the -169C/C genotype than in subjects with other genotypes (294.1 IU/ml vs. 145.5 IU/ml; n=120; P=0.026 by Student’s t-test), a conclusion not further established by regression analysis (P=0.12).
Table 4.
Association of SNP -169C/T with AITD and SLE
| Genotype
|
Recessive-trait comparison
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Disease | Number of subjects | CC | CT | TT | Allele C frequency | OR (95% CI) | χ2 | P |
| GD | 351 | 72 | 179 | 100 | 0.46 | 1.79 (1.34–2.39) | 15.7 | 0.000074 |
| HT | 158 | 30 | 74 | 54 | 0.42 | 1.62 (1.07–2.47) | 5.2 | 0.022 |
| AITD total | 509 | 102 | 253 | 154 | 0.45 | 1.74 (1.35–2.24) | 18.5 | 0.000017 |
| SLE | 564 | 100 | 259 | 205 | 0.41 | 1.49 (1.16–1.92) | 9.8 | 0.0017 |
| RAa+AITD+SLE | 2437 | 438 | 1167 | 832 | 0.42 | 1.52 (1.29–1.79) | 24.2 | 0.00000084 |
| Controls | 2037 | 257 | 995 | 785 | 0.37 | |||
RA represents sum of three sets (n=1364).
GD = Graves’ disease; HT = Hashimoto’s thyroiditis; AITD = Autoimmune thyroid disease; SLE = Systemic lupus erythematosus.
DISCUSSION
LD mapping of 1q21-23 in Japanese subjects revealed multiple LD blocks in the region, and 1 block containing FcRH3 displayed associations with RA. This association was replicated in a second Japanese case-control set. The RA-associated allele was also associated with increased risk of other autoimmune disorders, such as AITD (Graves’ disease and Hashimoto’s thyroiditis) and SLE. Recent reports on autoimmune disease-associated polymorphisms show that some disease-susceptible variants are often limited to specific ethnic groups12 while other polymorphisms are widely dispersed among Homo sapiens, but significantly associated in only specific ethnic groups41,42. We evaluated haplotypes consisting of 4 SNPs in FcRH3 in African American, European American, and Asian (Korean and Japanese) subjects. Weaker LD was identified in African Americans and substantial differences in allelic frequency were noted among three major ethnic groups (Supplementary Table 3 online).
Though the evidence presented herein for FcRH3 being an autoimmune susceptibility gene is very powerful, additional autoimmune disease-related genes may probably exist in this region in addition to FcRH3. For example, 1q23 represents a strong candidate locus for SLE-susceptibility6, particularly involving the association of the classical FcγR genes with SLE-susceptibility in the Japanese population19, although those variants are not in LD with -169C/T in our Japanese population (Δ<0.05, Fig.1a). Multiple SLE susceptibility genes are also syntenic to human 1q23 in murine models of SLE43.
Further evaluation of polymorphism associations showed that an SNP in the promoter region of FcRH3 alters expression of FcRH3 via NF-κB binding. Since higher expression of FcRH3 was observed in individuals with susceptible alleles, and augmented autoantibody production was associated with the susceptible genotype, important steps in the sequence of events leading to autoimmunity must proceed through FcRH3. That the susceptible allele is associated with HLA-DRB1 in RA is consistent with FcRH3 functioning in the context of HLA class II restriction, which is usually seen in the interaction between T-cells and antigen-presenting cells, including B-cells. Moreover, together with the dominant expression of FcRH3 on B-cells and the critical roles of B-cells suggested by a recent clinical trial of B-cell-depleting therapy44, the present findings might provide a genetic basis for B-cell abnormality in autoimmunity.
Although the precise function of FcRH3 is unknown, the predicted molecular structure suggests that FcRH3 is a membranous protein that conveys signals into cells through a cytoplasmic domain containing an immunoreceptor-tyrosine activation motif (ITAM) and an immunoreceptor-tyrosine inhibitory motif (ITIM)14. An in vitro study showing the binding of tyrosine kinases syk and ZAP70 to the ITAM region and tyrosine phosphatases SHP-1 and SHP-2 to the ITIM region17 supports the proposed signaling function of FcRH3. In a previous study examining in situ hybridization in human tonsil, FcRH3 was expressed in the germinal center, with particularly high expression in the light zone16, suggesting that FcRH3 functions predominantly in centrocytes. The present finding that CD40 stimulation, which is important in germinal center formation45, upregulates FcRH3 expression in B-cells could indicate that FcRH3 is specifically expressed in germinal center centrocytes under the influence of CD40 signals. In the light zone, centrocytes undergo clonal selection and affinity maturation regulated by positive and negative signals from antigen receptors and co-receptors46. High expression of FcRH3 and augmented autoantibody production in individuals with the disease-susceptible genotype is consistent with FcRH3 influencing the fate of B-cells and augments the emergence of self-reactive cells in the germinal center.
Besides a role of FcRH3 in lymphoid tissues, expression of FcRH3 in synovial tissue might explain the pathological connection between FcRH3 variants and RA. As we have shown, FcRH3 is strongly expressed in aggregated lymphocytes. Although our synovial samples showed only T-cell/B-cell aggregates, lymphocytes in RA synovial tissue are known to form a germinal center-like structure, called an “ectopic germinal center”, where T-cell dependent antibody production and affinity maturation occur36. Ectopic germinal center formation is also seen in tissues from patients with AITD and SLE, and FcRH3 might be involved in pathological autoimmune reaction in these disease-specific ectopic lymphocyte aggregates.
Considering that augmented expression of FcRH3 is associated with susceptibility to autoimmune disorders, and that FcRH3 expression is regulated in B-cells in the secondary lymphoid organ and is detected in lymphocytes of disease-specific tissues, FcRH3 very probably is functional molecule in immunity and potentially pathogenic in autoimmune disorders.
METHODS
Subjects
Three independent cohorts of RA patients (n=830, 217, and 323), a cohort of SLE patients (n=564), and a cohort of AITD patients (n=509; comprising Graves’ disease (n=351) and Hashimoto’s thyroiditis (n=158)), were enrolled for the study through several medical institutes in Japan. Four independent cohorts of unaffected control subjects (n=658, n=262, n=374 and n=752) were recruited at various sites in Japan. All subjects were Japanese. RA patients (84.2% women; mean age, 59.0 ±12.3 years; 75.0% rheumatoid factor-positive) satisfied the revised criteria of the American Rheumatism Association for RA47. SLE patients followed the criteria of the American College of Rheumatology for SLE48. Diagnosis of AITD was established based on clinical findings and results of routine examinations for circulating thyroid hormone and thyroid stimulating hormone (TSH) concentrations, serum levels of antibodies against thyroglobulin, thyroid microsomes and TSH receptors, ultrasonography, [99m]TCO4− (or [123I]) uptake and thyroid scintigraphy.
The first control cohort was used for evaluation of LD in 1q21-23 and was compared with the first RA cohort for initial identification of RA-associated LD block and SNPs. The second and third cohorts of RA and controls were used for replication test of the first cohort result. Graves’ disease, Hashimoto’s thyroiditis, SLE, combination of the two AITDs and combination of RA, SLE and AITDs were tested for associations using the total pool of controls. Control subjects in three other ethnic groups, Korean (n=100), African American (n=120) and European American (n=120), were enrolled for evaluation of FcRH3 haplotypes. Synovial tissues were sampled from individuals with RA who underwent arthroplastic surgery. All subjects provided informed consent to participate in the study, as approved by the ethical committee of the SNP Research Center, RIKEN.
SNPs and genotyping
SNPs were discovered in exons and 5′- and 3′-flanking regions of FcRH1, FcRH2, FcRH3 and FcRHψ4 (LOC343265) by direct sequencing of DNA from 24 patients. Other SNPs were selected from the JSNP database and Assay-On-Demand SNP database (Applied Biosystems). SNPs were genotyped using Invader and TaqMan assays41 as indicated by manufacturers. Probe sets for the Invader assay were designed and synthesized by Third Wave Technologies, and those for the TaqMan assay were obtained from Applied Biosystems. When assessing the results of SNP genotyping, we generally exclude successful call rates<0.95 and values of P<0.01 obtained by HWE testing in control subjects. The error rate of Invader assay was 0.0023, which was estimated by 11,092 assays in 2 replicates using 118 randomly selected SNPs (internal control data).
Luciferase assay
The promoter fragment of three haplotypes corresponding to nt −523 to +203 of FcRH3 was cloned into the pGL3-Basic vector (Promega). Oligonucleotides were generated using the allelic sequences of nt −189 to −160 of FcRH3 as follows: -169T (5′GGTGAGATTACGGGAAGTCCTTGATCTGTA3′); -169C (5′GGTGAGATTACGGGAAGTCCCTGATCTGTA3′); and Cont (GGAGTGTAATCGGGTTGACCAAGTACAGAT). A single copy or 4 tandem copies of these oligonucleotides were cloned into pGL3-Promoter vector (Promega). Raji cells (RCB1647; RIKEN Cell Bank) were grown in RPMI1640 medium supplemented with 10% fetal bovine serum and antibiotics. We electroporated (230 V and 975 μF) 1 ×107 cells with 5 pmol of constructs and 1 pmol of pRL-TK vector (internal control for transfection efficiency) in a 0.4-cm gap cuvette. After 48 h, cells were collected and luciferase activity measured using the Dual-Luciferase Reporter Assay System (Promega).
EMSA
EMSA and preparation of nuclear extract from Raji cells were performed as previously described49. Oligonucleotides -169T and -169C were labeled with digoxigenin -11-ddUTP using the DIG gel-shift kit (Roche). We incubated 5 μg of nuclear extract with 40 fmol of DIG-labeled nucleotide for 25 min at room temperature. For competition experiments, nuclear extract was pre-incubated with unlabeled oligonucleotide (100-fold excess) before adding digoxigenin-labeled oligonucleotide. For supershift assays, 4 μg of anti-p50, p52, p65, RelB and cRel antibodies and rabbit IgG (control antibody) (Santa Cruz Biotechnology) were incubated for 15 min at room temperature after incubation of the labeled probe. Protein-DNA complexes were separated on a non-denaturing 6% polyacrylamide gel in 0.5× Tris-Borate-EDTA buffer. The gel was transferred to a nitrocellulose membrane, and signals were detected using an LAS-3000 luminoimage analyzer (Fujifilm).
RNA extraction and cDNA preparation
Peripheral blood was collected from consented healthy volunteers to obtain CD19-positive lymphocytes. Polymorphonuclear cells were separated by differential centrifugation using Lymphoprep resolving solution (AXIS-FIELD). CD19-positive lymphocytes were isolated using the MACS system with CD19 microbeads (Miltenyi Biotec), and cell purity >95% was confirmed using flow cytometry. Cells were stimulated with anti-CD40 Ab (Cymbus Biotechnology), anti-IgM Ab (Jackson Immunoresearch), Il-4 (eBioscience), APRIL (PeproTech), BAFF (PeproTech) and LPS (Sigma) for 4 h. Total RNA was isolated using RNeasy Mini Kit (Qiagen). RNAs of other normal tissues were quantified using Premium Total RNA (Clontech). Total RNA was reverse transcribed using TaqMan Gold RT-PCR reagents with random hexamers (Applied Biosystems), in accordance with the instructions of the manufacturer.
Quantification of FcRH3 expression using real-time RT-PCR
Real-time quantitative PCR was performed using an ABI PRISM 7900 (Applied Biosystems) and Assay-on-Demand TaqMan probe and primers (Hs00364720_m1 for FcRH3), according to the manufacturer’s instructions. A standard curve was generated from the amplification data for FcRH3 primers using a dilution series of total RNA from Raji cells as templates, and data were normalized to GUS level.
ASTQ
Allele-specific quantification was performed as previously described34 with some modifications. Preparation of cDNA from B-cells was undertaken as described above. Both cDNA and genomic DNA were amplified by PCR for 37 cycles using primers specific for exon 2 of FcRH3 (Supplementary Table 4 online), and for an additional cycle using forward primer with Alexa Fluor 488 label at the 5′-end. Products were directly digested using Eag I by incubation at 37 °C for 12 h. Full digestion was monitored by the inclusion of PCR products from homozygote +358G/G. Digested products were then run on a 12.5% polyacrylamide gel, followed by quantification using an LAS-3000 analyzer.
In situ hybridization and immunohistochemistry
In situ hybridization was performed as previously described50. Probes were obtained from PCR products using the sequence of FcRH3 (nt 2052–2490, comprising the intracellular unique region that is poorly conserved among members of this family). An additional probe of the 5′-UTR sequence yielded similar results. Control probes were also examined, and yielded no specific hybridization (data not shown). Antibodies to CD3 (clone PS-1, Nichirei) and CD20 (clone L26, Zymed) were used for immunohistochemistry with an ABC Elite kit (Vector Labs) according to the manufacturer’s instructions. No specific staining was detected using mouse isotype IgG (data not shown).
Measurement of autoantibodies
Rheumatoid factor in sera of RA patients was measured using latex-enhanced immunonephelometric assay. Anti-DNA antibody in sera of SLE patients was measured by radioimmunoassay. RA patients (n=147, 81.1% women; mean age, 63.9 ±10.6 years; 87.8% rheumatoid factor positive; mean Steinbrocker radiographic stage, 3.2) and SLE patients (n=120, 92.6% woman; mean age, 36.6 ±12.7 years) were part of the cohorts and from a single medical institute, respectively. For each patient, the maximum value of rheumatoid factor and anti-DNA antibody during the treatment period in the medical center or outpatient clinic was used. Anti-cyclic citrullinated peptide antibody was detected at a single time point using enzyme-linked immunosorbent assay, as previously described38.
Statistical analysis
LD index Δ28 was calculated and Figure 1a drawn using Excel software (Microsoft). Haplotype frequencies were estimated using HAPLOTYPER software. The χ2 test was applied for contingency table tests for associations between allele/genotype distribution and phenotypes. FcRH3 expression in B-cells and autoantibody production were regressed on the number of susceptible alleles (coded 0, 1, and 2). All other statistical analyses, unless otherwise stated, were performed using STATISTICA software (StatSoft).
URLs
JSNP database (http://snp.ims.u-tokyo.ac.jp/index.html)
TRANSFAC (http://www.gene-regulation.com).
HAPLOTYPER (http://www.people.fas.harvard.edu/~junliu/Haplo/docMain.htm)
GenBank accession number
FcRH3 mRNA, NM_052939.
Acknowledgments
We wish to extend our thanks to: E. Kanno and other members of the Laboratory for Rheumatic Diseases for technical assistance; H. Kawakami for expertise in computer programming; S Yoshino, M. Yukioka, S. Tohma, Y. Nishioka, T. Matsubara, S. Wakitani, R. Teshima, N. Ishikawa, K. Ito, K. Ito, K. Kuma, H. Tamai and T. Akamizu for clinical sample collection; M. Ishikawa and Y. Amasaki for preparation of the 2nd population study; M. Nagashima and S. Yoshino for sampling RA synovium; and K. Nagatani and Y. Komagata for advice. This work was supported by a grant from the Japanese Millennium Project and the United States National Institutes of Health.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- 1.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 2.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 3.Newton JL, Harney SM, Wordsworth BP, Brown MA. A review of the MHC genetics of rheumatoid arthritis. Genes Immun. 2004;5:151–157. doi: 10.1038/sj.gene.6364045. [DOI] [PubMed] [Google Scholar]
- 4.Kochi Y, et al. Analysis of single-nucleotide polymorphisms in Japanese rheumatoid arthritis patients shows additional susceptibility markers besides the classic shared epitope susceptibility sequences. Arthritis Rheum. 2004;50:63–71. doi: 10.1002/art.11366. [DOI] [PubMed] [Google Scholar]
- 5.Seldin MF, Amos CI, Ward R, Gregersen PK. The genetics revolution and the assault on rheumatoid arthritis. Arthritis Rheum. 1999;42:1071–1079. doi: 10.1002/1529-0131(199906)42:6<1071::AID-ANR1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 6.Tsao BP. The genetics of human systemic lupus erythematosus. Trends Immunol. 2003;24:595–602. doi: 10.1016/j.it.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Bowcock AM, Cookson WO. The genetics of psoriasis, psoriatic arthritis and atopic dermatitis. Hum Mol Genet. 2004;13(Suppl 1):R43–55. doi: 10.1093/hmg/ddh094. [DOI] [PubMed] [Google Scholar]
- 8.Marrack P, Kappler J, Kotzin BL. Autoimmune disease: why and where it occurs. Nat Med. 2001;7:899–905. doi: 10.1038/90935. [DOI] [PubMed] [Google Scholar]
- 9.Ueda H, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 10.Becker KG, et al. Clustering of non-major histocompatibility complex susceptibility candidate loci in human autoimmune diseases. Proc Natl Acad Sci USA. 1998;95:9979–9984. doi: 10.1073/pnas.95.17.9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tokuhiro S, et al. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat Genet. 2003;35:341–348. doi: 10.1038/ng1267. [DOI] [PubMed] [Google Scholar]
- 12.Begovich AB, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004;75:330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis RS, Wang YH, Kubagawa H, Cooper MD. Identification of a family of Fc receptor homologs with preferential B cell expression. Proc Natl Acad Sci USA. 2001;98:9772–9777. doi: 10.1073/pnas.171308498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis RS, et al. Fc receptor homologs: newest members of a remarkably diverse Fc receptor gene family. Immunol Rev. 2002;190:123–136. doi: 10.1034/j.1600-065x.2002.19009.x. [DOI] [PubMed] [Google Scholar]
- 15.Hatzivassiliou G, et al. IRTA1 and IRTA2, novel immunoglobulin superfamily receptors expressed in B cells and involved in chromosome 1q21 abnormalities in B cell malignancy. Immunity. 2001;14:277–289. doi: 10.1016/s1074-7613(01)00109-1. [DOI] [PubMed] [Google Scholar]
- 16.Miller I, Hatzivassiliou G, Cattoretti G, Mendelsohn C, Dalla-Favera R. IRTAs: a new family of immunoglobulinlike receptors differentially expressed in B cells. Blood. 2002;99:2662–2669. doi: 10.1182/blood.v99.8.2662. [DOI] [PubMed] [Google Scholar]
- 17.Xu MJ, Zhao R, Cao H, Zhao ZJ. SPAP2, an Ig family receptor containing both ITIMs and ITAMs. Biochem Biophys Res Commun. 2002;293:1037–1046. doi: 10.1016/S0006-291X(02)00332-7. [DOI] [PubMed] [Google Scholar]
- 18.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 19.Kyogoku C, et al. Fcgamma receptor gene polymorphisms in Japanese patients with systemic lupus erythematosus: contribution of FCGR2B to genetic susceptibility. Arthritis Rheum. 2002;46:1242–1254. doi: 10.1002/art.10257. [DOI] [PubMed] [Google Scholar]
- 20.Capon F, et al. Fine mapping of the PSORS4 psoriasis susceptibility region on chromosome 1q21. J Invest Dermatol. 2001;116:728–730. doi: 10.1046/j.1523-1747.2001.01311.x. [DOI] [PubMed] [Google Scholar]
- 21.Dai KZ, et al. The T cell regulator gene SH2D2A contributes to the genetic susceptibility of multiple sclerosis. Genes Immun. 2001;2:263–268. doi: 10.1038/sj.gene.6363774. [DOI] [PubMed] [Google Scholar]
- 22.Jirholt J, et al. Genetic linkage analysis of collagen-induced arthritis in the mouse. Eur J Immunol. 1998;28:3321–3328. doi: 10.1002/(SICI)1521-4141(199810)28:10<3321::AID-IMMU3321>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 23.Sundvall M, et al. Identification of murine loci associated with susceptibility to chronic experimental autoimmune encephalomyelitis. Nat Genet. 1995;10:313–317. doi: 10.1038/ng0795-313. [DOI] [PubMed] [Google Scholar]
- 24.Teuscher C, et al. Evidence that Tmevd2 and eae3 may represent either a common locus or members of a gene complex controlling susceptibility to immunologically mediated demyelination in mice. J Immunol. 1997;159:4930–4934. [PubMed] [Google Scholar]
- 25.Podolin PL, et al. Congenic mapping of the insulin-dependent diabetes (Idd) gene, Idd10, localizes two genes mediating the Idd10 effect and eliminates the candidate Fcgr1. J Immunol. 1997;159:1835–1843. [PubMed] [Google Scholar]
- 26.Nieto A, et al. Involvement of Fcgamma receptor IIIA genotypes in susceptibility to rheumatoid arthritis. Arthritis Rheum. 2000;43:735–739. doi: 10.1002/1529-0131(200004)43:4<735::AID-ANR3>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 27.Radstake TR, et al. Role of Fcgamma receptors IIA, IIIA, and IIIB in susceptibility to rheumatoid arthritis. J Rheumatol. 2003;30:926–933. [PubMed] [Google Scholar]
- 28.Devlin B, Risch N. A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics. 1995;29:311–322. doi: 10.1006/geno.1995.9003. [DOI] [PubMed] [Google Scholar]
- 29.Cordell HJ, Clayton DG. A unified stepwise regression procedure for evaluating the relative effects of polymorphisms within a gene using case/control or family data: application to HLA in type 1 diabetes. Am J Hum Genet. 2002;70:124–141. doi: 10.1086/338007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sebastiani P, et al. Minimal haplotype tagging. Proc Natl Acad Sci USA. 2003;100:9900–9905. doi: 10.1073/pnas.1633613100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsunoda. T et al. Variation of gene-based SNPs and linkage disequilibrium patterns in the human genome. Hum. Mol. Genet. (2004). [DOI] [PubMed]
- 32.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pritchard JK, Rosenberg NA. Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet. 1999;65:220–228. doi: 10.1086/302449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaijzel EL, et al. Allele-specific quantification of tumor necrosis factor alpha (TNF) transcription and the role of promoter polymorphisms in rheumatoid arthritis patients and healthy individuals. Genes Immun. 2001;2:135–144. doi: 10.1038/sj.gene.6363747. [DOI] [PubMed] [Google Scholar]
- 35.Takemura S, et al. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167:1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 36.Weyand CM, Goronzy JJ. Ectopic germinal center formation in rheumatoid synovitis. Ann N Y Acad Sci. 2003;987:140–149. doi: 10.1111/j.1749-6632.2003.tb06042.x. [DOI] [PubMed] [Google Scholar]
- 37.Alarcon GS, et al. Suppression of rheumatoid factor production by methotrexate in patients with rheumatoid arthritis. Evidence for differential influences of therapy and clinical status on IgM and IgA rheumatoid factor expression. Arthritis Rheum. 1990;33:1156–1161. doi: 10.1002/art.1780330816. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki K, et al. High diagnostic performance of ELISA detection of antibodies to citrullinated antigens in rheumatoid arthritis. Scand J Rheumatol. 2003;32:197–204. doi: 10.1080/03009740310003677. [DOI] [PubMed] [Google Scholar]
- 39.Rantapaa-Dahlqvist S, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 40.Capon F, Semprini S, Dallapiccola B, Novelli G. Evidence for interaction between psoriasis-susceptibility loci on chromosomes 6p21 and 1q21. Am J Hum Genet. 1999;65:1798–1800. doi: 10.1086/302653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki A, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003;34:395–402. doi: 10.1038/ng1206. [DOI] [PubMed] [Google Scholar]
- 42.Barton A, et al. A functional haplotype of the PADI4 gene associated with rheumatoid arthritis in a Japanese population is not associated in a United Kingdom population. Arthritis Rheum. 2004;50:1117–1121. doi: 10.1002/art.20169. [DOI] [PubMed] [Google Scholar]
- 43.Jorgensen TN, Gubbels MR, Kotzin BL. New insights into disease pathogenesis from mouse lupus genetics. Curr Opin Immunol. 2004;16:787–793. doi: 10.1016/j.coi.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 44.Edwards JC, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 45.Gray D, et al. Observations on memory B-cell development. Semin Immunol. 1997;9:249–254. doi: 10.1006/smim.1997.0077. [DOI] [PubMed] [Google Scholar]
- 46.van Eijk M, Defrance T, Hennino A, de Groot C. Death-receptor contribution to the germinal-center reaction. Trends Immunol. 2001;22:677–682. doi: 10.1016/s1471-4906(01)02086-5. [DOI] [PubMed] [Google Scholar]
- 47.Arnett FC, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 48.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 49.Aikawa Y, Yamamoto M, Yamamoto T, Morimoto K, Tanaka K. An anti-rheumatic agent T-614 inhibits NF-kappaB activation in LPS- and TNF-alpha-stimulated THP-1 cells without interfering with IkappaBalpha degradation. Inflamm Res. 2002;51:188–194. doi: 10.1007/pl00000291. [DOI] [PubMed] [Google Scholar]
- 50.Hoshino M, et al. Identification of the stef gene that encodes a novel guanine nucleotide exchange factor specific for Rac1. J Biol Chem. 1999;274:17837–17844. doi: 10.1074/jbc.274.25.17837. [DOI] [PubMed] [Google Scholar]