

Abstract
The influenza A virus genome is composed of eight negative-sense RNA segments (called vRNAs), all of which must be packaged to produce an infectious virion. It is not clear whether individual vRNAs are packaged specifically or at random, however, and the total vRNA capacity of the virion is unknown. We have created modified forms of the viral nucleoprotein (NP), neuraminidase (NA), and nonstructural (NS) vRNAs that encode green or yellow fluorescent proteins and studied the efficiency with which these are packaged by using a plasmid-based influenza A virus assembly system. Packaging was assessed precisely and quantitatively by scoring transduction of the fluorescent markers in a single-round infectivity assay with a flow cytometer. We found that, under conditions in which virions are limiting, pairs of alternatively tagged vRNAs compete for packaging but do so in a nonspecific manner. Reporters representing different vRNAs were not packaged additively, as would be expected under specific packaging, but instead appeared to compete for a common niche in the virion. Moreover, 3 to 5% of transduction-competent viruses were found to incorporate two alternative reporters, regardless of whether those reporters represented the same or different vRNAs — a finding compatible with random, but not with specific, packaging. Probabilistic estimates suggest that in order to achieve this level of dual transduction by chance alone, each influenza A virus virion must package an average of 9 to 11 vRNAs.
The genome of influenza A virus consists of eight single-stranded, negative-sense RNA segments (vRNAs) that together encode the 11 known viral proteins (1, 13). To be infectious, an influenza A virus particle must package at least one copy of each segment. cis-Acting signals necessary for packaging appear to reside within the 3′ and 5′ untranslated regions (UTRs) of each vRNA (17), regions that also control vRNA transcription and replication by the viral RNA-dependent RNA polymerase (24). In the influenza virus strain A/WSN/33, for example, these UTRs include 12 and 13 bases at the extreme 3′ and 5′ ends, respectively, that are common to all vRNAs, as well as the adjacent 7 to 45 bases at each end whose sequence is unique to a given segment and highly conserved in other strains. When attached in appropriate pairs to the ends of a heterologous RNA, the UTR sequences are sufficient to direct packaging into influenza virus virions and also to support subsequent rounds of replication (17). At least some specific bases required for packaging occur within the region that mediates polymerase binding (9, 16, 28), which presumably implies that interaction with the viral polymerase is an essential requirement for packaging.
One fundamental question in influenza virus genetics is whether the virus discriminates among its segments during the packaging process. While a variety of mechanisms could be envisioned, the issue is often framed in terms of two simple, hypothetical models that are intended to represent the extreme alternatives. Under the first model, called random packaging, each virion selectively incorporates viral segments (as opposed to other viral or cellular RNAs) but does not differentiate among them, so that the likelihood of acquiring the full complement of vRNAs is determined entirely by chance. The second model, by contrast, asserts that the virus targets each of the eight segments individually and independently, a phenomenon called specific packaging (3). The evidence in support of either model is limited and inconclusive. Evidence for specific packaging, for example, is adduced mainly from the finding that the various vRNAs are equimolar within viral particles even though their concentrations in the infected cells may differ (27) and from several reports that defective interfering vRNA mutants can competitively inhibit packaging of their normal counterparts but not of other vRNAs (6, 19-21, 23).
The recent development of efficient, plasmid-based systems for assembling infectious influenza A virus virions now provides an opportunity to dissect the packaging mechanism in detail (8, 10, 22). As an initial step, we have developed a quantitative assay in which pairs of genetically marked vRNAs compete for packaging into transduction-competent particles. Here we report that, in this reverse genetic system, the influenza virus segments compete for packaging but do so in a random, nonspecific manner.
MATERIALS AND METHODS
Cells.
293T cells were grown in Dulbecco's modified Eagle's medium H-21 supplemented with penicillin and streptomycin, glutamine, and 10% heat-inactivated fetal calf serum (FCS). Uninfected MDCK cells were grown in Eagle's minimal essential medium (MEM) supplemented with penicillin and streptomycin, glutamine, nonessential amino acids (Gibco-BRL), and 10% FCS. After infection, MDCK cells were maintained in L-15 medium supplemented with penicillin and streptomycin, glutamine, nonessential amino acids, 15 mM HEPES (pH 7.5), 0.75 g of NaHCO3 per liter, and 0.125% (wt/vol) bovine serum albumin.
Construction of GFP and YFP reporters.
The 17-plasmid influenza A virus assembly system was a generous gift from Y. Kawaoka (University of Wisconsin) and used essentially as described before (22). We created five segment expression reporter plasmids by first deleting all vRNA sequences from the wild-type neuraminidase (NA) vRNA vector through PCR amplification with the primers5′-CGGCGGCGGGGTACCGTCTCCTCCCCCCCACCTTCGGAGGTCGACCAGT-3′ and 5′-CGGCGGCGGGGTACCGTCTCCAATAACCCGGCGGCCCAAAATGCC-3′, digesting with KpnI, and recircularizing by ligation. The resulting vector was equivalent to plasmid pHH21, described by Neumann et al. (10, 22). We cleaved this vector at its two BsmB1 sites and inserted coding sequences for the green (GFP) and yellow (YFP) fluorescent proteins of Aequorea victoria that had been PCR amplified from the plasmids EGFP-N1 and EYFP-N1 (Clontech), respectively, with primers that added flanking influenza A virus UTRs and a BsmB1 site at each end. The NA reporters encoding GFP and YFP were created with primers 5′-CGGCGGCGGCGTCTCGTATTAGTAGAAACAAGGAGTTTTTTGAACAAATTACTTGTACAGCTCGTCC-3′ and 5′-CGGCGGCGGCGTCTCGGGGAGCGAAAGCAGGAGTTTAAATGGTGAGCAA GGGCGAGGAGC-3′. Nucleoprotein (NP) vRNA reporters were made with primers 5′-CGGCGGCGGCGTCTCGTATTAGTAGAAACAAGGGTATTTTTCTTTACTTGTACAGCTCGTCC-3′ and 5′-CGGCGGCGGCGTCTCGGGGAGCAAAAGCAGGGTAGATAATCACTCACAGAGTGACATCGAAATCATGGTGAGCAAGGGCGAGGAGC-3′. The nonstructural (NS) vRNA reporter was made with primers 5′-CGGCGGCGGCGTCTCGTATTAGTAGAAACAAGGGTGTTTTTTATTATTACTTGTACAGCTCGTCC-3′ and 5′-CGGCGGCGGCGTCTCGGGGAGCAAAAGCAGGGTGACAAAGACATAATGGTGAGCAAGGGCGAGGAGC-3′. NA and NP “competitor” vectors, encoding red fluorescent protein (RFP) from Discosoma sp., were created in an identical manner with PCR primers that amplified the entire 680-nucleotide RFP coding sequence from pDsRed-N1 (Clontech).
Generation of infectious influenza virus particles.
293T cells were transfected in 35-mm dishes at approximately 50% confluency with Trans IT LT-1 transfection reagent (Panvera, Madison, Wis.). Following the protocol of Neumann et al. (22), every transfection included 1 μg each of the eight wild-type vRNA vectors; 1 μg each of expression vectors for PB1, PB2, NP, NA, HA, and NS2 proteins; 0.1 μg each of vectors for M2 and PA proteins; and 2 μg of the M1 protein vector. To this mixture, we added various combinations of vRNA reporters as indicated in the text, along with sufficient pHH21 to maintain total DNA input constant in a given experiment. Transfection reagent (2 μl per μg of DNA) was added to 100 μl of serum-free Opti-MEM, mixed, and incubated at room temperature for 15 min. DNA was then added, mixed by pipetting, and allowed to sit at room temperature for 15 min before being added to cells. Six hours later, the transfection mixture was removed, and the cells were washed once and then refed with Opti-MEM containing 0.3% bovine serum albumin and 0.01% FCS. Supernatants were collected at 48 h posttransfection, clarified by microcentrifugation for 5 min, and then used for infection of MDCK cells either immediately or after storage at −80°C.
Infection of MDCK cells.
Confluent monolayers of MDCK cells in six-well plates were infected with 293T supernatants. MDCK cells were washed once with phosphate-buffered saline containing calcium and magnesium (PBS+CM), inoculated with 250 μl of 293T supernatant, and incubated for 1 h at 37°C. Viral inoculum was removed, and cells were then washed once with PBS+CM and refed with L-15 medium.
Flow cytometry.
Infected cells were harvested 10 h postinfection by removing medium, washing once with calcium- and magnesium-free PBS, and digesting with trypsin until they detached from the plate. Cells were then washed once with PBS+CM, pelleted by centrifugation, and resuspended in 2 ml of ice-cold PBS+CM for analysis. Flow cytometry was performed with a FACSCalibur cytometer (Becton Dickinson) with a 525-nm short-pass dichroic filter to separate the signals and paired 550/30-nm and 510/20-nm bandpass filters to collect them.
Quantitative analysis of vRNAs in transfected 293T cells.
Reporter-derived vRNAs were quantified with an RNase protection assay (RPA) that detected antisense or sense forms of both the GFP and YFP sequences but not RFP or any authentic influenza virus vRNA. Primers 5′-CCCCCCAAGCTTGAAGGCTACGTCCAGGAGCGC-3′ and 5′-CCGCCGGAATTCTCCTCGATGTTGTGGCGG-3′ were used in a PCR with pEYFP-N1 (Clontech) as the template to amplify residues 271 to 521, which are common to both the GFP and YFP sequences. The PCR product was digested with EcoRI and HindII and cloned into pBluescript II KS+ (Stratagene). This RPA plasmid was linearized with EcoRI or HindIII and transcribed in vitro with T3 or T7 RNA polymerase and [α-32P]UTP. Total cellular RNA was isolated from 293T cells 48 h after transfection, with Trizol (Gibco-BRL), according to the manufacturer's instructions. The RPA III kit (Ambion) was used to perform RPA with 10 μg of input RNA and 80,000 cpm of probe per reaction.
RESULTS AND DISCUSSION
Influenza A virus will package and replicate heterologous RNA molecules that carry viral UTR sequences at their 3′ and 5′ ends. Such artificial segments can also be packaged by influenza virus-like particles assembled with plasmid-based transfection systems. For the present study, we used the assembly system developed by Neumann and colleagues (22), which is based on 17 plasmid expression vectors that together encode the eight wild-type vRNAs and nine viral proteins needed to assemble an influenza A virus virion. We confirmed that when 293T cells were transfected with these plasmids in the recommended stoichiometries (see Materials and Methods), infectious influenza A virus particles were efficiently produced, accumulating over 48 h to concentrations of roughly 106 PFU/ml in the 293T supernatant. Unless otherwise noted, this standard mixture of 17 vectors was included in all our transfections to provide a fixed supply of virion components, and other plasmids were added as detailed below.
The vRNA expression vectors are derivatives of pHH21 (10, 22), which uses a mammalian RNA polymerase I promoter and terminator to transcribe the appropriate viral coding and UTR sequences. We used this backbone to create five new reporter vectors (Table 1), each specifying a variant of one of the three vRNAs that normally encode the viral neuraminidase (NA), nucleoprotein (NP), or nonstructural (NS) proteins. These reporter vRNAs had the same UTRs as their wild-type counterparts but lacked any viral genes; instead, they contained the 719-base negative-sense coding sequences for either the green (GFP) or yellow (YFP) fluorescent protein, which are identical at all but 7 bases. We thus obtained two alternatively tagged NP reporters (NP-G and NP-Y), two NA reporters (NA-G and NA-Y), and a single NS reporter (NS-G). For the purposes of this study, pairs of reporters that have the same UTRs will be termed homologous, whereas those with dissimilar UTRs will be called nonhomologous reporters.
TABLE 1.
Reporter vRNAs used in this studya
| Construct | Sequence |
|---|---|
| NP-Y or NP-G | 3′-UCGUUUUCGUCCCAUCUAUUAGUGAGUGUCUCACUGUAGCUUUAG—antisense GFP or YFP—UCUUUUUAUGGGAACAAAGAUGA-5′ |
| NA-Y or NA-G | 3′-UCGCUUUCGUCCUCAAAUU—antisense GFP or YFP—AAACAAGUUUUUUGAGGAACAAAGAUGA-5′ |
| NS-G | 3′-UCGUUUUCGUCCCACUGUUUCUGUAU—antisense GFP—AUUUUUUGUGGGAACAAAGAUGA-5′ |
Sequences shown are for the vRNAs expressed from each of the five reporter plasmids under control of the human polymerase I promoter and mouse polymerase I terminator elements. Each contains the 719-base coding sequence of GFP or YFP in the antisense orientation, flanked by the 3′ and 5′ UTR sequences shown. The latter are derived from influenza virus A/WSN/33. The conserved 3′ and 5′ bases common to all influenza virus RNAs are underlined.
To verify that the reporters were biologically active, we transfected each into 293T cells along with the 17 wild-type vectors, collected supernatants 48 h later, and used them to infect MDCK cells at a ratio of 0.1 to 1.0 PFU/cell. Ten hours later (to allow time for primary infection but not for secondary spread), the MDCK cells were harvested and screened individually for fluorescence by two-color flow cytometry. As illustrated in Fig. 1A, roughly 3 to 8% of MDCK cells exposed to virions that had been assembled in the presence of any single reporter vRNA exhibited significant yellow or green fluorescence, implying that each of these cells had been infected by a virion carrying the reporter vRNA, which had then been transcribed by the viral polymerase to yield positive-sense mRNAs. The absolute number of transductants (i.e., fluorescent MDCK cells) obtained with a given reporter varied somewhat among experiments, presumably due to varying growth states of the cell populations, but it was highly reproducible within any single experiment (Fig. 1B). Moreover, when viruses were assembled with two homologous GFP and YFP reporters (e.g., NP-Y and NP-G) in combination, we could readily discriminate two subpopulations of transductants that each expressed only one fluorescent marker or the other, as well as a small minority that expressed both (Fig. 1A and B). These results confirmed that our reporter vRNAs could be packaged and transduced by influenza virus virions and that the fluorescence they conferred could be used to precisely enumerate transduced cells during the first round of infection.
FIG. 1.

Quantitative assay for influenza virus vRNA packaging. (A) Immunofluorescence flow cytometry of MDCK cells infected with plasmid-derived influenza virus virions. Cultured 293T cells were transfected with the 17-plasmid assembly system supplemented with 2 μg of NP-Y, with 2 μg of NP-G, with 1 μg each of NP-Y and NP-G, or with 2 μg of the control plasmid pHH21. Supernatants harvested 48 h later were used to infect MDCK cells, which were themselves harvested and analyzed by flow cytometry 10 h postinfection. Each point represents one of 20,000 viable MDCK cells from each population, analyzed for green (GFP) and yellow (YFP) fluorescence, each expressed on an exponential scale. The four quadrants of each plot represent (clockwise from lower left) nonfluorescent, yellow fluorescent, dual-colored, and green fluorescent MDCK cells, respectively. Data are from one representative experiment. (B) Quantitative transduction data for viruses prepared with the indicated amounts of various reporter plasmids. Each bar indicates the numbers of yellow, green, and dual-colored transductants obtained per 20,000 MDCK cells. Data are mean ± standard error of the mean for triplicate transfections in a single experiment. Statistically significant differences between yields from homologous reporters were observed in some individual experiments, as here, but were not reproducible (see Table 2).
To determine whether this property could be exploited in an assay for packaging, we first asked whether transduction frequency depended on the amount of reporter DNA introduced into the 293T producer cells, where all relevant packaging occurred. To that end, we transfected 293T cells with various amounts (0.5 to 5.0 μg) of NP-Y reporter along with the standard 17 plasmids and harvested the cells and their supernatants 48 h later. We then extracted RNA from these cells and probed it for reporter-derived sequences in an RPA. As shown in Fig. 2A (left), positive-sense reporter RNAs (i.e., mRNAs and cRNAs) were readily detectable in amounts that increased in proportion to vector DNA input up to the highest level tested (Fig. 2A). Similar results were also obtained with the NA-based reporters (data not shown). This result indicated that transfection efficiency and reporter transcription in the 293T cells were not limiting within this range of vector input. When we measured the transducing activity of supernatants from these cells (Fig. 2B), however, we found that the yield of fluorescent MDCK cells rose to a maximum with a transfection of 1 to 2 μg of reporter DNA and then appeared to plateau, suggesting that the capacity of the virions to incorporate reporter vRNAs had reached saturation. The yield of transductants then declined somewhat at the highest plasmid doses, perhaps indicating that highly overexpressed reporter vRNAs could competitively inhibit packaging of the wild-type vRNAs needed for transduction or infectivity. Consistent with that interpretation, we found that the plaque-forming titers of the same supernatants likewise tended to decline as reporter input increased (Fig. 2C).
FIG. 2.

Saturable packaging and transduction of reporter vRNA. (A) Dose-dependent expression of reporter transcripts. 293T cells were transfected with the indicated amounts of NP-Y reporter vector along with either all 17 standard plasmids (at left) or all except the wild-type (wt) NP vRNA vector (at right). Cellular RNA harvested 48 h posttransfection was probed for positive-sense reporter transcripts (mRNA and cRNA) by RPA. Control cells were transfected either with a GFP-mRNA expression vector (EGFP-N1) or with pHH21. (B) Virions isolated from supernatants of the same cells (whose transfections included the wild-type NP vRNA vector) were assayed for MDCK-transducing activity at 10 h postinfection by flow cytometry. (C) Total plaque-forming activities of the supernatants in panel B, assayed in MDCK monolayers.
Because our planned experiments involved cotransfecting various pairs of reporters into 293T cells, we next tested whether these vectors interfered with each other at the level of vRNA expression. With an RPA probe that detected negative-sense sequences common to both the GFP and YFP coding regions, we first measured the levels of vRNA produced by 1 μg of each of the five reporters transfected individually and found that they were similar (Fig. 3A). We then compared the effects of transfecting 1 μg of each reporter either alone or in combination with 1 μg of a second competitor vRNA vector in which UTRs from the NP or NA segments flanked the coding sequence of the RFP, which could not be detected by the RPA probe. In repeated transfections, we tested all possible pairwise combinations of the five reporters with the two competitor vectors and assayed reporter vRNA by RPA. Because we observed no significant differences in the effects of the two competitors or between homologous reporters, the data were pooled. As depicted in Fig. 3B (right), serial dilution of the input RNA confirmed that the RPA could readily detect a 50% decrease in reporter vRNA expression in the 293T cells. Nevertheless, we found no evidence that expression of any reporter vRNA was affected by cotransfecting a second vRNA vector, regardless of whether the two were homologous or nonhomologous (Fig. 3A, left). This was consistent with our earlier finding that omitting the wild-type NP vRNA vector from the standard transfection did not appreciably affect expression of NP-Y reporter RNA (Fig. 2A, right).
FIG. 3.

Reporter vRNAs are comparably and independently expressed in 293T cells. (A) RPA of reporter-specific negative-sense RNA (vRNA) in 293T cells transfected 48 h previously with 1 μg of each indicated reporter plasmid along with the 17-plasmid assembly system. (B) Reporter expression in 293T cells 48 h after transfection with 1 μg of each indicated reporter type either alone (alone) or in combination with an NA- or NP-based competitor plasmid encoding RFP (+ comp). Levels of vRNA were assayed by RPA with a probe that detects GFP and YFP but not RFP sequences and were quantified by phosphorimaging. Each of the five reporters was tested alone (in quadruplicate) and with each of the two competitors (in duplicate); data shown are mean ± standard error of the mean, pooled for homologous reporters. At right, titration of one representative cellular RNA sample to determine the sensitivity of the assay.
The foregoing results together indicated that, when transfected into 293T cells, all five reporters generated comparable amounts of genetically tagged vRNA that could be packaged and transduced. When the total amount of reporter plasmid introduced was on the order of 1 to 2 μg, these vRNAs appeared to be expressed independently, in that expression of any one reporter did not detectably influence the expression of other reporters in the 293T cells, regardless of whether they carried the same or different UTRs. The quantity of virions produced by the standard transfection of these cells, moreover, had a finite capacity to transduce any single reporter vRNA. This capacity was 70 to 80% saturated by 1 μg and fully saturated by 2 μg of input reporter plasmid (Fig. 2B) and presumably reflected the finite quantity of any single reporter vRNA that could be packaged into transduction-competent virions.
The saturable nature of this phenomenon offered the chance to ask whether nonhomologous reporters, i.e., those with dissimilar UTRs, competed for a common niche (as would be predicted by the random packaging model) or instead for independent, segment-specific niches (as expected for specific packaging) within the assembled virions. We addressed this question with a combinatorial approach, by comparing the transduction efficiency of each reporter when transfected either singly or together with any other reporters that carried the alternative fluorescence marker. In simultaneous transfections on a single day, all possible binary combinations of reporters were tested, which ensured that every YFP reporter was individually paired with every GFP and also that each reporter was paired with one homologous reporter as well as with one or two nonhomologous reporters. By repeating this entire experiment three times, we obtained a data set of triplicate measurements for each of the 12 possible reporter combinations and also for 1-μg and 2-μg transfections of each reporter alone. As in our preliminary studies (Fig. 2B), we found that 1 μg of any single reporter yielded 70 to 80% as many transductants as 2 μg (data not shown); mean values for the three experiments ranged from 72% for NP-G to 79% for NA-G. This confirmed that packaging of each of the five vectors was near saturation in these studies. We then analyzed this data set with regard to two separate parameters for which the random and specific packaging models gave divergent predictions.
In the first analysis, we compared the total yield of transductants produced by cotransfecting pairs of homologous or nonhomologous reporters. Under the random packaging model, in which all reporters compete equally for a common niche, the yield of transductants from 1 μg of any two reporters is expected to approximate the yield from 2 μg of either reporter alone, since 2 μg is sufficient to saturate the niche. (To the extent that the apparent niches for two reporters are not identical, the predicted value for the pair would lie in the range between their individual yields.) Under specific packaging, this prediction applies only to homologous vRNAs, whereas the yield from any pair of nonhomologous reporters, by contrast, is expected to equal the sum of the yields from 1 μg of each reporter alone.
Table 2 presents the relevant data from our study, with actual transductant yields listed alongside those predicted by the random and specific models. Among the 18 transfections listed, which include both homologous and nonhomologous vRNA pairings, 12 gave transduction yields that fell within the range predicted under the random model; of the remainder, 4 exceeded this range and 2 fell below it. Based on these results, the random model could not be excluded on statistical grounds, either for the entire group (P = 0.12 by sign test) or for either the homologous or nonhomologous groups individually (P = 0.31 and 0.12, respectively). Of the nonhomologous vRNA pairs, however, all 12 produced transductant yields lower than that predicted for specific packaging (P < 0.001). The results of this analysis are therefore statistically compatible with the random, but not with the specific, model.
TABLE 2.
Cotransfection of homologous and nonhomologous reporter pairs in near-saturating amounts
| Reporter pair | Expt. no. | Total transductant yielda
|
||
|---|---|---|---|---|
| Predicted
|
Actual | |||
| Random model | Specific model | |||
| Homologous | ||||
| NP-Y + NP-G | 1 | 2,331-2,334 | Same | 2,280 |
| 2 | 1,545-2,479 | Same | 1,769 | |
| 3 | 1,103-1,300 | Same | 1,206 | |
| NA-Y + NA-G | 1 | 1,013-1,308 | Same | 1,360 |
| 2 | 742-1,977 | Same | 1,077 | |
| 3 | 589-1,033 | Same | 1,229 | |
| Nonhomologous | ||||
| NP-Y + NA-G | 1 | 1,013-2,334 | 2,509 | 1,835 |
| 2 | 1,545-1,977 | 2,717 | 1,966 | |
| 3 | 589-1,103 | 1,270 | 1,125 | |
| NP-Y + NS-G | 1 | 848-2,334 | 2,349 | 1,930 |
| 2 | 1,486-1,672 | 2,708 | 1,589 | |
| 3 | 642-1,103 | 1,156 | 849 | |
| NP-G + NA-Y | 1 | 1,308-2,331 | 3,033 | 2,198 |
| 2 | 742-2,479 | 2,071 | 1,400 | |
| 3 | 1,033-1,300 | 1,576 | 999 | |
| NA-Y + NS-G | 1 | 848-1,308 | 1,741 | 1,312 |
| 2 | 742-1,672 | 2,076 | 1,050 | |
| 3 | 642-1,033 | 989 | 847 | |
Number of yellow, green, and dual-colored MDCK transductants per 20,000 cells. Actual yield was obtained by cotransfecting 1 μg each of the two plasmids indicated. Predictions under the random model were calculated as the range between the the yields obtained by transfecting 2 μg of either the GFP (underlined) or YFP plasmid alone in the same experiment. For statistical analysis, the sign test was performed with respect to the center (mean) of the range. For nonhomologous pairs, predictions under the specific model represent the sum of the yields obtained by transfecting 1 μg of each plasmid alone in the same experiment. For homologous pairs, the predictions are the same as under the random model.
Our second analysis focused on virions that copackaged and transduced two reporter vRNAs simultaneously, producing singly infected MDCK cells that expressed both yellow and green fluorescence. We had previously observed a small percentage of such dual transducers among virions carrying homologous pairs of NP or NA reporters (Fig. 1B); the low multiplicity of infection used in these studies made it unlikely that these were an artifact of dual infection. Neither the random nor the specific packaging model can be used to predict the absolute frequency of dual transductions a priori. Under the specific model at saturating conditions, however, copackaging would be expected to occur more frequently with nonhomologous than with homologous vRNAs, since only the latter would compete for packaging into a common niche. Random packaging, on the other hand, predicts equal frequencies with both sets of vRNAs. The actual frequencies of dual transductions from our data set are depicted in Fig. 4. The frequencies observed for homologous and nonhomologous reporters were statistically indistinguishable (P = 0.30 by Student's t test), though there was a nonsignificant trend toward lower frequencies with nonhomologous (mean, 2.9% ± 0.37% standard error of the mean [SEM]) compared to homologous (3.6% ± 0.56%) reporter pairs for the study as a whole.
FIG. 4.

Frequency of dual transductants. Dual transductant MDCK cells are expressed as a percentage of total transductants for combinations of homologous (Hom) and nonhomologous (Non-Hom) reporters. Data were pooled from three independent experiments.
Together, these results indicate that vRNAs may compete for packaging into influenza virus virions but that this competition is not vRNA specific. Certain limitations of our study must be noted. First, the virions that we examined were produced with a plasmid-directed assembly system. Although this system yields authentic, replication-competent virus, the first round of assembly and packaging events which we studied involved vRNAs and proteins expressed from heterologous promoters and may not accurately mimic those of a natural infection. While previous studies of influenza virus packaging have often focused on viral supernatants, which contain a preponderance of nonviable particles, ours scored only particles that could support at least one round of infection and transduction and hence had some level of biological activity. The proportion of those particles that were fully infectious and replication competent, however, is unknown and likely to be small (data not shown).
Recent data suggest that, at least for defective interfering mutants of one segment, stable propagation through multiple rounds of packaging requires additional 5′ sequences that were not included in our reporters (5). We do not know whether a single transduced copy of reporter vRNA can confer detectable fluorescence; if multiple copies must be transduced by one virion, specific packaging of the first reporter might be obscured in our analysis by subsequent copies packaged randomly. Our analysis, moreover, considered only three of the eight vRNAs and only the extreme alternatives of random or specific packaging. We cannot exclude more sophisticated models in which particular vRNAs might be packaged in a hierarchical, length-dependent, or interdependent manner.
Our results nevertheless add to the preponderance of evidence that influenza virus packages its segments nonspecifically. The recent finding that critical packaging signals map to within the UTR sequences common to all influenza virus vRNAs lends particularly strong support to this view (28). Though seemingly inefficient, a random packaging mechanism could in theory suffice if each influenza virus virion incorporates, on average, more than eight vRNAs. Other authors have calculated, for example, that random packaging of 10 to 12 vRNAs per particle would enable 2.8 to 9.3% of virions to acquire the full complement of eight (7, 26); those values are compatible with the estimated 5 to 10% ratio of infectious to noninfectious particles found in actual influenza virus populations (2, 14).
Two separate groups have isolated and characterized influenza virus strains carrying at least nine or ten vRNAs, respectively (7, 25), and the maximum number that can be accommodated stably is unknown. In this regard, we note that when we formed viruses in the presence of both GFP- and YFP-tagged versions of a given vRNA at equal concentrations, roughly 3 to 5% of transduction-competent virions incorporated both reporters (Fig. 1 and 4). Assuming that the two reporters were packaged equivalently, this implies that an additional 3 to 5% of virions carry two (or more) copies of each reporter alone, so that a total of 9 to 15% of the population carries at least two reporter vRNAs. Under a random packaging scheme, the probability (p) that a given virion will encapsidate r copies of any given vRNA can be calculated with the equation:
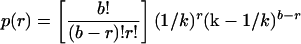 |
where b is the number of vRNAs acquired randomly by each virion and k is the number of unique vRNA species available for packaging. In our experiments, k = 9, as each virus selects from among eight wild-type vRNAs and one pair of homologous reporter vRNAs that are assumed to be equivalent. Under these conditions, five to seven segments must be packaged randomly into each virion in order to ensure that 10 to 15% will incorporate two or more copies of the reporter. (The calculated probabilities are 6.4, 9.8, 13.5, and 17.8% for b = 4, 5, 6, and 7, respectively.) Our assay scored only transduction-competent virions, however, all of which presumably must contain either three or four specific wild-type vRNAs (i.e., those that encode the three components of the viral polymerase and possibly NP), as these are likely required for efficient transcription and expression of the reporter (4, 11, 12, 15, 18). By virtue of the scoring process, these four would not be considered part of the randomly packaged pool, but rather must be added to the critical value of the five to seven segments determined above. Our results are therefore most compatible with the view that, on average, each influenza A virus virion packages 9 to 11 vRNAs and does so in a nonspecific manner.
Acknowledgments
We thank Y. Kawaoka and G. Hobom for the influenza virus plasmids, U. Koehn and P. Bacchetti for statistical advice, J. Gordon and S. Elmes for assistance with flow cytometry, and J. Regan for critical review of the manuscript.
This work was supported by NIH grant AI-40317.
REFERENCES
- 1.Chen, W., P. A. Calvo, D. Malide, J. Gibbs, U. Schubert, I. Bacik, S. Basta, R. O'Neill, J. Schickli, P. Palese, P. Henklein, J. R. Bennink, and J. W. Yewdell. 2001. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 7:1306-1312. [DOI] [PubMed] [Google Scholar]
- 2.Compans, R. W., and P. W. Choppin. 1975. Reproduction of myxoviruses, p. 179-252. In H. Fraenkel-Conrat and R. R. Wagner (ed.), Comprehensive virology, vol. 6. Plenum Press, New York, N.Y. [Google Scholar]
- 3.Compans, R. W., N. J. Dimmock, and H. Meier-Ewert. 1970. An electron microscopic study of the influenza virus-infected cell, p. 87-109. In R.D. Barry and B. W. J. Mahy (ed.), The biology of large RNA viruses. Academic Press, London, England.
- 4.de la Luna, S., J. Martin, A. Portela, and J. Ortin. 1993. Influenza virus naked RNA can be expressed upon transfection into cells coexpressing the three subunits of the polymerase and the nucleoprotein from simian virus 40 recombinant viruses. J. Gen. Virol. 74:535-539. [DOI] [PubMed] [Google Scholar]
- 5.Duhaut, S., and N. J. Dimmock. 2000. Approximately 150 nucleotides from the 5′ end of an influenza A virus segment 1 defective virion RNA are needed for genome stability during passage of defective virus in infected cells. Virology 275:278-285. [DOI] [PubMed] [Google Scholar]
- 6.Duhaut, S. D., and J. W. McCauley. 1996. Defective RNAs inhibit the assembly of influenza virus genome segments in a segment-specific manner. Virology 216:326-337. [DOI] [PubMed] [Google Scholar]
- 7.Enami, M., G. Sharma, C. Benham, and P. Palese. 1991. An influenza virus containing nine different RNA segments. Virology 185:291-298. [DOI] [PubMed] [Google Scholar]
- 8.Fodor, E., L. Devenish, O. G. Engelhardt, P. Palese, G. G. Brownlee, and A. Garcia-Sastre. 1999. Rescue of influenza A virus from recombinant DNA. J. Virol. 73:9679-9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez, S., and J. Ortin. 1999. Distinct regions of influenza virus PB1 polymerase subunit recognize vRNA and cRNA templates. EMBO J. 18:3767-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffmann, E., G. Neumann, Y. Kawaoka, G. Hobom, and R. G. Webster. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. USA 97:6108-6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang, T. S., P. Palese, and M. Krystal. 1990. Determination of influenza virus proteins required for genome replication. J. Virol. 64:5669-5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimura, N., M. Nishida, K. Nagata, A. Ishihama, K. Oda, and S. Nakada. 1992. Transcription of a recombinant influenza virus RNA in cells that can express the influenza virus RNA polymerase and nucleoprotein genes. J. Gen. Virol. 73:1321-1328. [DOI] [PubMed] [Google Scholar]
- 13.Lamb, R. A. 1983. The influenza virus RNA segments and their encoded proteins. Springer, New York, N.Y.
- 14.Lamb, R. A., and P. W. Choppin. 1983. The gene structure and replication of influenza virus. Annu. Rev. Biochem. 52:467-506. [DOI] [PubMed] [Google Scholar]
- 15.Lee, M. T., K. Bishop, L. Medcalf, D. Elton, P. Digard, and L. Tiley. 2002. Definition of the minimal viral components required for the initiation of unprimed RNA synthesis by influenza virus RNA polymerase. Nucleic Acids Res. 30:429-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li, M. L., B. C. Ramirez, and R. M. Krug. 1998. RNA-dependent activation of primer RNA production by influenza virus polymerase: different regions of the same protein subunit constitute the two required RNA-binding sites. EMBO J. 17:5844-5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luytjes, W., M. Krystal, M. Enami, J. D. Pavin, and P. Palese. 1989. Amplification, expression, and packaging of foreign gene by influenza virus. Cell 59:1107-1113. [DOI] [PubMed] [Google Scholar]
- 18.Mena, I., S. de la Luna, C. Albo, J. Martin, A. Nieto, J. Ortin, and A. Portela. 1994. Synthesis of biologically active influenza virus core proteins with a vaccinia virus-T7 RNA polymerase expression system. J. Gen. Virol. 75:2109-2114. [DOI] [PubMed] [Google Scholar]
- 19.Nakajima, K., M. Ueda, and A. Sugiura. 1979. Origin of small RNA in von Magnus particles of influenza virus. J. Virol. 29:1142-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nayak, D. P., T. M. Chambers, and R. M. Akkina. 1989. Structure of defective-interfering RNAs of influenza viruses and their role in interference, p. 269-317. In R. M. Krug (ed.), The influenza viruses. Plenum Press, New York, N.Y.
- 21.Nayak, D. P., N. Sivasubramanian, A. R. Davis, R. Cortini, and J. Sung. 1982. Complete sequence analyses show that two defective interfering influenza virus RNAs contain a single internal deletion of a polymerase gene. Proc. Natl. Acad. Sci. USA 79:2216-2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neumann, G., T. Watanabe, H. Ito, S. Watanabe, H. Goto, P. Gao, M. Hughes, D. R. Perez, R. Donis, E. Hoffmann, G. Hobom, and Y. Kawaoka. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. USA 96:9345-9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Odagiri, T., and K. Tobita. 1990. Mutation in NS2, a nonstructural protein of influenza A virus, extragenically causes aberrant replication and expression of the PA gene and leads to generation of defective interfering particles. Proc. Natl. Acad. Sci. USA 87:5988-5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parvin, J. D., P. Palese, A. Honda, A. Ishihama, and M. Krystal. 1989. Promoter analysis of influenza virus RNA polymerase. J. Virol. 63:5142-5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scholtissek, C., W. Rohde, E. Harms, R. Rott, M. Orlich, and C. B. Boschek. 1978. A possible partial heterozygote of an influenza A virus. Virology 89:506-516. [DOI] [PubMed] [Google Scholar]
- 26.Sekellick, M. J., S. A. Carra, A. Bowman, D. A. Hopkins, and P. I. Marcus. 2000. Transient resistance of influenza virus to interferon action attributed to random multiple packaging and activity of NS genes. J. Interferon Cytokine Res. 20:963-970. [DOI] [PubMed] [Google Scholar]
- 27.Smith, G. L., and A. J. Hay. 1982. Replication of the influenza virus genome. Virology 118:96-108. [DOI] [PubMed] [Google Scholar]
- 28.Tchatalbachev, S., R. Flick, and G. Hobom. 2001. The packaging signal of influenza virus RNA molecules. RNA 7:979-989. [DOI] [PMC free article] [PubMed] [Google Scholar]