

Abstract
Evidence that NSP2 plays a role in packaging and replication comes from studies on tsE(1400), a rotavirus mutant with a temperature-sensitive (ts) lesion in the NSP2 gene. Cells infected with tsE and maintained at nonpermissive temperature contain few replication-assembly factories (viroplasms) or replication intermediates and produce virus particles that are mostly empty. Sequence analysis has indicated that an A152V mutation in NSP2 is responsible for the ts phenotype of tsE. To gain insight into the effect of the mutation on the octameric structure and biochemical activities of tsE NSP2, the protein was expressed in bacteria and purified to homogeneity. Analytical ultracentrifugation showed that tsE NSP2 formed octamers which, like those formed by wild-type (wt) NSP2, undergo conformational change into more compact structures upon binding of nucleotides. However, exposure to Mg2+ and the nonpermissive temperature caused disruption of the tsE octamers and yielded the formation of polydisperse NSP2 aggregates, events not observed with wt octamers. Biochemical analysis showed that the RNA-binding, helix-destabilizing and NTPase activities of tsE NSP2 were significantly less at the nonpermissive temperature than at the permissive temperature. In contrast, these activities for wt NSP2 were higher at the nonpermissive temperature. Our results indicate that the octamer is the fully functional form of NSP2 and the form required for productive virus replication. The propensity of tsE NSP2 to form large aggregates provides a possible explanation for the inability of the protein to support packaging and/or replication in the infected cell at the nonpermissive temperature.
Rotaviruses are a significant cause of diarrheal disease in humans and other animals (12). These viruses are members of the Reoviridae and contain genomes consisting of 11 segments of double-stranded RNA (dsRNA) that encode six structural proteins and six nonstructural proteins (7). The rotavirion is a triple-layer icosahedron formed by the outer-layer proteins, VP7 and VP4, the middle-layer protein, VP6, and the core matrix protein, VP2 (25). Closely associated with the VP2 matrix are the RNA-dependent RNA polymerase, VP1 (40), and the multifunctional capping enzyme, VP3 (4). During the replication cycle, viral mRNAs serve as templates for the synthesis of minus-strand RNA to form the dsRNA genome (22). Synthesis of the genome segments occurs concurrently with the packaging of the mRNAs into core-like replication intermediates (RIs) consisting of not only the structural but also the nonstructural proteins (9). The formation of RIs and the replication of the dsRNA genome occur in large cytoplasmic inclusions (viroplasms) that form in infected cells.
The roles of the nonstructural proteins in RNA packaging and replication are unknown. However, studies of tsE(1400), a simian SA11 rotavirus mutant with a temperature-sensitive (ts) lesion mapping to the gene for NSP2 (10), indicate that this protein plays an important role in these processes. Specifically, at the nonpermissive temperature (39°C), despite the production of viral proteins, tsE-infected cells are defective in the production of viral single-stranded RNA (ssRNA) and dsRNA (3) and produce virus particles that are mostly empty (28). Likewise, the formation of viroplasms is defective at the nonpermissive temperature in tsE-infected cells (28). The essential nature of NSP2 in the formation of viroplasms was demonstrated by the observation that expression of NSP2 and NSP5 together, but not individually, in uninfected cells generates viroplasm-like inclusions (8). The function of the interaction of NSP2 with NSP5 is not clear, but the event does cause the hyperphosphorylation of NSP5 (1, 41) and in doing so may modulate its activity. Protein-protein cross-linking studies have suggested that in addition to NSP5, NSP2 interacts with the viral polymerase VP1 (14).
NSP2 (mass, 35 kDa) is a nonspecific ssRNA-binding protein (13, 39) that self-assembles into stable octamers, probably formed by the interaction of NSP2 tetramers (36). The octamers bind to ssRNA in a cooperative fashion, giving rise to higher-order complexes composed of a single RNA molecule and multiple copies of the octamer (39). NSP2 octamers also possess a nucleotide- and Mg2+-independent helix-destabilization activity (38), a function that has been suggested to remove RNA-RNA duplexes in viral mRNAs that may inhibit RNA packaging and replication (26). Besides RNA-binding activity, NSP2 octamers display an associated Mg2+-dependent NTPase activity that can catalyze the hydrolysis of each of the four nucleoside triphosphates (NTPs) (39) and that may provide energy for packaging. In the presence of Mg2+ and nucleotides, NSP2 octamers undergo a conformational change into a more condensed form (36). The presence of Mg2+ alone promotes the dissociation of the octamers into tetramers (36).
To provide a better understanding of the role of NSP2 in rotavirus replication, the ts form of the protein encoded by tsE(1400) (tsE NSP2) was expressed in bacteria and purified to homogeneity. Analysis of tsE NSP2 by analytical ultracentrifugation indicated that the protein formed octamers similar to those of the wild-type (wt) protein. However, unlike wt NSP2, tsE NSP2 in the presence of Mg2+ underwent strong temperature-dependent aggregation, most likely representing the inappropriate interaction of tetramers. Furthermore, unlike the wt protein, tsE NSP2 displayed a decrease in ssRNA-binding, helix-destabilizing, and NTPase activities at temperatures that are restrictive for the growth of tsE in vivo. The loss of the structural integrity of the tsE NSP2 octamers with increasing temperatures correlates well with the loss of its associated biochemical activities. We propose that the inability of tsE NSP2 to support packaging and dsRNA synthesis at the nonpermissive temperature in vivo is due to the failure of the protein to form the octameric unit.
MATERIALS AND METHODS
Cells and viruses.
The tsE(1400) virus was generated by chemical mutagenesis of SA11 rotavirus (27). The ts lesion was mapped to the gene encoding NSP2 (gene 8 for SA11) by analysis of the ts phenotypes of reassortant viruses produced upon coinfection with tsE(1400) and the Wa strain of rotavirus (10).
To obtain the revertants RtsE(1400)-R1 and RtsE(1400)-R2, individual plaque isolates of tsE(1400) were amplified by propagation at the permissive temperature (31°C) in MA104 cells. The virus clones were then independently plated at the nonpermissive temperature (39°C), and a revertant plaque for each clone was picked. Revertant viruses were subjected to two additional rounds of plaque purification prior to amplification to high-titer stocks. The phenotype of the revertants was confirmed by plaque assay at 31 and 39°C.
Cloning of gene 8 of tsE.
Gene 8 cDNAs of tsE(1400), RtsE(1400)-R1, and RtsE(1400)-R2 were prepared from viral mRNAs made by transcriptionally active double-layer virus particles (17) or from purified genomic dsRNA (24). The reverse-transcription and amplification reaction mixtures contained oligonucleotide primers representing the 5′ and 3′ ends of SA11 gene 8 RNA (GenBank accession number P03537). The products of PCRs were ligated into the vector pCR1000 (Invitrogen), producing [pCRg8(tsE)], or pCR2.1-Topo and transformed into Escherichia coli DH5α. Bacteria containing the appropriate plasmid were identified based on antibiotic resistance, plasmid size, and restriction enzyme digestion. The sequences of the gene 8 RNA of tsE(1400) were derived from five independently generated gene 8 cDNAs. Two independently generated cDNAs were used to obtain the sequences of the gene 8 RNAs of each tsE revertant. Sequences were determined by automated sequencing using an ABI 310 or 3100 Genetic Analyzer (PE Applied Biosystems) or with a Sequenase 2.0 DNA sequencing kit (Amersham) and suitable oligonucleotide primers (30).
The tsE gene 8 cDNA insert was recovered from PCRg8(tsE) by digestion with EcoRI and HindIII and was ligated into the EcoRI and HindIII sites of SP72. Following transformation of E. coli DH5α, bacteria containing the appropriate plasmid [pSP72g8(tsE)] were identified based on antibiotic resistance, plasmid size, and restriction enzyme digestion.
Construction of the expression vector, pQE60g8(tsE).
The tsE gene 8 insert in the vector [pSP72g8(tsE)] was subcloned into the vector pQE60 (Qiagen) following the same protocol described previously for the construction of pQE60g8 (39). The accuracy of the tsE gene 8 sequence in pQE60 was confirmed by automated sequencing. In pQE60g8(tsE), the open reading frame encoding tsE NSP2 is situated immediately upstream from six in-frame codons for His. Thus, the recombinant tsE NSP2 expressed from pQE60g8(tsE) contains a C-terminal His tag.
Expression and purification of tsE NSP2.
tsE NSP2 (R127, V152, I200) was expressed in E. coli M15(pREP4) containing pQE60g8(tsE) and purified using a Ni2+-nitrilotriacetic acid-agarose column as previously described (36). The protein eluted from the column was dialyzed extensively against 10 mM Tris-HCl, pH 7.2, 10 mM NaCl, 0.5 mM dithiothreitol (DTT), and 0.5 mM EDTA. Analysis of the protein sample by electrophoresis on sodium dodecyl sulfate (SDS)-polyacrylamide gels and Coomassie blue staining indicated that its purity was close to 100%. The concentration of the purified recombinant protein was determined by coelectrophoresis with known amounts of bovine serum albumin.
Purified 35S-labeled wt and tsE NSP2 was produced in bacteria as described above except that protein expression was induced in Cys-free, Met-free Dulbecco's modified Eagle's medium containing 20 μCi of 35S-amino acids (35S-Express [NEN]; 1,175 Ci/mmol) per ml of medium.
Sedimentation equilibrium.
Sedimentation equilibrium studies were conducted with a Beckman Optima XL-I ultracentrifuge, using an An50 Ti eight-hole rotor and interference optics. Double-sector charcoal-filled epon centerpieces were filled with 110 μl of sample at concentrations of 2 to 5 μM. Sedimentation equilibrium was performed at a rotor temperature of 8°C and at several rotor speeds between 3,000 and 16,000 rpm. The partial-specific volume of the protein and the buffer density and viscosity were calculated with the program Sednterp (16). No thermodynamic nonidealities were observed, and the data analysis was based on Boltzmann distributions of ideal species in the centrifugal field.
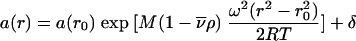 |
where a(r) denotes the experimentally measured fringe displacement at the distance r from the center of rotation, M and 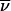 denote the protein molar mass and partial specific volume, ρ denotes the solvent density, ω denotes the angular velocity of the rotor, T denotes the absolute temperature, R denotes the gas constant, and r0 denotes an arbitrary reference radius (37).
denote the protein molar mass and partial specific volume, ρ denotes the solvent density, ω denotes the angular velocity of the rotor, T denotes the absolute temperature, R denotes the gas constant, and r0 denotes an arbitrary reference radius (37).
Sedimentation velocity.
Sedimentation velocity experiments were conducted using an An50 Ti eight-hole rotor and an interference or absorbance optical detection system. Double-sector charcoal-filled epon centerpieces were filled with 350 μl of sample at concentrations between 1 and 10 μM. The rotor temperature was set between 18 and 22°C, chosen to minimize temperature changes during a 1- to 2-h equilibration period of the rotor in the vacuum chamber prior to the start of the run. The rotor was then accelerated to the experimental rotor speeds of between 30,000 and 50,000 rpm at maximal rate. Fringe displacement profiles were acquired in intervals of 30 to 40 s.
The analysis of the sedimentation velocity profiles was performed by direct boundary modeling by solutions of the Lamm equation:
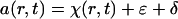 |
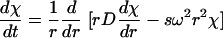 |
with a(r,t) denoting the experimentally measured absorbance, ɛ and δ denoting the systematic and random signal offset, s and D denoting the sedimentation and diffusion coefficients of the protein, and χ denoting its concentration at position r and time t (15). This was combined with the algebraic calculation of systematic time-invariant and radial-invariant noise components ɛ (33). Because these systematic signals are arbitrary offsets introduced from the detection system, calculated systematic offsets can be subtracted from the raw data without introduction of bias, if the degrees of freedom in the analysis are not reduced. Therefore, the final calculated best-fit offsets are subtracted from the raw data for clarity of presentation. The Lamm equation solutions were calculated by using the finite-element and moving-frame-of-reference method as described previously (5, 31, 34), at a radial increment of approximately 0.001 cm. The finite element simulations were modified to include the consideration of the acceleration phase of the rotor (36). In the discrete species models, the location of the meniscus was first graphically initialized and then treated as a floating parameter and optimized in the nonlinear regression of the data. The resulting meniscus position was then used in the continuous size-distribution analysis (see below). Models with discrete species were fit globally to sedimentation velocity data acquired at different rotor speeds, treating the species concentrations as local parameters and their molar mass and sedimentation coefficient values as global parameters.
The sedimentation coefficient distributions were calculated with the c(s) method by direct modeling with distributions of Lamm equation solutions:
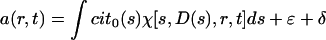 |
(where c0(s)ds is the loading concentration of species with a sedimentation coefficient between s and s + ds). Following the procedure outlined in detail elsewhere (32, 35), the integral was solved by discretization into 250 to 300 s values between 4 S and 18 S (unless noted otherwise), and a value of 1.3 for the frictional ratio f/f0 was used for estimating the average diffusional broadening of the sedimentation boundaries D(s). Maximum entropy regularization was used to calculate the simplest size distribution within a confidence level of 0.68 of the best-fit distribution (32). Analysis of the continuous sedimentation coefficient distribution was performed with the software Sedfit.
In vitro synthesis of RNAs.
The DNA template for synthesis of the Luc200 RNA probe was generated by amplifying a portion of the luciferase gene in the plasmid pGL2 (Promega) with Taq DNA polymerase (Invitrogen) and the positive-sense primer, 5′-TAATACGACTCACTATACCATGGAAGACGCCAAAAACATAAAGAAAGG-3′ (the T7 promoter sequence is underlined), and the negative-sense primer, 5′-CATTTCGAAGTACTCAGC-3′. 32P-labeled Luc200 probe was transcribed from the amplified DNA with an Ambion MEGAshortscript kit according to the protocol of the manufacturer except that the concentration of cold UTP was reduced to 1.875 mM and that 10 μCi of [α-32P]UTP (800 Ci/mmol) was included per 20 μl of reaction mixture (21). The 32P-labeled RNA probes were purified by electrophoresis on and elution from 8% polyacrylamide gels containing 7 M urea (20).
To produce the A11-StyI RNA, SP65g8.5′-3′SacII (21) was linearized with StyI, blunt ended by treatment with T4 DNA polymerase, and transcribed with T7 RNA polymerase using an Ambion MAXIscript kit (23). After DNase treatment, the RNA products were purified by phenol-chloroform extraction and isopropanol precipitation. The A11-StyI RNA was purified by electrophoresis on and elution from 8% polyacrylamide gels containing 7 M urea (20). RNA concentrations were calculated from optical densities at 260 nm.
Gel shift assays.
The procedure used for analysis of rNSP2-RNA interactions by gel shift assay was similar to that described earlier (21). wt or tsE NSP2 was preincubated in low-salt buffer (LSB) (2 mM Tris-HCl (pH 7.2), 0.5 mM EDTA, 0.5 mM DTT) in a final volume of 15 μl for 30 min at 31 or 39°C in the presence or absence of 100 mM NaCl. After addition of 32P-labeled Luc200 RNA, the reactions were incubated for an additional 30 min. The reaction mixtures were analyzed by electrophoresis for 3 to 4 h at 175 V on nondenaturing 6% polyacrylamide gels in Tris-glycine buffer (21). Protein-probe complexes were detected on the gel by autoradiography, and the intensity of the radiolabeled bands was quantified with a Molecular Dynamics PhosphorImager.
Preparation of DNA-RNA duplexes for strand-displacement assays.
To prepare the 5′-end-labeled DNA oligonucleotide 18AD (5′-GCCGCTCAAACGGCGACTGAGGAT-3′), a 50-μl reaction mixture containing 100 pmol of the oligonucleotide, 10 μCi of [γ-32P]ATP (3,000 Ci/mmol; NEN), and 20 U of T4 polynucleotide kinase (Invitrogen) was incubated at 37°C for 1 h. The reaction was terminated by incubation at 65°C for 20 min. Unincorporated nucleotides were removed from the reaction mixture by centrifugation though Sephadex G-25 spin columns (Roche). Following electrophoresis on 20% polyacrylamide gels containing 7 M urea, the concentrations of the end-labeled oligonucleotides were estimated using a phosphorimager.
The following procedure was used to prepare the DNA-RNA duplex, A11-StyI-18AD. Twenty picomoles of 5′-end-labeled DNA oligonucleotide, 18AD, was mixed with 40 to 200 pmol of unlabeled A11-StyI RNA in buffer containing 10 mM Tris-HCl, pH 8.0, and 200 mM NaCl. After heating to 95°C for 5 min, the mixture was cooled gradually to 25°C over a 5-h period. The quality of the duplexes was assessed by electrophoresis on nondenaturing 20% polyacrylamide gels in Tris-glycine buffer (19). Concentrations of duplexes resolved by electrophoresis were determined by comparison with known amounts of radiolabeled oligonucleotides using a phosphorimager.
Strand-displacement assay.
Fifty or 200 pmol of either wt or tsE NSP2 was preincubated for 30 min at 31 or 39°C in buffer containing 25 mM HEPES-KOH [pH 7.5], 20 mM NaCl, and 1 mM DTT (38). After addition of 0.25 pmol of a radiolabeled DNA-RNA duplex, incubation was continued for another 30 min. The reaction mixtures were incubated with 40 μg of proteinase K for 15 min at 37°C, and digestion was terminated by the addition of an equal volume of sample buffer (50 mM Tris-HCl [pH 8.8], 50 mM glycine, 20 mM EDTA, 0.2% SDS, 0.04% Triton X-100, 25% glycerol, bromophenol blue). The reaction products were analyzed by electrophoresis on nondenaturing 20% polyacrylamide gels, visualized by autoradiography, and quantitated with a phosphorimager.
NTPase assay.
wt or tsE NSP2 in 50 mM Tris-HCl (pH 7.5) and 5 mM MgCl2 in a final volume of 20 μl were preincubated for 30 min at 31 or 39°C. After addition of 10 μCi of [α-32P]UTP (3,000 Ci/mmol; Amersham), the reaction mixtures were incubated for another 30 min (39). One microliter of 0.5 mM EDTA was added to the mixtures, and the samples were then deproteinized by phenol-chloroform extraction. A 1-μl aliquot of each reaction mixture was spotted onto PEI-cellulose F sheets (EM Science), and UTP, UDP, and Pi were resolved by ascending thin-layer chromatography in 1.2 M LiCl. Radiolabeled spots on the sheets were detected by autoradiography and quantitated with a phosphorimager. UDP and UMP markers were made by hydrolysis of 10 μCi of [α-32P]UTP with 5 U of calf intestinal phosphatase (Invitrogen) and 5 U of tobacco acid pyrophosphatase (Epicentre), respectively.
RESULTS
Sequence analysis of tsE NSP2.
Sequencing of gene 8 cDNAs showed that NSP2 encoded by the tsE(1400) mutant (tsE NSP2) differed from that encoded by the parental strain of SA11 virus (wt NSP2) (Q03243) by two (A152V and V200I) and, less frequently, three amino acids (H127R, A152V, and V200I) (L20901). Inspection of all wt NSP2 amino acid sequences of group A rotaviruses reported to date (simian SA11 [Q03242, P03537], human Wa [Q03245], DS1 [Q03240],IS2 [X94562], A [S30581], and KU [AB022770], porcine OSU [P09366], bovine BRV [S49005], UK [P03538], and NCDV [Q03241], and avian PO-13 [AB009625] and Ty-1 [Q03244]) revealed that A152 was strictly conserved and that H127 was present in all mammalian isolates but was an alanine in avian isolates. The amino acid at position 200 was variable in identity. While most commonly a valine or serine, in some cases a lysine (avian) or alanine (porcine) was at position 200.
The fact that sequencing indicated that the H127R mutation was not a common feature of all NSP2 produced by tsE suggested that this amino change was not responsible for the ts phenotype of the virus. Further insight into the amino acid changes responsible for the phenotype of tsE was gained by sequencing gene 8 cDNAs prepared from the tsE revertant viruses RtsE-R1 and RtsE-R2. The analysis showed that NSP2 of both revertants contained the wild-type A152 residue instead of the V152 found in tsE NSP2. In contrast, the consensus amino acid sequence of NSP2 encoded by both revertants included the same V200I mutation found in tsE NSP2. The NSP2 consensus sequences of both revertants included the wild-type H127 residue and were otherwise identical to the sequence of wt NSP2. Collectively, these data indicate that it is the A152V mutation and not other changes in tsE NSP2 which are responsible for the phenotype of tsE.
Expression of recombinant tsE NSP2.
tsE NSP2 and wt NSP2 were each expressed with a C-terminal histidine tag in E. coli using isopropyl-β-d-thiogalactopyranoside-inducible expression vectors. Following induction, bacteria were maintained in medium containing [35S]methionine at 31°C, the permissive temperature for tsE, or at 37°C, close to the 39°C nonpermissive temperature for tsE. After expression, the recombinant proteins were purified from the soluble fraction of the bacterial lysates under nondenaturing conditions by Ni2+-affinity chromatography. The protein eluted from the columns was evaluated by SDS-polyacrylamide gel electrophoresis and autoradiography (Fig. 1). Under identical expression and purification conditions, the amount of recombinant protein recovered from bacteria expressing tsE NSP2 at 37°C was threefold less than from bacteria expressing wt NSP2, while at 31°C the difference was only ∼1.5-fold. These differences implied that tsE NSP2 may be more prone to aggregation at the higher temperature. Hence, tsE NSP2 and wt NSP2 expressed from bacteria at 31°C were used in the subsequent experiments described in the study.
FIG. 1.

Purification of recombinant wt and tsE NSP2. wt and tsE NSP2 were expressed in E. coli at 31°C in the presence of 35S-labeled methionine and purified by Ni2+-affinity chromatography. Portions of the soluble and insoluble fractions of the bacterial lysate and the purified protein (column eluate) were analyzed by SDS-polyacrylamide gel electrophoresis and autoradiography.
tsE NSP2 forms octamers in solution.
We showed previously that purified recombinant wt NSP2 is octameric and that Mg2+ induces dissociation of NSP2 octamers into tetramers (36). Higher sedimentation rates were reported for wt NSP2 in the presence of nucleotides, suggesting that the cofactor causes conformational changes in the octamer. To gain insight into the impact of the ts lesion on the structure of tsE NSP2, the oligomeric status of the purified recombinant protein was evaluated by velocity sedimentation at 20°C. The analysis showed that tsE NSP2 formed a predominant peak of 12.23 S, a result analogous to that obtained for wt NSP2 sedimented in the same run (Fig. 2). A comparison of the sedimentation coefficient [c(s)] distributions in the presence and absence of 0.75 mM ADP for wt and tsE NSP2 at 20°C showed that both proteins responded similarly to the presence of nucleotides (Fig. 2). Overall, the similarity of s values and responses to nucleotide binding indicates that tsE NSP2 is octameric and indistinguishable from wt NSP2. Analysis of the protein by equilibrium sedimentation at several centrifugation speeds in the absence of Mg 2+ and nucleotides showed profiles that fit well into a single species model with a molar mass of 308 kDa (data not shown), which is close to the 302 kDa expected for the octameric protein.
FIG. 2.

Multimeric status of tsE NSP2 and impact of ADP on sedimentation. Sedimentation coefficient distribution c(s) from the analysis of the sedimentation velocity profiles at 45,000 rpm (data not shown) in a solution containing 4 mM Tris-HCl (pH 7.2)-10 mM NaCl at 20°C for tsE NSP2 with (solid line) and without (dotted line) ADP. For comparison, the c(s) distribution for wt NSP2 with (dashed line) and without (dash-dotted line) ADP from the same run are also shown
Close inspection of the c(s) distribution profiles indicated that a few percent of the tsE NSP2 protein existed in forms smaller and larger than an octamer, suggesting that some of the protein had dissociated or aggregated. This possibility was further evaluated in a sequential sedimentation velocity experiment in which tsE NSP2 was initially sedimented at 45,000 rpm, followed 3 h later with mixing of the sample volume contained in the centrifugation cell and a second velocity run at 30,000 rpm (Fig. 3). An excellent global fit (root mean square deviation [rmsd] of 0.0043) of the results was obtained with a model for three species with the octamer as the main component, possessing a best-fit sedimentation coefficient of 12.24 S and a molar mass of 302 kDa. For the smaller species, we obtained an s value of 9.8 S, with a molar mass consistent with that of a hexamer, and for the larger species, the global fit converged to an apparent molar mass of 180 kDa and an s value of 17.2 S. The unphysical combination of the latter values clearly indicates that the aggregated material is heterogeneous. In earlier sedimentation velocity studies with wt NSP2, an ∼7-S tetramer species was also identified, whose presence was enhanced by exposure of the wt NSP2 octamers to Mg2+ (36). In addition, the earlier study identified an 8- to 10-S species of wt NSP2. The 8- to 10-S species was speculated to be a hexamer derived by the slow dissociation of the NSP2 octamer (36) and appears analogous to the 9.8-S species identified for tsE NSP2. Although the relative abundance of the 9.8- and 17.2-S species in the tsE NSP2 preparation was very small, it appeared to slowly increase with time. Thus, tsE NSP2 may have a greater tendency to aggregate than wt NSP2.
FIG. 3.

Global sedimentation velocity analysis of tsE NSP2. Direct boundary fit of sedimentation velocity data obtained during the first 7,200 s of sedimentation at 45,000 rpm (A and B) and during the first 15,200 s at 30,000 rpm (C and D) with tsE NSP2 in a solution containing 4 mM Tris-HCl (pH 7.2)-10 mM NaCl at 20°C. For clarity, only every 10th scan and 10th radial data point is shown (circles). Best-fit distributions are shown as solid lines and are based on a model for an octamer of 12.24 S, a hexamer of 9.8 S, and a large species of 17.2 S. Partial concentrations resulted in ∼96% of octamer, ∼2% of 9.7-S species, and 2% of large aggregate at 45,000 rpm, and 88% octamer, ∼10% of 9.8-S species, ∼2% of large aggregate at 30,000 rpm, respectively. Residuals are shown in panels B and D, with a global rmsd of 0.0043 fringes.
Aggregation of tsE NSP2 in the presence of Mg2+.
A qualitatively new sedimentation behavior of tsE NSP2 was observed when the protein was incubated with 4 mM MgCl2 at 20°C for 2 h prior to centrifugation. As illustrated in Fig. 4A, this treatment of tsE NSP2 produced a very broad c(s) distribution that included a number of separate peaks. Although, in general, the interpretation of peaks in broad c(s) distributions can be difficult because of possible regularization artifacts (35), in the present case the assignment can be made with confidence, as we have prior knowledge of the s values of the different oligomers. Accordingly, we observed that exposure to Mg2+ caused the dissociation of most tsE NSP2 octamers and generated tetramers (∼7 S), hexamers (9.8 S), and a series of higher oligomers of up to 30 S. If tsE NSP2 is incubated with Mg2+ as described above, except that 0.75 mM ADP is also present, the c(s) distribution was much more confined to the octameric state, with an s value shifted to ∼13 S (Fig. 4A, dotted line). This shows that ADP, as has been observed previously for wt NSP2 (36), acts to stabilize the protein in the octameric state.
FIG. 4.

Aggregation of tsE NSP2 in the presence of Mg2+. (A) Sedimentation coefficient distributions c(s) of tsE NSP2 in a solution containing 4 mM Tris-HCl (pH 7.2)-10 mM NaCl with 4 mM MgCl2 at 20°C, 45,000 rpm (solid line). Calculated c(s) distributions under the same conditions, but with 0.75 mM ADP, are shown as a dotted line. (B) Sedimentation coefficient distributions of tsE NSP2 from sedimentation velocity experiments at 45,000 rpm after incubation in a solution containing 4 mM Tris-HCl (pH 7.2)-10 mM NaCl with 4 mM MgCl2 for 1 h at 25°C (data offset by 0.15), 2 h at 20°C (data offset by 0.05), and 2 h at 37°C (data without offset). For comparison, the c(s) distribution obtained in the absence of MgCl2 (dashed line, data offset by 0.3) is shown. The vertical axis is in arbitrary units.
Temperature dependence of tsE NSP2 aggregation.
The possibility that the aggregation of tsE NSP2 may be a temperature-dependent event was then explored by comparing the c(s) distributions produced by velocity runs of the protein after incubation in the presence of 4 mM MgCl2 at different temperatures and for different times. As shown in Fig. 4B, a 1-h incubation at 25°C resulted in dissociation of ∼50% of octamers into tetramers, with some intermediate hexamers also visible. Incubation for 2 h at 20°C resulted in the further loss of octamers and the appearance of aggregates larger than the octamer. Incubation at higher temperature (37°C) for 2 h caused the formation of a broad distribution of large aggregates and a near-complete loss of octamers and tetramers.
To compare the effect of temperature on wt and tsE NP2, the proteins in 4 mM Tris-HCl (pH 7.2)-10 mM NaCl with 4 mM MgCl2 were incubated for 2 h on ice or at 37°C and then analyzed by sedimentation velocity at 20°C. The results showed that at both incubation temperatures, most wt NSP2 octamers were dissociated into tetramers (Fig. 5C and D). While most tsE NSP2 octamers incubated at the low temperature were also dissociated into tetramers, the extent of their dissociation was greater than that observed for wt NSP2 octamers (Fig. 5B). In contrast, when tsE NSP2 was incubated at the high temperature, octamers and tetramers were lost, and very large complexes sedimenting at 20 to >50 S appeared (Fig. 5A). Some evidence could be found for the presence of smaller species of 2.6 S in the preparation, presumably a tsE NSP2 dimer. A closer inspection of wt NSP2 incubated at 37°C (Fig. 5C) also revealed some partial aggregation, but only to a very limited extent. It is noteworthy that any octamer remaining after incubation at low or high temperature in the presence of Mg2+ does sediment, forming a separate boundary, and could be modeled by a multiple noninteracting species model, reconfirming our previous observation (36) that the time scale of NSP2 self-assembly is slow compared to results of the sedimentation velocity experiments.
FIG. 5.

Mg2+-induced aggregation of tsE NSP2 is temperature dependent. Velocity data during the first 2,200 s of sedimentation of tsE NSP2 (A and B) and wt NSP2 (C and D) at 45,000 rpm in a solution containing 4 mM Tris-HCl (pH 7.2)-10 mM NaCl with 4 mM MgCl2 at 20°C, after incubation of the samples on ice (B and D) or at 37°C (A and C) for a period of 2 h before sedimentation. Shown are data for tsE NSP2 (A and B) and for wt NSP2 (C and D) derived under identical conditions. The data are corrected for time-invariant noise as calculated by c(s) analysis (data not shown).
Overall, solution structure analysis of tsE NSP2 showed that although the protein was capable of forming stable octamers, it exhibited a tendency to aggregate. In the presence of Mg2+, dissociation of octamers into tetramers produced significant temperature-dependent aggregation of tsE NSP2, which may be due to inappropriate interactions between tsE NSP2 tetramers. Subsequent experiments were designed to assess if the tendency of tsE NSP2 to aggregate at the nonpermissive temperature affected the biochemical activities of the protein.
tsE NSP2 exhibits temperature-sensitive RNA binding.
The interaction of NSP2 with ssRNA leads to the formation of higher-order RNA-protein complexes, in which each ssRNA is bound by more than one NSP2 octamer (39). To determine whether the ability of NSP2 to bind ssRNA is affected by the ts lesion, increasing amounts (2 to 28 pmol) of tsE or wt NSP2 were each preincubated for 30 min at 31 or 39°C. Afterwards, 1 pmol of 32P-labeled Luc200, a nonviral RNA probe of 200 nucleotides was mixed with the proteins. To increase the stringency of RNA-protein interactions, 100 mM NaCl was also added to some of the mixtures. After incubation for an additional 30 min at the respective temperatures, NSP2-probe complexes in the mixtures were identified by nondenaturing gel electrophoresis and autoradiography. For each reaction mixture, the fraction of the probe bound by the protein was quantitated using a phosphorimager and plotted as a function of the amount of protein that was added to the reaction mixture.
As shown in Fig. 6A and B and Fig. 7A, when incubated at 31°C, tsE NSP2 bound ssRNA to form RNA-protein complexes indistinguishable from those formed by wt NSP2 at the same protein concentration. In contrast, when incubated at 39°C, tsE NSP2 bound less probe at any given concentration than wt NSP2 and exhibited a reduced ability to form higher-order RNA-protein complexes (Fig. 6C and D, comparing lanes 9 to 15, and Fig. 7C). These observed differences in the RNA-binding activity of the two proteins at the nonpermissive temperatures to form higher-order complexes were even more pronounced when the stringency of the reaction was increased by inclusion of NaCl in reaction mixtures (Fig. 6G and H, lanes 9 to 15, and Fig. 7D). Notably, the formation of lower-order RNA-protein complexes by tsE NSP2 at 39°C in reaction mixtures was relatively less affected by salt than the formation of higher-order complexes. However, regardless of whether or not NaCl was included in reaction mixtures incubated at 39°C, fewer lower-order RNA-protein complexes were formed by tsE NSP2 than by wt NSP2 (Fig. 6, C versus D and G versus H). Thus, tsE NSP2 was deficient in both the formation of lower- and higher-order RNA-protein complexes at the restrictive temperature.
FIG. 6.

Formation of large aggregates by tsE NSP2 at nonpermissive temperature affects RNA binding. Increments of 2 pmol of wt NSP2 (A, C, E, and G) or tsE NSP2 (B, D, F, and H) in LSB were incubated for 30 min in the presence (E, F, G, and H) or absence (A, B, C, and D) of 100 mM NaCl at 31°C (A, B, E, and F) or at 39°C (C, D, G, and H). Afterwards, 1 pmol of 32P-labeled Luc200 RNA was added to each reaction mixture, and incubation continued at the respective temperatures for an additional 30 min. RNA-protein complexes in the mixtures were resolved by nondenaturing gel electrophoresis and detected by autoradiography. Lane 1 of each panel represents 1 pmol of the radiolabeled Luc200 RNA resolved in the absence of protein. Lane 2 of each panel represents a reaction mixture with 2 pmol of protein. ori, gel origin; fp, free probe; RNP-I, lower-order RNA-protein complexes; RNP-II, higher-order RNA-protein complexes.
FIG. 7.

Quantitation of the ssRNA-binding activity of tsE and wt NSP2. From the results presented in Fig. 6, the fraction of the total radiolabeled RNA bound to protein in each reaction mixture was quantitated using a phosphorimager and plotted as a function of protein concentration. Open squares and diamonds represent the results of assays performed with wt NSP2 and tsE NSP2, respectively, in the absence of NaCl; closed squares and diamonds represent the results of assays performed with wt NSP2 and tsE NSP2 proteins, respectively, in the presence of 100 mM NaCl.
The presence of the radiolabeled probe at the origin of the gels shown in Fig. 6D and H suggests that a fraction of tsE NSP2 when incubated at 39°C forms very large RNA-protein complexes that are not able to migrate into the gel under the electrophoretic conditions. This conclusion is consistent with the results of the velocity sedimentation experiments, which indicated that higher temperatures promote the aggregation of tsE NSP2.
A quantitative comparison of the ssRNA-binding activities of tsE and wt NSP2 was done by calculating the relative RNA-binding activities of each protein at the permissive and nonpermissive temperatures in the presence or absence of NaCl. The relative RNA-binding activity was defined as the concentration of the protein at which half the RNA probe in the reaction was bound to the protein. As shown in Table 1, the relative RNA-binding activities of wt NSP2 and tsE NSP2 were similar at 31°C in both the presence and absence of 100 mM NaCl. However, a pronounced difference existed in the relative RNA-binding activities of wt NSP2 and tsE NSP2 at 39°C, in the presence and absence of NaCl. Specifically, binding of 50% of the RNA probe at 39°C in reaction mixtures without NaCl required a concentration of tsE NSP2 that was 38% higher than that of wt NSP2. When 100 mM NaCl was present in the reaction mixtures, the concentration of tsE NSP2 required to bind 50% of the probe was 50% higher than that of wt NSP2. These results show that at the nonpermissive temperature, tsE NSP2 fails to bind ssRNA as efficiently as wt NSP2.
TABLE 1.
Relative RNA-binding activities of wt and tsE NSP2
| Preincubation condition(s) | Concn (μM) required for 50% RNA bindinga
|
|
|---|---|---|
| wt NSP2 | tsE NSP2 | |
| 31°C | 6.5 | 7.5 |
| 31°C + 100 mM NaCl | 9.0 | 9.0 |
| 39°C | 6.5 | 9.0 |
| 39°C + 100 mM NaCl | 7.0 | 10.5 |
Concentration of wt or tsE NSP2 required to bind 50% of the 32P-labeled Luc 200 RNA in the reaction mixture.
Reduced helix destabilization by tsE NSP2 at the nonpermissive temperature.
NSP2 possesses a Mg2+ and ATP-independent helix-destabilizing activity (38). The activity imparts to the protein, under saturating conditions, the ability to disrupt both DNA-RNA and RNA-RNA duplexes. Characterization of this activity suggested that NSP2 destabilizes a helix not by an enzymatic process but by a passive process that is driven by the affinity of the protein for single-stranded regions of a partial duplex. If this proposed mechanism of helix destabilization is correct, then the reduced ssRNA-binding affinity of tsE NSP2 relative to wt NSP2 at the nonpermissive temperature (Table 1) should consequently decrease the helix-destabilizing activity of the protein.
The helix-destabilizing activity of tsE NSP2 was compared to that of wt NSP2 using the strand-displacement assay. The assay was performed by adding either 50 or 200 pmol of wt or tsE NSP2, preincubated at 31 or 39°C for 30 min, to 0.1 pmol of the radiolabeled partial duplex All-StyI-18AD. The duplex was constructed by annealing the 5′-end-labeled 27-mer DNA oligonucleotide, 18AD, to the unlabeled 48-mer RNA, A11-StyI. The reaction mixtures were incubated for 30 min at 31 or 39°C and then treated with proteinase K. The samples were diluted into buffer containing 0.1% SDS and analyzed by electrophoresis on nondenaturing 20% polyacrylamide gels and autoradiography (Fig. 8A). The extent of strand displacement was quantified with a phosphorimager (Fig. 8B). The analysis showed that the levels of helix-destabilizing activity at 31°C were similar in reaction mixtures containing larger amounts (200 pmol) of tsE NSP2 or wt NSP2. However, in reaction mixtures containing smaller amounts (50 pmol) of protein and also incubated at 31°C, the level of helix destabilization stimulated by tsE NSP2 was less than that stimulated by wt NSP2. These results suggest that even at permissive temperature, the helix destabilization activity of tsE NSP2 may be somewhat defective but this defect can be overcome by increasing the amount of the ts protein present relative to that of its RNA substrate. With an increase in the preincubation and reaction temperatures to 39°C, the helix-destabilizing activity of tsE NSP2 was 50% of that of wt NSP2 regardless of the amount of protein used in the assay (Fig. 8B). Taken together, these data indicate that tsE NSP2 contains a ts lesion that inhibits the ability of the protein to mediate helix destabilization at the nonpermissive temperature.
FIG. 8.

tsE NSP2 exhibits reduced helix-destabilizing activity. Either 50 (lanes 4, 6, 8, and 10) or 200 (lanes 5, 7, 9, and 11) pmol of wt NSP2 (lanes 4 to 7) or tsE NSP2 (lanes 8 to 11) were incubated at 31 or 39°C for 30 min. After the addition of 0.25 pmol of 32P-end-labeled DNA-RNA duplex, A11-StyI-18AD, the mixtures were incubated for an additional 30 min at the respective temperatures. Duplex and single-stranded radiolabeled oligonucleotides in the reaction mixtures were resolved by electrophoresis on a nondenaturing 20% polyacrylamide gel and detected by autoradiography (A). To calculate the helix-destabilizing activity of the protein in each mixture, the amount of single-stranded (unannealed) 32P-labeled oligonucleotide was quantitated and reported as the percentage of the total amount of radiolabeled oligonucleotide (annealed plus unannealed) in the mixture. The final values were corrected for background activity in control assays performed without protein (lane 2) and normalized (taken as 100%) to that of the reaction where the duplex was heat denatured (lane 3 and panel B). The error bars represent the standard error of mean calculated by the PRISM program (GraphPad Software, San Diego, Calif.) from the results of two independent experiments.
Deficiency in the NTPase activity of tsE NSP2.
To compare the NTPase activities of wt and tsE NSP2, various amounts of each protein were preincubated for 30 min at 31 or 39°C. Afterwards, the proteins were combined with [α-32P]UTP and incubated at these temperatures for an additional 30 min. UTP and UDP in the reaction mixtures were then resolved by thin-layer chromatography (Fig. 9A and B). The percentage of UTP hydrolyzed in the reaction mixtures was calculated and used to determine the specific UTPase activity of wt NSP2 and tsE NSP2 at 31 and 39°C (Fig. 9C). Unexpectedly, the results showed that the UTPase activity of tsE NSP2 was one-half that of wt NSP2 at 31°C, the permissive temperature for the tsE virus. At 39°C, the nonpermissive temperature, tsE NSP2 exhibited an even greater decrease (four- to fivefold) in activity relative to wt NSP2. With an increase in temperature from 31 to 39°C, the UTPase activity of wt NSP2 increased almost twofold, in a manner typical of most enzymes. In contrast, tsE NSP2 showed an ∼twofold decrease in its UTPase activity when its incubation temperature was increased from 31 to 39°C. These results indicate that the UTPase, and most likely the NTPase, activity of tsE NSP2 is moderately defective even at the permissive temperature. However, the results also show that the protein exhibits a temperature-dependent phenotype, in that the UTPase activity of tsE NSP2 is significantly less at the permissive temperature than at the nonpermissive temperature.
FIG. 9.

Temperature-sensitive UTPase activity of tsE NSP2. Reaction mixtures containing no added protein or containing 1.5, 3, 4.5, or 6 μg of either wt NSP2 (lanes 4 to 7) or tsE NSP2 (lanes 8 to 11) were incubated at 31°C (A) or 39°C (B) for 30 min. After the addition of 10 μCi of [α-32P]UTP to each reaction mixture, incubation was continued at the respective temperatures for an additional 30 min. UTP and UDP in the reaction mixtures were resolved by thin-layer chromatography and detected by autoradiography (A and B). The positions of UDP and UMP were determined by cochromatography of markers prepared by digestion of [α-32P]UTP with calf intestinal phosphatase (lane 1) or tobacco alkaline pyrophosphatase (lane 2). The percent of UTP hydrolyzed in each reaction mixture was calculated, corrected for background levels (in the absence of added protein, lane 3 in each panel), and divided by the quantity of wt NSP2 or tsE NSP2 used in the assay (1.5, 3, 4.5, or 6 μg). The values for each protein were then averaged, yielding the specific activity (C). Each error bar represents the standard error of the mean calculated from the results of two independent experiments.
DISCUSSION
Characterization of ts mutants of rotaviruses and other members of the Reoviridae has provided valuable insight into the role of the viral proteins in replication (3, 6, 18), assembly (11, 17, 29), and morphogenesis (2, 28). With the current lack of a reverse genetic system for the rotaviruses that would allow rationally designed mutant proteins to be introduced into the virus, studies on ts mutants have the potential to provide unique insights into the viral life cycle and the role of viral proteins. With the goal to understand the structure and function of the NSP2 protein, we have expressed and studied the solution structure of NSP2 encoded by the rotavirus ts mutant, tsE. Cells infected with tsE and maintained at nonpermissive temperature are defective in the formation of viroplasms and RIs and produce larger numbers of empty particles, indicating that NSP2 plays an essential role in genome packaging and replication (3, 28).
Recent studies with wt NSP2 have shown that the protein self-assembles into octamers and can undergo conformational changes induced by binding of Mg2+ and nucleotides (36). In the present study, we found that tsE NSP2 can also exist as octamers, which show hydrodynamic behavior identical to that of the wt protein. This is consistent with the earlier finding that tsE NSP2 is electrophoretically indistinguishable from the wt protein (28). Also, sedimentation velocity showed that the tsE octamer exhibited an increase in sedimentation rate upon nucleotide binding that was similar to that of the wt octamer. Thus, the octameric form of tsE NSP2 appears to be identical to octamers formed by the wt protein. The major difference that we observed between tsE and wt NSP2 was the slow and highly temperature-dependent aggregation of the ts protein in the presence of Mg2+. As the mutation of the tsE protein results from the replacement of one or more amino acids with those that are more hydrophobic (H127R, A152V, and V200I), an entropically driven aggregation at higher temperature seems to be a very plausible origin for the temperature sensitivity of the tsE phenotype. Because temperature-dependent aggregation could also be observed, although only to a very low degree, with wt NSP2 (Fig. 5C), it appears that the ts and wt proteins are structurally very similar and that the main differences are in the energetics of the different protein states. This appears plausible, considering the viability of the tsE mutant when grown at 31°C. Structurally, the aggregates formed by tsE NSP2 appear to be highly elongated, possibly fibers, as suggested by the modest increase in the total intensity of the scattered light compared to the large increase of the hydrodynamic radius (or large decrease of the diffusion coefficient).
Previously, by analytical ultracentrifugation and circular dichroism, it was shown that Mg2+ induces a conformational change in the wt octamer, leading to its dissociation to tetramers (36). Other studies showed that the strand-displacement activity of NSP2 can be inhibited with increasing Mg2+ concentration (38), consistent with the view that the octamer is the functional unit of NSP2 for its interactions with nucleic acids and that the octamer is destabilized by Mg2+. Since Mg2+ is a component of the intracellular environment, such a transition between tetrameric and octameric forms of the protein may well represent a functionally important property of the protein. For example, the transition between forms may be required in the possible role of NSP2 as a molecular motor involved in the transport of mRNA through the channels of core RIs (39). In this context, our observations from the c(s) distributions measured at different stages in the aggregation process, which indicate that tsE NSP2 octamers first dissociate into smaller subunits that then are subject to aggregation, may be of relevance. One can hypothesize that the hydrophobic residues introduced by the tsE mutation are exposed to the protein surface in the Mg2+-linked tetramer conformation of the protein and that, therefore, a temperature-dependent aggregation of tetramers via hydrophobic patches takes place. In this picture, the lethal condition may arise either because the tsE NSP2 protein is locked into the Mg2+-induced (tetramer-like) conformation of the NSP2 protein or because the resulting aggregates do not allow sufficient access for efficient binding of ligands and thereby interfere with protein function.
The results from the electrophoretic mobility shift assays indicated that tsE octamers have reduced affinity for RNA at the nonpermissive temperature, in comparison to wt octamers. Also, the mobility shift assays indicated that the ts protein formed large aggregates at the nonpermissive temperature that were not able to migrate into native polyacrylamide gels. In contrast, such evidence for aggregation was not detected for tsE NSP2 analyzed at the permissive temperature or for wt NSP2 analyzed at either the permissive or nonpermissive temperature. Therefore, like the sedimentation velocity experiments, the mobility shift assays suggest that tsE NSP2 is subject to temperature-dependent aggregation. Furthermore, the data reveal evidence that aggregation and the loss of the octamer unit are correlated with a decrease in the ability of NSP2 to bind RNA.
Previous results indicate that the saturative and high-affinity binding of NSP2 octamers to ssRNA drives the destabilization of partial helixes, by a process that is Mg2+ and ATP independent (38). Support for this mechanism of helix destabilization comes from the observation that the reduction in the ssRNA binding activity of tsE NSP2 at the nonpermissive temperature is accompanied by a reduction in its helix-destabilizing activity. The correlation between the decrease of helix-destabilizing activity and the aggregation of the protein at the nonpermissive temperature, noted by sedimentation velocity, supports a conclusion that efficient RNA binding and helix destabilization require the octameric form of NSP2. The helix-destabilizing activity of NSP2 could facilitate packaging by removing RNA secondary structures that may otherwise impede passage of mRNAs into cores (26). Defects in the helix-destabilizing activity of tsE NSP2 at the nonpermissive temperature, due to aggregation of the protein, may result in inefficient movement of mRNAs into cores, leading to an increase in the formation of empty virus particles, a property that is characteristic of tsE-infected cells (28).
The NTPase activity of NSP2 is able to use any of the nucleotides as a substrate, requires Mg2+ as a cofactor, and functions in the absence of RNA (39). The ability of NSP2 to support helix destabilization in the absence of Mg2+ and nucleotide suggests that the NTPase and helix-destabilizing activities of the protein may operate independently of each other. Our analysis showed that the NTPase activity of tsE NSP2 was 50% and 20 to 25% of that of wt NSP2 at the permissive and nonpermissive temperatures, respectively. The decrease in activity for tsE NSP2 is best explained by the observation that Mg2+, a component of the NTPase assay, has a greater destabilizing effect on tsE octamers than wt octamers, even at the permissive temperature (Fig. 4) (36). The destabilizing effect of Mg2+ on tsE octamers is temperature dependent, which probably accounts for the decrease in NTPase activity that was observed as the assay temperature was increased from 31 to 39°C. These results support a hypothesis that maximal levels of NTPase activity are associated with the octameric form of NSP2 and that the octamer represents the fully functional form of NSP2.
How a defect in the hydrolysis of NTP by NSP2 could affect the role of the protein in the viral life cycle is unclear. Presumably, such a defect could prevent the generation of energy necessary for the putative molecular motor of NSP2 to drive packaging. Alternatively, a defect in NTP hydrolysis may interfere with the productive interaction of NSP2 and NSP5. These two proteins cooperate in the assembly of viroplasms (8), and the interaction of two proteins results in the hyperphosphorylation of NSP5 (1, 41). Importantly, hyperphosphorylation is necessary for the localization of NSP5 to viroplasms (8). The hyperphosphoryation of NSP5 has been proposed to result from the transfer of phosphate moieties generated by the NTPase activity of NSP2 to NSP5 (41). Thus, by interfering with its NTPase activity, the ts lesion of tsE NSP2 may prevent the efficient phosphorylation of NSP5 at the nonpermissive temperature and thereby inhibit the formation of viroplasms. Since it is at these sites that RNA packaging and replication occur, the inability to form viroplasms in the infected cell would result in a decrease in the formation of RIs and a relative increase in the assembly of empty particles, both of which are characteristics of tsE-infected cells maintained at the nonpermissive temperature. Thus, it is perhaps not just defects in the ability of tsE NSP2 to bind RNA and to destabilize helixes at the nonpermissive temperature that contribute to the phenotype of this mutant virus but also the ability of the protein to hydrolyze NTP.
Acknowledgments
Z.F.T. and P.S. contributed equally to this study.
We thank Maha Kattoura for cloning and sequencing gene 8 cDNAs of the tsE virus.
REFERENCES
- 1.Afrikanova, I., E. Fabretti, M. C. Miozzo, and O. R. Burrone. 1998. Rotavirus NSP5 phosphorylation is up-regulated by interaction with NSP2. J. Gen. Virol. 79:2679-2686. [DOI] [PubMed] [Google Scholar]
- 2.Becker, M. M., M. I. Goral, P. R. Hazelton, G. S. Baer, S. E. Rodgers, E. G. Brown, K. M. Coombs, and T. S. Dermody. 2001. Reovirus óNS protein is required for nucleation of viral assembly complexes and formation of viral inclusions. J. Virol. 75:1459-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen, D., J. L. Gombold, and R. F. Ramig. 1990. Intracellular RNA synthesis directed by temperature-sensitive mutants of simian rotavirus SA11. Virology 178:143-151. [DOI] [PubMed] [Google Scholar]
- 4.Chen, D., C. L. Luongo, M. L. Nibert, and J. T. Patton. 1999. Rotavirus open cores catalyze 5′-capping and methylation of exogenous RNA: evidence that VP3 is a methyltransferase. Virology 265:120-130. [DOI] [PubMed] [Google Scholar]
- 5.Claverie, J.-M., H. Dreux, and R. Cohen. 1975. Sedimentation of generalized systems of interacting particles. I. Solution of systems of complete Lamm equations. Biopolymers 14:1685-1700. [DOI] [PubMed] [Google Scholar]
- 6.Coombs, K. M. 1996. Identification and characterization of a double-stranded RNA− reovirus temperature-sensitive mutant defective in minor core protein μ2. J. Virol. 70:4237-4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Estes, M. K. 2001. Rotaviruses and their replication, p. 1747-1785. In D. Knipe, M. Howley, et al. (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 8.Fabbretti, E., I. Afrikanova, F. Vascotto, and O. R. Burrone. 1999. Two non-structural rotavirus proteins, NSP2 and NSP5, form viroplasm-like structures in vivo. J. Gen. Virol. 80:333-339. [DOI] [PubMed] [Google Scholar]
- 9.Gallegos, C. O., and J. T. Patton. 1989. Characterization of rotavirus replication intermediates: a model for the assembly of single-shelled particles. Virology 172:616-627. [DOI] [PubMed] [Google Scholar]
- 10.Gombold, J. L., M. K. Estes, and R. F. Ramig. 1985. Assignment of simian rotavirus SA11 temperature-sensitive mutant groups B and E to genome segments. Virology 143:309-320. [DOI] [PubMed] [Google Scholar]
- 11.Hazelton, P. R., and K. M. Coombs. 1999. The reovirus mutant tsA279 L2 gene is associated with generation of a spikeless core particle: implications for capsid assembly. J. Virol. 73:2298-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kapikian, A. Z., Y. Hoshino, and R. M. Chanock. 2001. Rotaviruses, p. 1787-1833. In D. Knipe, M. Howley, et al. (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 13.Kattoura, M., L. L. Clapp, and J. T. Patton. 1992. The rotavirus non-structural protein, NS35, is a nonspecific RNA-binding protein. Virology 191:698-708. [DOI] [PubMed] [Google Scholar]
- 14.Kattoura, M. D., X. Chen, and J. T. Patton. 1994. The rotavirus RNA binding protein NS35 (NSP2) forms 10S multimers and interacts with the viral RNA polymerase. Virology 202:803-813. [DOI] [PubMed] [Google Scholar]
- 15.Lamm, O. 1929. Die Differentialgleichung der Ultrazentrifugierung. Ark. Mat. Astr. Fys. 21B:1-4. [Google Scholar]
- 16.Laue, T. M., B. D. Shah, T. M. Ridgeway, and S. L. Pelletier. 1992. Computer-aided interpretation of analytical sedimentation data for proteins, p. 90-125. In S. E. Harding, A. J. Rowe, and J. C. Horton (ed.), Analytical ultracentrifugation in biochemistry and polymer science. The Royal Society of Chemistry, Cambridge, United Kingdom.
- 17.Mansell, E. A., R. F. Ramig, and J. T. Patton. 1994. Temperature-sensitive lesions in the capsid proteins of the rotavirus mutants tsF and tsG that affect virion assembly. Virology 204:69-81. [DOI] [PubMed] [Google Scholar]
- 18.Munoz, M., M. Rios, and E. Spencer. 1995. Characteristics of single- and double-stranded RNA synthesis by a rotavirus SA-11 mutant thermosensitive in the RNA polymerase gene. Intervirology 38:256-263. [DOI] [PubMed] [Google Scholar]
- 19.Patton, J. T. 1986. Synthesis of simian rotavirus SA11 double stranded RNA in a cell-free system. Virus Res. 6:217-223. [DOI] [PubMed] [Google Scholar]
- 20.Patton, J. T. 1996. Rotavirus VP1 alone specifically binds to the 3′ end of viral mRNA but the interaction is not sufficient to initiate minus-strand synthesis. J. Virol. 70:7940-7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patton, J. T., and D. Chen. 1999. RNA-binding and capping activities of proteins in rotavirus open cores. J. Virol. 73:1382-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patton, J. T., and E. Spencer. 2000. Genome replication and packaging of segmented double-stranded RNA viruses. Virology 277:217-225. [DOI] [PubMed] [Google Scholar]
- 23.Patton, J. T., M. Wentz, J. Xiaobo, and R. F. Ramig. 1996. cis-acting signals that promote genome replication in rotavirus mRNA. J. Virol. 70:3961-3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patton, J. T., Z. Taraporewala, D. Chen, V. Chizhikov, M. Jones, A. Elhelu, M. Collins, K. Kearney, M. Wagner, Y. Hoshino, and V. Gouvea. 2001. Effect of intragenic rearrangement and changes in the 3′ consensus sequence on NSP1 expression and rotavirus replication. J. Virol. 75:2076-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prasad, B. V. V., G. J. Wang, J. P. M. Clerx, and W. Chiu. 1988. Three-dimensional structure of rotavirus. J. Mol. Biol. 199:269-275. [DOI] [PubMed] [Google Scholar]
- 26.Qiao, X., J. Qiao, and L. Mindich. 1995. Interference with bacteriophage phi6 genomic RNA packaging by hairpin structures. J. Virol. 69:5502-5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramig, R. F. 1982. Isolation and genetic characterization of temperature-sensitive mutants of simian rotavirus SA11. Virology 120:93-105. [DOI] [PubMed] [Google Scholar]
- 28.Ramig, R. F., and B. L. Petrie. 1984. Characterization of temperature-sensitive mutants of simian rotavirus SA11: protein synthesis and morphogenesis. J. Virol. 49:665-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandino, A. M., J. Fernandez, J. Pizarro, M. Vasquez, and E. Spencer. 1994. Structure of rotavirus particle: interaction of the inner capsid protein VP6 with the core polypeptide VP3. Biol. Res. 27:39-48. [PubMed] [Google Scholar]
- 30.Sanger, F., S. Nicklen, and A. R. Coulsen. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schuck, P. 1998. Sedimentation analysis of noninteracting and self-associating solutes using numerical solutions to the Lamm equation. Biophys. J. 75:1503-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schuck, P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78:1606-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuck, P., and B. Demeler. 1999. Direct sedimentation analysis of interference-optical data in analytical ultracentrifugation. Biophys. J. 76:2288-2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuck, P., C. E. MacPhee, and G. J. Howlett. 1998. Determination of sedimentation coefficients for small peptides. Biophys. J. 74:466-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schuck, P., M. A. Perugini, N. R. Gonzales, G. J. Howlett, and D. Schubert. 2002. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys. J. 82:1096-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schuck, P., Z. Taraporewala, P. McPhie, and J. T. Patton. 2001. Rotavirus nonstructural protein NSP2 self-assembles into octamers that undergo ligand-induced conformational changes. J. Biol. Chem. 276:9679-9687. [DOI] [PubMed] [Google Scholar]
- 37.Svedberg, T., and K. O. Pederson. 1940. The ultracentrifuge. Oxford University Press, London, United Kingdom.
- 38.Taraporewala, Z., D. Chen, and J. T. Patton. 1999. Multimers formed by the rotavirus nonstructural protein NSP2 bind to RNA and have nucleoside triphosphatase activity. J. Virol. 73:9934-9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taraporewala, Z. F., and J. T. Patton. 2001. Identification and characterization of the helix-destabilizing activity of rotavirus nonstructural protein NSP2. J. Virol. 75:4519-4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valenzuela, S., J. Pizarro, A. M. Sandino, M. Vasquez, J. Fernandez, O. Hernandez, J. Patton, and E. Spencer. 1991. Photoaffinity labeling of rotavirus VP1 with 8-azido-ATP: identification of the viral RNA polymerase. J. Virol. 65:3964-3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vende, P., Z. F. Taraporewala, and J. T. Patton. 2002. RNA-binding activity of the rotavirus phosphoprotein NSP5 includes affinity for double-stranded RNA. J. Virol. 76:5291-5299. [DOI] [PMC free article] [PubMed] [Google Scholar]