

Abstract
Human immunodeficiency virus type 1 (HIV-1) Gag protease cleavage sites (CS) undergo sequence changes during the development of resistance to several protease inhibitors (PIs). We have analyzed the association of sequence variation at the p7/p1 and p1/p6 CS in conjunction with amprenavir (APV)-specific protease mutations following PI combination therapy with APV. Querying a central resistance data repository resulted in the detection of significant associations (P < 0.001) between the presence of APV protease signature mutations and Gag L449F (p1/p6 LP1′F) and P453L (p1/p6 PP5′L) CS changes. In population-based sequence analyses the I50V mutant was invariably linked to either L449F or P453L. Clonal analysis revealed that both CS mutations were never present in the same genome. Sequential plasma samples from one patient revealed a transition from I50V M46L P453L viruses at early time points to I50V M46I L449F viruses in later samples. Various combinations of the protease and Gag mutations were introduced into the HXB2 laboratory strain of HIV-1. In both single- and multiple-cycle assay systems and in the context of I50V, the L449F and P453L changes consistently increased the 50% inhibitory concentration of APV, while the CS changes alone had no measurable effect on inhibitor sensitivity. The decreased in vitro fitness of the I50V mutant was only partially improved by addition of either CS change (I50V M46I L449F mutant replicative capacity ≈ 16% of that of wild-type virus). Western blot analysis of Pr55 Gag precursor cleavage products from infected-cell cultures indicated accumulation of uncleaved Gag p1-p6 in all I50V viruses without coexisting CS changes. Purified I50V protease catalyzed cleavage of decapeptides incorporating the L449F or P453L change 10-fold and 22-fold more efficiently than cleavage of the wild-type substrate, respectively. HIV-1 protease CS changes are selected during PI therapy and can have effects on both viral fitness and phenotypic resistance to PIs.
Chemotherapy of human immunodeficiency virus type 1 (HIV-1) infection with drug combinations containing inhibitors of the virus aspartyl class protease has been reported to reduce clinical disease progression and mortality of HIV infection (11, 15, 36). However, suboptimal therapy permitting ongoing virus replication in the presence of protease inhibitors (PIs) results in the emergence of virus escape variants that have lost sensitivity to inhibition by the administered PI. Characteristically, an accumulation over time of primary and secondary amino acid substitutions in the protease is observed in these viruses (4, 27). Some of the secondary variations found in mutant proteases also occur in HIV-1 isolates from patients who never received PI treatment. Non-subtype B proteases often display a number of such sequence deviations, leading to altered enzyme inhibition characteristics with currently available inhibitors (18, 20, 38, 44, 45). Primary and secondary changes are thought to act in a concerted fashion by modifying molecular inhibitor contacts at the protease active site and by maintaining functionality of the enzyme (10, 26).
Orchestrated cleavage by the protease at the different Gag, Gag-Pol (17, 21, 34, 46), and Nef (13, 47) recognition sites needs to occur for virus replication of drug-resistant mutants to be retained. Evolution at multiple cleavage sites (CS) in PI-resistant variants in vivo has been described, but only the effects of CS alterations at the Gag p7/p1 and p1/p6 sites have been the subject of detailed investigations (5, 9, 49). The p7-p1-p6 polyprotein represents a p55 processing intermediate. Cleavage at the p7/p1 and p1/p6 scissile bonds occurs late and requires the presence of p15 RNA, suggesting a complex and potentially rate-limiting step in viral assembly and maturation (34, 39, 40, 43, 46). p7/p1 AlaP2Val (Gag A431V) and p1/p6 LeuP1′Phe (Gag L449F) mutations have been shown to partially compensate for decreased replicative capacity caused by primary PI resistance mutations affecting the protease active site (9, 23, 49). We describe here novel associations of p1/p6 CS mutations with PI resistance in viruses isolated from patients treated with the PI amprenavir (APV). In vitro development of reduced susceptibility to this drug has been demonstrated to be associated with an I50V primary mutation (25, 29, 41). The I50V, I54L or -M, V32I plus I47V, and I84V protease substitutions are pathways selected by APV-based combination therapy in HIV-infected patients with incomplete suppression of plasma viremia. The I50V and I54L or -M mutants and the V32I I47V double mutant are not commonly observed with other PIs in vivo (22, 42). We show here that changes at Gag positions 449 and 453 can lead to significant decreases in susceptibility to APV when present with APV-specific primary protease mutations. We provide evidence to support the concept of distinguishable effects of gag mutations on (i) in vitro viral fitness and (ii) increased resistance to PIs. We use infected-cell culture data and enzymatic data to elucidate the molecular basis of these observations.
MATERIALS AND METHODS
Cells and viruses.
Standard recombinant virus assays were carried out with MT-4 cells using HIV-1 HXB2 as a virus control. For single-replication cycle assays, 293 human embryonic kidney cells were used with a reference virus containing HIV-1 NL4-3 protease and reverse transcriptase (RT) sequences, as described previously (33).
Generation of mutant recombinant HIV-1.
A plasmid vector containing the HXB2 DNA sequence corresponding to the C terminus of Gag and the entire protease was used as a template for site-specific mutagenesis with the QuikChange kit (Stratagene Cloning Systems, La Jolla, Calif.) and custom-synthesized oligonucleotides in order to generate Gag and protease mutant viruses. A PCR product was generated from the mutated plasmids which covered the p7/p1 and p1/p6 cleavage sites and the PR deletion in the RVA vector pHXBΔCSPR (37). PCR products were cotransfected with linearized vector pHXBΔCSPR into MT-4 cells. Supernatants containing mutant viruses were harvested at peak cytopathic effect. The presence of the desired mutations was confirmed by nucleotide sequencing of mutated plasmids and of recombinant virus as described below.
Determination of susceptibility to PIs.
Saquinavir was kindly provided by I. Duncan (Roche Products Ltd., Welwyn Garden City, United Kingdom), and all other PIs were synthesized in-house. The concentrations of all currently licensed antiretrovirally active PIs that inhibit 50% of virus replication (IC50 values) were determined with recombinant viruses bearing protease and Gag mutations by using a tetrazolium-based colorimetric assay (31). Twofold serial drug dilutions were prepared in 96-well plates, and 4 × 104 MT-4 cells infected with 100 50% tissue culture infectious doses (TCID50) were added to each well. After incubation for 5 days, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added. The amount of conversion to the blue formazan product was assessed by reading the optical absorbance at 590 nm. The concentration of drug required to increase the absorbency to levels 50% of those in uninfected control cells was calculated by regression analysis.
In addition, IC50 values for PIs were determined with mutant viruses by using a single-cycle assay, as described previously (33). Briefly, 293 cells were trypsinized 16 h after transfection and were added to 96-well plates containing serial dilutions of PIs. Virus stocks generated in the presence of PIs were used to infect fresh 293 cells in the absence of inhibitors. Virus replication was monitored by measuring luciferase activity approximately 48 h after infection.
In vitro growth comparison assays.
The replicative capacities of site-specific mutants were determined by using the single-cycle assay described above. Luciferase activities in cultures of different recombinants were determined and expressed in relation to those for the HIV-1 NL4-3 control virus. Replicative capacity (relative light units [RLU] after infection) was normalized for transfection efficiency in transfected cells (33, 50).
Clinical resistance database and statistical methods.
A central repository of resistance data (Oracle database) was queried to retrieve all available Gag and protease amino acid sequences. Matching Gag sequences (amino acids 428 to 453) and protease sequences were available for 1,060 viral isolates from 432 subjects from four studies, PROA3001, PROAB3006, CNAA2007, and PROAB3004 (12, 32). Before the data were analyzed, data for any subjects for whom we had multiple samples were collapsed to one observation, which flagged whether or not the subjects had a given mutation of interest in at least one of their virus isolates. Fisher's exact tests were then carried out in order to identify significant (P < 0.05) pairwise associations between the presence or absence of I50V, V32I plus I47V, I54L, I54M, and I84V changes in protease and the presence or absence of non-consensus B substitutions in Gag residues 428 to 453. All statistical tests were two sided and were performed by using a 5% alpha level. All data manipulation, tabulations, and calculations were performed with the SAS 6.12 system (SAS Institute, Cary, N.C.) under UNIX.
Population and clonal sequencing of HIV gag and pol genome regions.
cDNA was synthesized by RT-PCR from plasma viral extracts as described previously (19). Viral nucleotide sequences were determined for the entire protease gene and the gag region encoding Gag amino acids 428 to 453 (p7/p1 P5 to p1/p6 P5′) using the PRISM BigDye terminator reagent kit (Applied Biosystems [ABI], Foster City, Calif.). Reaction products were separated and analyzed with an ABI 377 DNA automated sequencer. Viral protease RT-PCR products were cloned by insertion into a plasmid vector that was used for transformation of competent Escherichia coli cells (pCR2.1-TOPO; TOPO TA cloning kit; Invitrogen, Carlsbad, Calif.). PCR products generated from resultant overnight colonies (10 to 12 colonies per isolate) were sequenced as described above.
Western blots of infected-cell culture supernatants.
MT-4 cells (5 × 105 cells/ml) were infected with HIV-1HXB2 or mutant viruses at a multiplicity of infection of 10−4 (viral titers were determined on the basis of TCID50) and maintained in 5 ml of supplemented medium. Cells were harvested 5 days postinfection and centrifuged at 400 × g for 5 min. Viral supernatants were centrifuged at 50,000 × g for 80 min to pellet virus particles. Precipitates were resuspended in 2× Laemmli buffer (2% sodium dodecyl sulfate [SDS], 10% glycerol, 5% β-mercaptoethanol, 0.002% bromophenol blue, 0.0625 M Tris-HCl [pH 6.8, Sigma]) and heated at 80°C for 10 min before loading. Samples and protein molecular weight standards (Invitrogen) were separated on SDS-10% polyacrylamide gels (Novex) and then transferred onto polyvinylidene difluoride membranes (Immobilon P; Millipore) by electroblotting (Promega). Membranes were incubated with blocking reagent (10% skim milk-0.05% Tween in phosphate-buffered saline) for 1 h at room temperature and then hybridized with an anti-p6 monoclonal antibody (diluted 1:1,000; Advance Bioscience Laboratories, Inc.) overnight at 4°C. Upon being washed, membranes were incubated with peroxidase-conjugated rabbit anti-rat antiserum (diluted 1:8,000; Sigma). Immune complexes were visualized with the ECL system (Amersham) according to the manufacturer's instructions.
Membranes were stripped by incubation in 100 mM β-mercaptoethanol-2% SDS-62.5 mM Tris-HCl, pH 6.8, for 30 min at 50°C, and washing with phosphate-buffered saline-Tween. Membranes were rehybridized with an anti-p24 antibody (diluted 1:5,000; DAKO); rabbit anti-sheep horseradish peroxidase (diluted 1:1,000) was used as secondary antibody for detection of immune complexes.
In vitro cleavage of peptide substrates by wild-type (wt) and I50V proteases.
Decapeptides labeled with Dabcyl and Edans moieties at the amino and carboxyl termini were purchased from SynPep; their purity was greater than 95%. Peptides were dissolved in dimethyl sulfoxide to yield solutions that were between 5 and 10 mM peptide based on mass. These solutions of peptides were stored at −20°C. The concentrations of peptides were calculated from the absorbance of the Dabcyl moiety (ɛ430 = 23 mM−1 cm−1 in methanol [16]). HIV-1 proteases E(wt) and E(I50V) were purified from E. coli containing the appropriate constructs as described previously (35). The concentration of enzyme was determined by titration of enzymatic activity with APV, whose concentration was determined spectrophotometrically (ɛ265 = 16.7 mM−1 cm−1 in 0.1 N NaOH). Enzyme concentrations were expressed in terms of APV binding sites (35). Concentrations of E(wt) and E(I50V) were 5.8 and 7.5 μM, respectively, in stock solutions of 50 mM HEPES (pH 7.0), 10% glycerol, 5% ethylene glycol, 175 mM imidazole, and 1 mM β-mercaptoethanol. The enzyme was stored as 100-μl samples at −80°C.
The standard buffer consisted of 0.1 M MES (morpholineethanesulfonic acid)-Tris and 1.25 M NaCl, pH 6.0. The standard temperature was 25°C. Steady-state fluorescence data were collected with a Kontron SFM 25 spectrofluorometer. In a typical assay, 20 nM peptide (peptide solubility was estimated to be greater than 100 nM) was equilibrated in the standard buffer in a quartz cuvette in the spectrofluorometer at 25°C. The reaction was initiated with 50 to 750 nM enzyme. Product formation was monitored with an excitation λ of 370 nm and an emission λ of 500 nm.
The substrates were completely hydrolyzed with E(wt) to determine the total fluorescence change associated with the hydrolysis reaction. The fluorescence changes were normalized to the total fluorescence change such that the time course for the observed fluorescence change increased from a value of zero to a value of 1. The equation
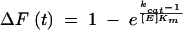 |
(where F = fluorescence, E = enzyme, and t = time) was fitted to these data to give an estimated value for kcat/Km. The validity of this equation requires that an insignificant fraction of the substrate be present as the enzyme-substrate complex during the reaction. This was demonstrated to be the case by the finding that the value of kcat/Km was not dependent on enzyme concentration with the p66/INT peptide. Further support for this assumption was the finding that the hydrolysis of the p1/p6gag peptide [a very inefficient substrate for E(I50V)] by 116 nM E(wt) was not inhibited by 750 nM E(I50V). This indicated that the Km of E(I50V) for p1/p6gag was much greater than 2,000 nM.
RESULTS
Association between protease and p7/p1 and p1/p6 CS substitutions in clinical HIV-1 isolates with reduced susceptibility to APV.
We investigated the coevolution of Gag and protease sequences in viruses from patients presenting with detectable plasma viremia while receiving APV combination therapy and investigated the association of HIV-1 protease substitutions with changes at the p7/p1 and p1/p6 CS. In a central clinical virology data repository, 1,060 viral isolates from 432 subjects enrolled into four clinical trials with APV were identified. Matching protease and p7/p1/p6 sequences spanning Gag amino acids 428 to 453 were available for all of these samples.
Gag P453L has recently been reported to be associated with the appearance of the I84V mutant (1). We found a significant difference (P < 0.01) in prevalence of P453L between viral isolates derived from PI-naive patients (7 of 167, 4.2%) and isolates from PI-experienced patients (34 of 156, 22%). In viruses with APV-selected protease mutants, several deviations at the Gag CS from the HIV-1 consensus B sequence were found to be associated with I50V, I54L, and I84V changes (Table 1). Highly significant associations (P < 0.001) among one or more of these protease mutations and nonconsensus residues at three p1/p6 positions, L449 (P1′), R452 (P4′), and P453 (P5′), were noted.
TABLE 1.
HIV-1 Gag-protease sequence associations for APV-specific protease mutants
| Protease mutation(s) |
Pa for non consensus B amino acid changes at indicated Gag residue
|
|||||
|---|---|---|---|---|---|---|
| p7/p1 CS
|
p1/p6 CS
|
|||||
| R429 (P4) | A431 (P2) | N447 (P2) | L449 (P1′) | R452 (P4′) | P453 (P5′) | |
| I50V | NSb | NS | NS | <0.001 | <0.001 | <0.001 |
| I54L | NS | NS | 0.005 | <0.001 | <0.001 | <0.001 |
| I84V | 0.034 | <0.001 | NS | 0.018 | NS | <0.001 |
| V32I + I47V | NS | NS | 0.033 | NS | NS | NS |
| I54M | NS | NS | NS | NS | 0.006 | NS |
By the two-sided Fisher exact test.
NS, not significant.
We next investigated evolution at the p1/p6 site in viruses from patients who developed the I50V signature mutant during therapy with APV in study PROAB3006 of nucleoside RT inhibitor-experienced, PI-naive HIV-1-infected patients. A total of 504 patients were enrolled in this study. Virus from nine patients contained variants with I50V. All of these viruses also developed either L449F (I50V L449F; n = 4) or P453L (I50V P453L; n = 4) or each CS change consecutively (I50V P453L followed by I50V L449F in later samples [n = 1], described in detail below). All mutants specified contained other resistance-associated mutations in both protease and RT (data not shown).
Taken together these observations suggested a very close and possibly invariable linkage of the I50V change in the protease to an alteration in the Gag p6 protein in vivo. To further corroborate this hypothesis with evidence collected in vitro, we identified samples from early dose escalation studies conducted with APV (41). In these experiments, APV was added to MT-4 cell cultures initially infected with HIV-1 HXB2 at <0.01 TCID50 per cell. Virus was then passaged in increasing concentrations of inhibitor. We retrospectively evaluated nucleotide sequences corresponding to the Gag p7/p1 and p1/p6 CS from different passages. Population sequencing indicated the early presence of L449F at passage 1 (200 nM APV). Passage 2 virus (400 nM APV) consisted of I50V L449F sequences. The same double mutants were observed during subsequent passages 3 and 4. At passage 6 (1,600 nM APV) I50V M46L L449F mutants were predominant.
Clonal sequence data from the same selection experiment were then analyzed (12 clones/passage). Only one of three I50V-containing sequences also showed the L449F mutation in passage 1 virus, and vice versa only one of two L449F sequences contained I50V. Linkage of I50V and L449F in all sequences (data from 11 clones) was detected no earlier than in passage 6. Eight out of 11 clones coded for M46L, 2 of 11 coded for M46I, and 1 of 11 was wt.
Evolution of protease and p1/p6 CS from an APV-treated patient with detectable viremia monitored over 68 weeks.
We next determined in vivo virus evolution by examining viral genomes circulating in plasma from patient A. This patient participated in clinical study PROAB3006 and initially received APV-zidovudine (ZDV)-lamivudine (3TC) combination therapy (Table 2). A loss of virologic control was first evident at week 20. This was accompanied by the presence of the I50V mutation in all sequences. The prevailing Gag CS change was initially P453L (92% of clones). Position 46 in protease was known from previous in vitro and clinical studies to be critical for further decreasing phenotypic susceptibility to APV. M46L clones, which were first detected in this patient at week 32, were replaced by M46I-containing variants at later time points. The gradual replacement of M46L by M46I was accompanied by the appearance of L449F. Therefore, I50V M46L P453L mutants dominated at earlier time points and were replaced by I50V M46I L449F viruses in the week 68 sample. Virologic control in this patient had been partially regained after study week 24 by exchanging both nucleoside RT inhibitors and by adding indinavir (IDV) to the regimen.
TABLE 2.
Clonal analysis of protease- and Gag-coding regions of virus from PRO3006 patient A through 68 weeks of therapy
| Sample | Treatmenta | Plasma HIV-1 RNA (copies/ml) | No. of clones with indicated mutation/no. tested (%)b
|
||||
|---|---|---|---|---|---|---|---|
| P453L | L449F | I50V | M46L | M46I | |||
| Day 1 | APV-ZDV-3TC | 29,896 | |||||
| Week 4 | APV-ZDV-3TC | 18,879 | |||||
| Week 8 | APV-ZDV-3TC | 1,030 | NDc | ND | ND | ND | ND |
| Week 12 | APV-ZDV-3TC | 1,213 | |||||
| Week 16 | APV-ZDV-3TC | 644 | |||||
| Week 20 | APV-ZDV-3TC | 4,391 | 11/12 (92) | 1/12 (8) | 12/12 (100) | ||
| Week 24 | APV-ZDV-3TC | 22,250 | 12/12 (100) | 11/12 (92) | |||
| Week 28 | APV-DDI-D4T | 16,556 | 12/12 (100) | 12/12 (100) | |||
| Week 32 | APV-DDI-D4T | 4,828 | 12/12 (100) | 12/12 (100) | 11/12 (92) | ||
| Week 36 | APV-DDI-D4T | 3,580 | 12/12 (100) | 12/12 (100) | 1/12 (8) | ||
| Week 44 | APV-IDV-DDI-D4T | 3,704 | 6/12 (50) | 6/12 (50) | 12/12 (100) | 9/12 (75) | |
| Week 48 | APV-IDV-DDI-D4T | 752 | ND | ND | ND | ND | ND |
| Week 52 | APV-IDV-DDI-D4T | <400 | ND | ND | ND | ND | ND |
| Week 56 | APV-IDV-DDI-D4T | 582 | ND | ND | ND | ND | ND |
| Week 60 | APV-IDV-DDI-D4T | <400 | ND | ND | ND | ND | ND |
| Week 64 | APV-IDV-DDI-D4T | 2,630 | ND | ND | ND | ND | ND |
| Week 68 | APV-IDV-DDI-D4T | 3,292 | 12/12 (100) | 12/12 (100) | 12/12 (100) | ||
D4T, stavudine; DDI, dideoxyinosine.
Other deviations from the HIV-1 consensus B comparator were detected in all clones.
ND, not determined.
Susceptibility to PIs of HIV-1 recombinant viruses with alterations in protease and p1/p6 CS.
The effect of mutations in the gag CS-coding regions have so far been characterized as compensatory, restoring the fitness defects conferred by primary changes in protease. We were interested in obtaining sensitivity data for the APV-specific I50V protease mutant in connection with the distinct Gag substitution patterns identified in vitro and in the clinical samples. We used multiple- and single-cycle assay technologies to enable better discrimination between drug susceptibility and viral growth kinetics parameters (Tables 3 and 4). APV susceptibilities were decreased threefold by the I50V mutant, as expected. A further consistent and reproducible decrease of virus sensitivity to APV was measured for I50V P453L and I50V L449F mutant viruses, resulting in a 10-fold rise in IC50 compared to that for the wt control in the multiple-cycle assay. The addition of M46I to these sequences yielded mutants with the highest levels of resistance observed in this study (15-fold-reduced sensitivity to APV). In both test systems, P453L and L449F Gag changes alone did not measurably diminish APV susceptibilities.
TABLE 3.
Drug susceptibilities of HIV-1 protease and Gag mutant viruses determined by multiple-cycle sensitivity assay
| Virus | Mean IC50 (nM) ± SDa of:
|
|||||
|---|---|---|---|---|---|---|
| APV | IDV | LPV | NFV | RTV | SQV | |
| HXB2 | 75.7 ± 8.4 (1.0) | 33.1 ± 10.6 (1.0) | 24.4 ± 2.9 (1.0) | 21.6 ± 11.8 (1.0) | 58.5 ± 21.9 (1.0) | 7.0 ± 0.9 (1.0) |
| I54V | 39.5 ± 3.0 (0.5) | 9.4 ± 1.2 (0.4) | ||||
| I54M | 342.5 ± 38.1 (4.5) | |||||
| I54L | 217.7 ± 14.2 (2.9) | |||||
| I50V | 230.9 ± 32.2 (3.0) | 12.9 ± 0.0 (0.4) | 41.9 ± 7.8 (1.7) | 9.1 ± 2.3 (0.4) | 99.2 ± 11.9 (1.7) | 3.6 ± 1.0 (0.5) |
| L449F | 65.0 ± 21.6 (0.9) | 22.0 ± 2.3 (0.7) | 17.2 ± 1.1 (0.7) | 14.5 ± 4.0 (0.7) | 34.8 ± 4.1 (0.6) | 5.4 ± 1.6 (0.8) |
| P453L | 43.0 (0.6) | |||||
| I50V M46I | 816.1 ± 66.2 (10.8) | 190.4 ± 56.3 (6.4) | ||||
| I50V L449F | 772.5 ± 104.1 (10.2) | 22.0 ± 0.8 (0.7) | 84.4 ± 6.3 (3.5) | 16.1 ± 8.0 (0.7) | 157.4 ± 3.3 (2.7) | 4.6 ± 0.9 (0.7) |
| I50V P453L | 815.0 ± 106.5 (10.8) | 43.8 ± 21.4 (1.3) | 116.8 ± 24.0 (4.8) | 28.5 ± 2.1 (1.3) | 216.2 ± 12.7 (3.7) | 6.7 ± 1.7 (1.0) |
| I50V M46I L449F | 1,150.9 ± 41.5 (15.2) | 59.2 ± 21.9 (1.8) | 189.7 ± 24.1 (7.8) | 81.4 ± 45.4 (3.8) | 396.8 ± 149.7 (6.8) | 7.2 ± 0.6 (1.0) |
| I50V M46I P453L | 1,203.2 ± 70.0 (15.9) | 53.4 ± 24.3 (1.6) | 189.5 ± 14.9 (7.8) | 71.6 ± 42.1 (3.3) | 402.6 ± 179.8 (6.9) | 7.3 ± 1.4 (1.0) |
All experiments were done in triplicate. All APV IC50s for I50V-containing mutants are the results of five or more independent experiments. Numbers in Parentheses are fold shifts in sensitivities relative to that for HXB2. NFV, nelfinavir; SQV, saquinavir.
TABLE 4.
Relative drug susceptibilities and replicative capacities of HIV-1 protease and Gag mutant viruses determined by a single-cycle sensitivity assaya
| Virus | Repli- cative capacity (%) | Susceptibilityb to:
|
|||||
|---|---|---|---|---|---|---|---|
| APV | IDV | LPV | NFV | RTV | SQV | ||
| I50V | <1 | ND | ND | ND | ND | ND | ND |
| L449F | 68 | 1.3 | 0.8 | 0.9 | 1.0 | 1.2 | 0.9 |
| P453L | 82 | 0.9 | 0.8 | 0.8 | 0.9 | 1.2 | 0.8 |
| I50V M46I | 6 | 5.4 | 0.4 | 3.2 | 1.0 | 2.3 | 0.4 |
| I50V L449F | 1 | 5.9 | 0.5 | 2.5 | 0.7 | 1.9 | 0.5 |
| I50V P453L | <1 | 4.2 | 0.4 | 1.8 | 0.7 | 2.2 | 0.4 |
| I50V M46I L449F | 16 | 9.8 | 0.5 | 4.4 | 1.1 | 3.4 | 0.6 |
| I50V M46I P453L | 6 | 7.7 | 0.6 | 3.8 | 1.0 | 2.5 | 0.5 |
| I50V L449F P453L | 1 | 3.4 | 0.5 | 2.2 | 0.7 | 1.9 | 0.3 |
| I50V M46I L449F P453L | 14 | 8.5 | 0.6 | 6.7 | 1.3 | 4.3 | 0.7 |
In vitro replicative capacities and drug susceptibilities were determined as described in Materials and methods and as described previously (33).
Values are factors by which susceptibility differs from that for the reference virus. NFV, nelfinavir; SQV, saquinavir. ND, not determined.
The degree to which inhibitory potency was lost varied between the single- and multiple-cycle assays, with noticeably higher IC50 increases seen in the multiple-cycle assay. However, there was good overall concordance between the test systems regarding the ranking of mutants according to their sensitivities to inhibition by APV, which has the following proposed order: wt ≈ P453L ≈ L449F > I50V > I50V L449F ≈ I50V P453L ≈ I50V M46I > I50V M46I P453L ≈ I50V M46I L449F.
The coexistence of both Gag changes could not be demonstrated in any of the clones or population sequences analyzed in this study. We were intrigued by this observation and tested the hypothesis that the CS double mutant would not produce viable virus in vitro. A I50V M46I L449F P453L quadruple-mutant construct was generated and could be grown in both assays. This recombinant displayed a level of APV resistance similar to that observed for both of the triple mutants with only one CS change (Tables 3 and 4).
Cross-resistance to other currently available PIs conferred by the APV-specific mutants was also determined in both sensitivity assays. In general, some effect on viral susceptibility phenotypes was detected with lopinavir (LPV) and ritonavir (RTV). These findings are in concordance with earlier observations for RTV (42) and the level of susceptibility to LPV demonstrated was within the clinically relevant range currently applied for this compound (Kaletra; U.S. prescribing information, Abbott Laboratories reference 03-5061, 2000). Fold shifts in viral sensitivities to other PIs were higher in the multiple-cycle assay than in the single-cycle assay (Tables 3 and 4).
Replicative capacity of HIV-1 recombinant viruses with alterations in protease and Gag p6.
The effect exerted by protease and Gag mutations on viral replicative capacity was determined by measuring relative luciferase activities in the single-cycle assay (Table 4). The I50V single mutant did not produce a sufficient luminescence signal for quantification of the fitness level of this virus. Addition of P453L to the I50V mutant yielded virus replication levels sufficient to perform drug sensitivity assays. A slight improvement in viral growth was detected with the I50V L449F mutant. Inclusion of the M46I change in this background resulted in further restoration of viral growth, which was, however, less than 20% that of the wt control. The ranking of in vitro viral fitness for the mutants tested in this study was I50V ≈ I50V P453L < I50V L449F < I50V M46I ≈ I50V M46I P453L < I50V M46I L449F < P453L ≈ L449F < wt.
Effect of protease and CS alterations on Gag protein processing in vitro.
To further characterize the molecular basis of the observed effects of I50V CS mutants on virus replication and drug sensitivities, we investigated the composition of Gag proteins in cell cultures infected with different recombinant protease and CS mutant viruses. In the absence of P453L or L449F, recombinants expressing I50V showed a defect in cleavage of the p1/p6 site, as demonstrated by the accumulation of p15 and a p7 (p1-p6) product in the supernatants of both I50V and I50V M46I viruses (Fig. 1). The latter protein was recognized by the anti-p6 antibody and was not observed in variants without I50V or in variants with I50V plus one of the CS changes. It should be noted that significantly smaller amounts of p7 (p1-p6) were detected in overexposed Western blots of I50V P453L culture supernatants. Due to the observed variability of protein production, it was not possible to carry out a quantitative comparison of Gag proteins among the mutants investigated. The presence of nonspecific bands upon incubation with the anti-p6 antibody has been observed by others previously (9). Incubation of the identical blots with the anti-p24 antibody resulted in demonstration of p55 and p24 proteins in all viruses, but relative amounts of p24 in relation to p55 appeared to be highest in wt, P453L, and L449F culture supernatants (Fig. 1 and densitometric analysis; data not shown).
FIG. 1.

Analysis of Gag cleavage products from protease and Gag p1/p6-substituted HIV-1 recombinants. Supernatants from cell cultures infected with the indicated mutants were analyzed for differences in Gag processing. (Top) Immunoblot of SDS-polyacrylamide gel electrophoresis product incubated with an anti-p6 antibody. (Middle) Magnified and overexpressed low-molecular-weight section of the blot shown in the top panel. (Bottom) Identical blot as in the top and middle panels except that the product was incubated with an anti-p24 antibody. •, unprocessed p1-p6 intermediate (p7); ∗, processed p6; cc, cell control.
Cleavage of wt and mutant peptide substrates by wt and I50V protease in vitro.
We confirmed the defect in cleavage at the p1/p6 site by incubation of purified recombinant HIV-1 I50V or wt protease with synthetic decameric peptides mimicking the Gag p1/p6 CS. kcat/Km values were obtained for the HIV-1 clade B consensus peptide and altered oligomers specifying sequences with either P453L or L449F substitutions (Table 5). In this assay, the rate of cleavage of the consensus peptide by the I50V enzyme (0.00006 μM−1 s−1) was 0.3% of the rate observed with the unaltered protease (0.0198 μM−1 s−1). Enhanced enzymatic activity was observed with both proteases in the presence of mutant peptides, but the I50V L449F enzyme-substrate combination, which appeared to be favored both in vitro and in patients, was still less efficient than the wt (∼14-fold difference in kcat/Km).
TABLE 5.
Catalytic efficiencies of HIV-1 wt and I50V proteases with wt and p1/p6 mutant peptide substrates
| Peptide | Sequence |
kcat/Km (μM−1 s−1) for:
|
|
|---|---|---|---|
| E(wt) | E(I50V) | ||
| MA/CA | Dabcyl-γAbu VSQNY/PIVQNγAbu-Edans | 0.162 ± 0.005 | 0.0028 ± 0.0001 |
| CA/p2 | Dabcyl-γAbu KARVL/AEAMSγAbu-Edans | 0.27 ± 0.01 | 0.0130 ± 0.0004 |
| P2/NC | Dabcyl-γAbuSATIM/MORGNγAbu-Edans | 0.53 ± 0.01 | 0.0070 ± 0.0001 |
| NC/p1 | Dabcyl-γAbuERQAN/FLGKIγAbu-Edans | 0.00051 ± 0.00001 | 0.00002 ± 0.00001 |
| NC/TFP | Dabcyl-γAbuERQAN/FLREDγAbu-Edans | 0.00010 ± 0.00001 | <0.00001 |
| TFP/P6pol | Dabcyl-γAbuEDLAF/LQGKAγAbu-Edans | 0.0022 ± 0.0001 | 0.000060 ± 0.000002 |
| P1/p6gag | Dabcyl-γAbuRPGNF/LQSRPγAbu-Edans | 0.0198 ± 0.0001 | 0.00006 ± 0.00001 |
| Dabcyl-γAbuRPGNF/LQSRLγAbu-Edans (P453L) | 0.0620 ± 0.0006 | 0.00061 ± 0.00002 | |
| Dabcyl-γAbuRPGNF/FQSRPγAbu-Edans (L449F) | 0.1660 ± 0.0030 | 0.00139 ± 0.00002 | |
| P6pol/PR | Dabcyl-γAbuVSFNF/PQVTLγAbu-Edans | 0.115 ± 0.004 | 0.0049 ± 0.0001 |
| PR/RT | Dabcyl-γAbuCTLNF/PISPIγAbu-Edans | 0.028 ± 0.001 | 0.00067 ± 0.00002 |
| p51/p66 | Dabcyl-γAbuGAETF/YVDGAγAbu-Edans | 0.150 ± 0.002 | 0.0078 ± 0.0001 |
| p66/INT | Dabcyl-γAbuIRKVL/FLDGIγAbu-Edans | 0.235 ± 0.008 | 0.0101 ± 0.0003 |
| NEF | Dabcyl-γAbuAACAW/LEAQEγAbu-Edans | 0.076 ± 0.001 | 0.0024 ± 0.0001 |
The I50V enzyme showed lower catalytic efficiency than the wt enzyme with all wt peptides tested, and three of these wt substrates (NC/TFP, NC/p1, and TFP/p6) had kcat/Km values with I50V protease that were lower than the value obtained with wt p1/p6 peptide and this enzyme (Table 5).
DISCUSSION
We studied the significance of changes at the HIV-1 Gag p1/p6 CS for development of resistance to APV. Of particular interest and novelty is the finding that CS changes L449F and P453L, when present with substitutions in the protease, not only had an impact on the in vitro fitness of the respective variants but also consistently decreased the level of phenotypic sensitivity to APV.
Altered p1/p6 CS were found in all viruses from patients with the I50V substitution in the protease. Retrospective sequence analysis of in vitro-selected APV-resistant viruses confirmed a parallel sequence evolution at position 50 of the protease and at the Gag site. Replicative capacity was dramatically reduced with the I50V single mutant. Mutations at both codon 46 of the protease gene and the p1/p6 CS partially restored this defect. In all cell cultures infected with site-directed mutant HIV-1 expressing I50V without a CS alteration, a processing defect of the p15 precursor to the final p7, p1, and p6 products was evident and could be demonstrated by the accumulation of uncleaved p1-p6 protein. Accumulation of p15 and p7 (p1-p6) has been observed by others using different protease mutant viruses. In these studies, the addition of CS (p6/p1 and p7/p1) mutations also resulted in efficient processing of these precursors (9, 23). Microscopic evaluation of infected cells suggested differences in the morphology of the viral cytopathic effect, with I50V cultures showing larger syncytia and giant cells than viruses that contained substitutions at the CS in addition to I50V (data not shown).
In this study, the observed mutations in gag have been related to their effects on the Gag p1/p6 CS. It should be noted that these mutations may have other consequences for assembly and particle maturation: a proline-rich sequence at the N terminus of HIV-1 Gag p6 has been identified previously to be essential for efficient particle release. This region includes the PTAPP motif and has been shown to be involved in viral maturation events such as packaging of processed Pol proteins during late assembly (14, 28). P453 is part of this proline-rich sequence and is located two amino acids upstream from the first proline in PTAPP. Of note, a P453R P455Q mutated virus is growth restricted in primary monocytes, and this phenotype has been shown to be accompanied by a greatly reduced virion incorporation of Pol proteins (8). In addition, the mutation causing the Gag L449F change also leads to an S9F alteration in p6pol, whereas the P453L mutant is silent with respect to the p6 pol protein sequence. Therefore, both mutations may affect viral maturation steps other than cleavage of Gag p1/p6.
The block in viral processing and assembly was confirmed by the poor ability of purified I50V protease to process the wt p1/p6 substrate, whereas peptides specifying either of the CS changes observed in vitro and in patients improved the ability of the mutant protease to cleave this substrate. These data suggest that the virus escapes from drug pressure because the protease and Gag adapt in a concerted manner at multiple sites as follows: (i) by decreasing the affinity of the protease active site for APV and (ii) by increasing the ability of the virus to replicate efficiently. The CS changes contributed to both a drug resistance phenotype and viral fitness in the absence of a drug. Compared to I50V M46I, I50V P453L, or I50V L449F double mutants, the I50V M46I L449F mutant had higher resistance at a significantly increased fitness level. Neither L449F nor P453L measurably decreased the susceptibility to any PI when these Gag substitutions were present as single mutations. The increased efficiency of wt protease in cleaving both L449F and P453L peptides is likely to be suboptimal in terms of the coordinated Gag precursor processing needed for virus assembly in the absence of drug pressure. These data are in concordance with previously published results, which also showed an increased ability of wt and mutant proteases to catalyze cleavage of altered p1/p6 and p7/p1 peptides found in PI-resistant viruses (9). A recent study investigated the specificity of clinically derived protease mutants (7). By using substrate libraries mimicking sequence elements of the p7/p1 CS it could be shown that many drug-resistant proteases exhibit altered substrate specificities. At the P2 substrate position a high preference for Ile and Val was found, with the latter residue corresponding to the A431V CS mutation. These findings suggest that altered CS evolve in order to maintain the substrate specificity of proteases that have undergone sequence changes as a result of drug pressure (7).
The role of CS changes has been discussed in the context of improved viral replication with primary substitutions in the protease. The contribution of the altered CS to increased IC50s of the PIs against resistant viruses could not be demonstrated with the mutants investigated (6, 9, 23, 48, 49). However, Mammano et al. (24) used a single-cycle infectivity assay to quantify the effects of various combinations of protease and Gag substitutions on viral replication in the presence of different inhibitor concentrations. It was shown that the addition of the Gag p7/p1 A431V mutation to a protease I54V V82A double mutant increased the efficiency of replication only at intermediate RTV concentrations, not in the presence of high concentrations of RTV or in the absence of the drug (24). In another study, site-directed HIV-1 mutants resistant to the PI ABT-378 (LPV) were tested for their inhibitor sensitivities in the presence or absence of CS mutations. One out of three virus pairs analyzed showed a fourfold difference in IC50 when incubated with ABT-378, with a higher degree of resistance when Gag L449F was added to the L10F V32I M46I I47V I84V T91S protease background (3).
The impact of CS changes on phenotypic resistance may depend on the nature of protease and CS changes, as well as the test system used to determine drug susceptibility. Nevertheless, the data presented here suggest an influence not only of the protease sequence but also of the Gag p1/p6 protease substrate sequence on the ability of HIV-1 protease to bind APV in the single- and multiple-cycle assay systems used in this study. The observed changes in the CS likely cause higher affinity of the substrate for the protease, thereby increasing the IC50 measured with APV. Of note, a CS change at the distant P5′ position (P453L) can have an effect on viral fitness and inhibitor sensitivity similar to that of a change at the P1′ (L449F) position, the latter being located directly adjacent to the scissile peptide bond.
There is a question as to whether, in the specific situation of APV resistance mediated by I50V variants, the CS alterations must first be present in order for the I50V substitution to be viable. The in vitro passage data presented show the presence of I50V and L449F on the same and on separate sequences, indicating independent primary formation of both mutations. One could hypothesize that initially the copresence of I50V L449 and wt L449F viruses in a dually infected cell would be required to produce heterozygous progeny virions. I50V-L449F linkage would then occur via homologous recombination during reverse transcription in a newly infected cell.
The appearance of nonconsensus CS sequences in viruses from PI-treated patients can partly be explained by the occurrence of natural polymorphisms in Gag. The P453L mutation has been shown to occur in HIV-1 protease from untreated subjects and influenced the nature of protease mutants selected in patients failing therapy. In one study, I84V was significantly associated with the presence of P453L before the start of therapy, and deviations from V82 were negatively associated with P453L (1). The copresence of I84V and P453L in viruses from two patients who virologically failed multiple-PI therapy was described in a different study (2). We confirmed a highly significant association of I84V and P453L in a clinical virologic data repository. Moreover, considering all I50V isolates from a defined clinical trial conducted with APV, all mutants that did not contain L449F contained P453L. We observed the shift from I50V M46L P453L populations to I50V M46I L449F populations in one patient. L449F conferred higher replicative capacity than P453L in a I50V M46I protease background. L449F in the Gag p1/p6 peptide improved catalytic efficiency of the I50V protease to a greater extent than P453L. Taken together, these data lead to the conclusion that I50V in HIV-1 protease does not confer viability in vivo without a concurrent change at the Gag p1/P6 CS. L449F appears to be ultimately selected due to favorable molecular interactions with the mutant enzyme: the I50 side chain has been shown to make extensive contacts spanning the P2-P2′ positions of synthetic substrates (30). Hydrophobic interactions and van der Waals contacts are lost with the smaller Val residue present in I50V (R. Xu, W. Andrews, A. Spaltenstein, D. Danger, W. Dallas, L. Carter, M. Hanlon, L. Wright, and E. Furfine, 5th Int. Workshop HIV Drug Resist. Treat. Strateg., Scottsdale, Ariz., 4 to 8 June 2001, abstr. 54). With L449F, the aromatic Phe in the P1′ position of the substrate is likely to result in additional hydrophobic interactions with active-site residues.
Development of reduced sensitivity to certain PIs such as APV involves multiple structural adaptations of both protease and Gag proteins. We have studied amino acid substitutions at the Gag p1/p6 CS. With the exception of the p7/p1 site, the influence of other protease CS inside and outside Gag on PI resistance has not been investigated in great detail. However, one study on viral genotypes of 28 PI-treated patients who harbored PI cross-resistant viruses demonstrated evolution at other CS under PI selective pressure. For example, mutations affecting the Gag p2/p7 CS were significantly more frequent in viruses from the PI-treated patients than in those from a control group (5). The data presented here show reduced efficiency of an I50V mutant enzyme in cleaving all known wt CS substrates. Taken together, these findings suggest that there is selective pressure on CS other than p7/p1 and p1/p6 in the context of some drug-resistant proteases.
In conclusion, the consequences of amino acid changes L449F and P453L at the Gag p1/p6 CS include partial restoration of viral fitness through more-efficient cleavage by mutant protease and reduction of inhibitor sensitivity. The clinical and biochemical characteristics of Gag P453L have been described in detail for the first time in this study, and the biological implications of this substitution may extend to late-stage assembly events such as Pol protein packaging. These findings warrant further studies of CS substrate substitutions in PI-resistant HIV-1 to improve the understanding of viral maturation as well as molecular mechanisms of inhibitor escape.
Acknowledgments
We thank Andrew Theaker and Marcus Oxer for their continuous help with in silico analyses of clinical resistance data and Luke Carter for preparation of purified HIV-1 protease.
REFERENCES
- 1.Bally, F., R. Martinez, S. Peters, P. Sudre, and A. Telenti. 2000. Polymorphism of HIV type 1 gag p7/p1 and p1/p6 cleavage sites: clinical significance and implications for resistance to protease inhibitors. AIDS Res. Hum. Retroviruses 16:1209-1213. [DOI] [PubMed] [Google Scholar]
- 2.Bleiber, G., M. Munoz, A. Ciuffi, P. Meylan, and A. Telenti. 2001. Individual contributions of mutant protease and reverse transcriptase to viral infectivity, replication, and protein maturation of antiretroviral drug-resistant human immunodeficiency virus type 1. J. Virol. 75:3291-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carrillo, A., K. D. Stewart, H. L. Sham, D. W. Norbeck, W. E. Kohlbrenner, J. M. Leonard, D. J. Kempf, and A. Molla. 1998. In vitro selection and characterization of human immunodeficiency virus type 1 variants with increased resistance to ABT-378, a novel protease inhibitor. J. Virol. 72:7532-7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Condra, J. H., W. A. Schleif, O. M. Blahy, L. J. Gabryelski, D. J. Graham, J. C. Quintero, A. Rhodes, H. L. Hobbins, E. Roth, M. Shivaprakash, D. L. Titus, T. Yang, H. Teppler, K. E. Squires, P. J. Deutsch, and E. A. Emini. 1994. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature 374:569-571. [DOI] [PubMed] [Google Scholar]
- 5.Côté, H. C. F., Z. L. Brumme, and P. R. Harrigan. 2001. Human immunodeficiency virus type 1 protease cleavage site mutations associated with protease inhibitor cross-resistance selected by indinavir, ritonavir, and/or saquinavir. J. Virol. 75:589-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Croteau, C., L. Doyon, D. Thibeault, G. McKercher, L. Pilote, and D. Lamarre. 1997. Impaired viral fitness of human immunodeficiency virus type 1 variants with high-level resistance to protease inhibitors. J. Virol. 71:1089-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dauber, D. S., R. Ziermann, N. Parkin, D. J. Maly, S. Mahrus, J. L. Harris, J. A. Ellman, C. Petropoulos, and C. S. Craik. 2002. Altered substrate specificity of drug-resistant human immunodeficiency virus type 1 protease. J. Virol. 76:1359-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dettenhofer, M., and X. F. Yu. 1999. Proline residues in human immunodeficiency virus type 1 p6Gag exert a cell type-dependent effect on viral replication and virion incorporation of Pol proteins. J. Virol. 73:4696-4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doyon, L., G. Croteau, D. Thibeault, F. Poulin, L. Pilote, and D. Lamarre. 1996. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J. Virol. 70:3763-3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erickson, J. W., S. V. Gulnik, and M. Markowitz. 1999. Protease inhibitors: resistance, cross-resistance, fitness and the choice of initial and salvage therapies. AIDS 13:S189-S204. [PubMed] [Google Scholar]
- 11.Eron, J. J. 2000. HIV-1 protease inhibitors. Clin. Infect. Dis. 30(Suppl. 2):S160-S170. [DOI] [PubMed] [Google Scholar]
- 12.Falloon, J., M. Ait-Khaled, D. A. Thomas, C. L. Brosgart, J. J. Eron, J. Feinberg, T. P. Flanigan, S. M. Hammer, P. W. Kraus, R. Murphy, R. Torres, H. Masur, and the CNAA2007 Study Team. 2002. HIV-1 genotype and phenotype correlate with virological response to abacavir, amprenavir and efavirenz in treatment-experienced patients. AIDS 16:387-396. [DOI] [PubMed] [Google Scholar]
- 13.Freund, J., R. Kellner, J. Konvalinka, V. Wolber, H.-G. Kräusslich, and H. R. Kalbitzer. 1994. A possible regulation of negative factor (Nef) activity of human immunodeficiency virus type 1 by the viral protease. Eur. J. Biochem. 223:589-593. [DOI] [PubMed] [Google Scholar]
- 14.Göttlinger, H. G., T. Dorfman, J. G. Sodroski, and W. A. Haseltine. 1991. Effects of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Aad. Sci. USA 88:3195-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gulick, R. M., J. W. Mellors, D. Havlir, J. J. Eron, A. Meibohm, J. H. Condra, F. T. Valentine, D. McMahon, C. Gonzalez, L. Jonas, E. A. Emini, J. A. Chodakewitz, R. Isaacs, and D. D. Richman. 2000. 3-year suppression of HIV viremia with indinavir, zidovudine, and lamivudine. Ann. Intern. Med. 133:35-39. [DOI] [PubMed] [Google Scholar]
- 16.Haugland, R. P. 1996. Handbook of fluorescent probes and research chemicals, 6th ed., p 232. Molecular Probes, Eugene, Oreg.
- 17.Henderson, L. E., M. A. Bowers, R. C. Sowder, S. A. Serabyn, D. G. Johnson, J. W. Bess, L. O. Arthur, D. K. Bryant, and C. Fenselau. 1992. Gag proteins of the highly replicative MN strain of human immunodeficiency virus type 1: posttranslational modifications, proteolytic processing, and complete amino acid sequences. J. Virol. 66:1856-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozal, M. J., N. Shah, N. Shen, R. Yang, R. Fucini, T. C. Merigan, D. D. Richman, D. Morris, E. Hubbell, M. Chee, and T. R. Gingeras. 1996. Extensive polymorphisms observed in HIV-1 clade B protease gene using high-density oligonucleotide arrays. Nat. Med. 2:753-759. [DOI] [PubMed] [Google Scholar]
- 19.Larder, B. A., S. D. Kemp, and R. A. Harrigan. 1995. Potential mechanism for sustained antiretroviral efficacy of AZT-3TC combination therapy. Science 269:696-699. [DOI] [PubMed] [Google Scholar]
- 20.Lech, W. L., G. Wang, Y. L. Lang, Y. Chee, K. Dorman, D. McCrae, L. C. Lazzeroni, J. W. Erickson, J. S. Sinsheimer, and A. H. Kaplan. 1996. In vivo sequence diversity of the protease of human immunodeficiency virus type 1: presence of protease inhibitor-resistant variants in untreated subjects. J. Virol. 70:2038-2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lightfoote, M. M., J. E. Coligan, T. M. Folks, A. S. Fauci, M. A. Martin, and S. Venkatesan. 1986. Structural characterization of reverse transcriptase and endonuclease polypeptides of the acquired immunodeficiency syndrome retrovirus. J. Virol. 60:771-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maguire, M., D. Shortino, A. Klein, W. Harris, V. Manohitharajah, M. Tisdale, R. Elston, J. Yeo, S. Randall, F. Xu, H. Parker, J. May, and W. Snowden. 2002. Emergence of resistance to protease inhibitor amprenavir in human immunodeficiency virus type 1-infected patients: selection of four alternative viral protease genotypes and influence of viral susceptibility to coadministered reverse transcriptase nucleoside inhibitors. Antimicrob. Agents Chemother. 46:731-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mammano, F., C. Petit, and F. Clavel. 1998. Resistance-associated loss of viral fitness in human immunodeficiency virus type 1: phenotypic analysis of protease and Gag coevolution in protease inhibitor-treated patients. J. Virol. 72:7632-7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mammano F., V. Trouplin, V. Zennou, and F. Clavel. 2000. Retracing the evolutionary pathways of human immunodeficiency virus type 1 resistance to protease inhibitors: virus fitness in the absence and in the presence of drug. J. Virol. 74:8524-8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markland, W., B. G. Gao, J. D. Parsons, J. Black, L. Zuchowski, M. Tisdale, and R. Tung. 2000. Structural and kinetic analyses of the protease from amprenavir-resistant human immunodeficiency virus type 1 mutant rendered resistant to saquinavir and resensitized to amprenavir. J. Virol. 74:7636-7641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller, V. 2001. Resistance to protease inhibitors. J. Acquired Immune Defic. Syndr. 26:S34-S50. [DOI] [PubMed] [Google Scholar]
- 27.Molla, A., M. Korneyeva, Q. Gao, S. Vasavanonda, P. J. Schipper, H.-M. Mo, M. Markowitz, T. Chernyavskiy, P. Niu, N. Lyons, A. Hsu, R. Granneman, D. D. Ho, C. A. B. Boucher, J. M. Leonard, D. W. Norbeck, and D. J. Kempf. 1996. Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nat. Med. 2:760-766. [DOI] [PubMed] [Google Scholar]
- 28.Parent, L. J., R. P. Bennett, R. C. Craven, T. D. Nelle, N. K. Krishna, J. B. Bowzard, C. B. Wilson, B. A. Puffer, R. C. Montelaro, and J. W. Wills. 1995. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J. Virol. 69:5455-5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Partaledis, J. A., K. Yamaguchi, M. Tisdale, E. D. Blair, C. Falcione, B. Maschera, R. E. Myers, S. Pazhanisamy, O. Futer, A. B. Cullinan, C. M. Stuver, R. A. Byrn, and D. L. Livingston. 1995. In vitro selection and characterization of human immunodeficiency virus type 1 (HIV-1) isolates with reduced sensitivity to hydroxyethylamino sulfonamide inhibitors of HIV-1 aspartyl protease. J. Virol. 69:5228-5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pashanizamy, S., C. M. Stuver, A. B. Cullinan, N. Margolin, B. G. Rao, and D. J. Livingston. 1996. Kinetic characterization of human immunodeficiency virus type-1 protease-resistant variants. J. Biol. Chem. 271:17979-17985. [DOI] [PubMed] [Google Scholar]
- 31.Pauwels, R., J. Balzarini, M. Baba, R. Snoeck, D. Schols, P. Herdewijn, J. Desmyter, and E. deClercq. 1988. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Methods 20:309-321. [DOI] [PubMed] [Google Scholar]
- 32.Pedneault, L., C. Brothers, G. Pagano, P. Tymkewycz, J. Yeo, J. Millard, and A. Fetter. 2000. Safety profile and tolerability of amprenavir in the treatment of adult and paediatric patients with HIV infection. Clin. Ther. 22:1378-1394. [DOI] [PubMed] [Google Scholar]
- 33.Petropoulos, C. J., N. T. Parkin, K. L. Limoli, Y. S. Lie, T. Wrin, W. Huang, H. Tian, D. Smith, G. A. Winslow, D. C. Capon, and J. M. Whitcomb. 2000. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 44:920-928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pettit, S. F., S. F. Michael, and R. Swanstrom. 1993. The specificity of the HIV-1 protease. Perspect. Drug Discov. Des. 1:69-83. [Google Scholar]
- 35.Porter, D. J. T., M. H. Hanlon, L. H. Carter III, D. P. Danger, and E. S. Furfine. 2001. Effectors of HIV-1 protease peptidolytic activity. Biochemistry 40:11131-11139. [DOI] [PubMed] [Google Scholar]
- 36.Richman, D. D. 2001. HIV chemotherapy. Nature 410:995-1001. [DOI] [PubMed] [Google Scholar]
- 37.Robinson, L. R., R. E. Myers, B. W. Snowden, M. Tisdale, and E. D. Blair. 2000. HIV type 1 protease cleavage site mutations and viral fitness: implications for drug susceptibility phenotyping assays. AIDS Res. Hum. Retroviruses 16:1149-1156. [DOI] [PubMed] [Google Scholar]
- 38.Shafer, R. W., P. Hsu, A. K. Patick, C. Craig, and V. Brendel. 1999. Identification of biased amino acid substitution patterns in human immunodeficiency virus type 1 isolates from patients treated with protease inhibitors. J. Virol. 73:6197-6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheng, N., and S. Erickson-Viitanen. 1994. Cleavage of p15 protein in vitro by human immunodeficiency virus type 1 protease is RNA dependent. J. Virol. 68:6207-6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheng, N., S. C. Pettit, R. J. Tritch, D. H. Ozturk, M. R. Rayner, R. Swanstrom, and S. Erickson-Viitanen. 1997. Determinants of the human immunodeficiency virus type 1 p15NC-RNA interaction that affect enhanced cleavage by the viral protease. J. Virol. 71:5723-5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tisdale, M., R. E. Myers, B. Maschera, N. R. Parry, N. M. Oliver, and E. D. Blair. 1995. Cross-resistance analysis of human immunodeficiency virus type 1 variants individually selected for resistance to five different protease inhibitors. Antimicrob. Agents Chemother. 39:1704-1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tisdale, M., R. Myers, S. Randall, M. Maguire, M. Ait-Khaled, R. Elston, and W. Snowden. 2000. Resistance to the HIV protease inhibitor amprenavir in vitro and in clinical studies—a review. Clin. Drug Investig. 20:267-285. [Google Scholar]
- 43.Tritch, R. J., Y. S. E. Cheng, F. H. Yin, and S. Erickson-Viitanen. 1991. Mutagenesis of protease cleavage sites in the human immunodeficiency virus type 1 Gag polyprotein. J. Virol. 65:922-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Velasquez-Campoy, A., M. J. Todd, S. Vega, and E. Freire. 2001. Catalytic efficiency and vitality of HIV-1 proteases from African viral subtypes. Proc. Natl. Acad. Sci. USA 98:6062-6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vergne, L., M. Peeters, E. Mpoudi-Ngole, A. Bourgeois, F. Liegois, C. Toure-Kane, S. Mboup, C. Mulanga-Kabeya, E. Saman, J. Jourdan, J. Reynes, and E. Delaporte. 1998. Genetic diversity of protease and reverse transcriptase sequences in non-subtype B human immunodeficiency virus type 1 strains: evidence of many minor drug resistance mutations in treatment-naive patients. J. Clin. Microbiol. 38:3919-3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Veronese, F. D., R. Rahman, T. D. Copeland, S. Oroszlan, R. C. Gallo, and M. G. Sarngadharan. 1987. Immunological and chemical analysis of p6, the carboxy-terminal fragment of HIV p15. AIDS Res. Hum. Retroviruses 3:253-264. [DOI] [PubMed] [Google Scholar]
- 47.Welker, R., H. Kottler, H. R. Kalbitzer, and H. G. Kräusslich. 1996. Human immunodeficiency virus type 1 Nef protein is incorporated into virus particles and specifically cleaved by the viral proteinase. Virology 219:228-236. [DOI] [PubMed] [Google Scholar]
- 48.Zennou, V., F. Mammano, S. Paulous, D. Mathez, and F. Clavel. 1998. Loss of viral fitness associated with multiple Gag and Gag-Pol processing defects in human immunodeficiency virus type 1 variants selected for resistance to protease inhibitors in vivo. J. Virol. 72:3300-3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang, Y.-M., H. Imamichi, T. Imamichi, H. C. Lane, J. Falloon, M. B. Vasudevachari, and N. P. Salzman. 1997. Drug resistance during indinavir therapy is caused by mutations in the protease gene and its Gag substrate cleavage sites. J. Virol. 71:6662-6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ziermann, R., K. Limoli, K. Das, E. Arnold, C. J. Petropoulos, and N. T. Parkin. 2000. A mutation in human immunodeficiency virus type 1 protease, N88S, that causes in vitro hypersensitivity to amprenavir. J. Virol. 74:4414-4419. [DOI] [PMC free article] [PubMed] [Google Scholar]