

Abstract
Among single-spanning transmembrane receptors (sTMRs), two guanylyl cyclase receptors, GC1 and GC2, are critically important during phototransduction in vertebrate retinal photoreceptor cells. Ca2+-free forms of guanylyl cyclase-activating proteins (GCAPs) stimulate GCs intracellularly by a molecular mechanism that is not fully understood. To gain further insight into the mechanism of activation and specificity among these proteins, for the first time, several soluble and active truncated GCs and fusion proteins between intracellular domains of GCs and full-length GCAPs were generated. The GC activity of myristoylated GCAP–437–1054GC displayed typical [Ca2+] dependence, and was further enhanced by ATP and inhibited by guanylyl cyclase inhibitor protein (GCIP). The myristoyl group of GCAP1 appeared to be critical for the inhibition of GCs at high [Ca2+], even without membranes. In contrast, calmodulin (CaM)–437–1054GC1 fusion protein was inactive, but could be stimulated by exogenous GCAP1. In a series of experiments, we showed that the activation of GCs by linked GCAPs involved intra- and intermolecular mechanisms. The catalytically productive GCAP1–437–1054GC1 complex can dissociate, allowing binding and stimulation of the GC1 fusion protein by free GCAP1. This suggests that the intramolecular interactions within the fusion protein have low affinity and are mimicking the native system. We present evidence that the mechanism of GC activation by GCAPs involves a dimeric form of GCs, involves direct interaction between GCs and GCAPs, and does not require membrane components. Thus, fusion proteins may provide an important advance for further structural studies of photoreceptor GCs and other sTMRs with and without different forms of regulatory proteins.
The intracellular regulation of single transmembrane receptors (sTMRs) by soluble proteins is at the heart of many fundamental cellular processes. For example, tyrosine autophosphorylation of growth hormone sTMRs and other related receptors modulate their own kinase activity. Consequently, autophosphorylated sTMRs recruit intracellular proteins to plasma membranes, including phospholipase C and ras GTPase-activating protein, or proteins containing Src homology domains (1). In vertebrate retina, sTMR guanylyl cyclases GC1 and GC2 are responsible for the production of the internal messenger cGMP, which is depleted via photoactivated rhodopsin/G-protein-mediated stimulation of phosphodiesterase. The increase production of cGMP occurs concomitantly with lower intracellular [Ca2+], which is detected by small Ca2+-binding proteins (CBPs), termed guanylyl cyclase-activating proteins (GCAP1–3), that in turn regulate GCs (2-4). GCAP1 and GCAP3 are more closely related than GCAP2; however, these three proteins share strong similarity in overall topology. They are myristoylated and contain four EF-hand motifs with EF1 nonfunctional, as a consequence of the lack of key residues involved in Ca2+ coordination. GCAP1 and GCAP2 are rod- and cone-specific proteins (2-4), while GCAP3 is only expressed in cones (Imanishi et al., unpublished results).
Regulation of GCs could be considered as a model system for understanding the molecular mechanisms of intracellular interactions between sTMRs and intracellular CBPs. In addition, the malfunction of this regulation may lead to severe signal transduction abnormalities. For example, missense mutations constitutively activating GCAP1 or disabling mutations of GC1 lead to retina dystrophies and blindness (5-9).
Photoreceptor GCs are single-spanning plasma or internal membrane receptors and are thought to be associated with cytoskeletal structures of rod and cone outer segments (10, 11). For photoreceptor GCs, the intracellular regulation by GCAPs is so far the only known mechanism that controls enzymatic activity. In contrast, for other nonretinal GCs, the best-characterized mechanism of regulation of enzymatic activity is through binding of a peptide to the extracellular domain of GCs (12). All GCs display a significant relationship to vertebrate adenylyl cyclases (ACs) with respect to the catalytic mechanisms of cyclic nucleotide synthesis, including a conserved set of key residues involved in enzymatic catalysis (13, 14), although the transmembrane segments of ACs and GCs are dissimilar (15-17). Various AC isozymes are also regulated in a subtype-specific manner by intracellular proteins, such as G-protein α- and βγ-subunits, calmodulin (CaM)(18), and, interestingly, by a GCAP-like protein, termed VILIP1 (19, 20). Here, we use novel fusion proteins between photoreceptor GCs and CBPs to understand the mechanism and regulation of cGMP production. We found that activation by GCAPs involves a dimeric form of GC, direct interaction between GCs and GCAPs, and that the membrane components are dispensable during this process. This novel approach may provide an important step for further structural studies, not only for photoreceptor GCs with and without different forms of regulatory proteins, but also for other sTMRs.
MATERIALS AND METHODS
Cloning of the Intracellular Domain of Bovine GC1 and GC2. PCR products were cloned into pCRII-TOPO vector (TOPO TA Cloning Kit, Invitrogen) and sequenced by the dyedeoxyterminator method (ABI-Prism, Perkin-Elmer).
A KpnI site was introduced at the N-terminus of the intracellular domain of bovine GC1 by PCR with primers GCa (5′-CGGTACCAGGCACCGGCTGCTTCACATCCA-3′) and GCb (5′-CCCTCTTCACCACTTCCTCGGGA-3′). The intracellular domain of bovine GC1 (amino acids 437–1054) was then transferred between sites KpnI and BamHI of the pET30b vector (Novagen) in two fragments, KpnI–XmnI (PCR fragment) and XmnI–BamHI, covering the C-terminus of bovine GC1.
The intracellular domain of retGC2 (amino acids 437–1054) was amplified from a bovine retina cDNA library in two fragments with primers GC6 (5′-GGTACCAGACATCGTATAAATAAAATCCAG-3′) and GC9 (5′-CAAAGCTATAGACGTCTCCTG-3′) and primers GC8 (5′-CAGGAGACGTCTATAGCTTTG-3′) and GC10 (5′-GTCGACTCACCAGCTGCTTTTCTGC-3′). These KpnI–AatII and AatII–SalI fragments covering the intracellular domain of retGC2 were then transferred between the KpnI and SalI sites of pET30b.
The 437–1054GC1 and 437–1054GC2 fused to a His6-tag/thrombin/S-tag/enterokinase peptide linker (36 amino acids long, MHHHHHHSSGLVPRGSGMKETAAAKFERQHMDSPDL) were transferred as a XbaI–XhoI fragment into the pFastBac1 expression vector (Bac-to-Bac system, Life Technologies, Inc.). SF9 insect cells were transfected with the recombinant bacmids using cationic liposome-mediated transfection (CellFECTIN reagent, Life Technologies, Inc.). Expression of recombinant proteins was tested 3 days after infection.
Cloning of the Truncated Intracellular Domain of GC1. Truncated GC1 was amplified from a bovine retina cDNA library into two fragments. 468–1054GC1 was amplified by PCR with primers PL4 (5′-ATGGGGAATTCTCGAAAGGTGGCCCAG-3′) and PL7 (5′-GGATCAGATCTTCCAGGTTACTG-3′) and primers PL6 (5′-ATGGAAGATCTGATCCGGGAGCGCACA-3′) and XE2 (5′-GAATTCTCACTTCCCAGAAAAC-3′). 514–1054GC1 was cloned by PCR with primers GC19 (5′-GATATCCCCAGGAGATCGGCATATAG-3′) and PL7 and primers PL6 and XE2. These EcoRI–BglII or EcoRV–BglII fragments, for 468–1054GC1 and 514–1054GC1, respectively, were cloned with the BglII–EcoRI fragment into pET30b-opened EcoRI or EcoRV–EcoRI.
Cloning of GCAP–GC Fusion Proteins. Bovine GCAP1, GCAP2, or CaM was fused to the intracellular domain of bovine GC1 or GC2 in the pET30b vector. The coding sequences of bovine GCAP1, GCAP2, and CaM were amplified by PCR to remove the stop codon using primers FH23 (5′-CATATGGGGAACATTATGGAC-3′) and LN1 (5′-CATATGACCGTCGGCCTCCGCG-3′) for GCAP1, primers GCAP2U (5′-CACGGATCCATGGGGCAGCAGTTCAGCTGGGAG-3′) and LN2 (5′-CATATGGAACATGGCACTTTT-3′) for GCAP2, and primers FH30 (5′-CATATGGCTGACCAACTCAC-3′) and KP142 (5′-CATATGCTTAGCCGTCATCATTTG-3′) for CaM. The PCR was heated for 5 min at 94 °C, followed by 25 cycles at 94 °C for 30 s, primer specific temperatures for 30 s, and 68 °C for 2 min followed by 7 min at 68 °C. GCAP1 and CaM were then cloned into the NdeI site, GCAP2 between the XbaI and NdeI sites of pET30b vector upstream of the 36 amino acid long linker–437–1054GC1, –468–1054GC1, –514–1054GC1, or –437–1054GC2. The GCAP–linker–GC fusion proteins were then transferred as fragments XbaI–XhoI into the pFastBac1 vector and expressed in insect cells as described above.
Cloning of His-tag–, PPE-tag–, and Strep-tag–GCAP–GC Fusion Proteins. NcoI and NdeI restriction sites were inserted by PCR, respectively, on the ATG or before the stop codon of bovine GCAP1. Annealed oligonucleotides (5′-CTAGATTATGGCCCCGCGTGGACCGGACCGACCCGAGGGGATCGAGGAGGC-3′ and 5′-CATGGCCTCCTCGATCCCCTCGGGTCGGTCCGGTCCACGCGGGGCCATAAT-3′) encoding the “PRGPDRPEGIEE” HIV peptide fragment (PPE–GC1), that introduced XbaI and NcoI protruding ends, were cloned with the fragment NcoI–NdeI encoding GCAP1 between XbaI and NdeI in pET30b upstream of the 36 amino acid long linker–437–1054GC1.
The His6-tag was inserted at the GCAP1 5′-end by PCR with primers FH210 (5′-CATATGCACCATCACCATCACCATGGGAACATTATGGACGGTAAG-3′) and LN1 (5′-CATATGACCGTCGGCCTCCGCG-3′) that also introduced NdeI sites. The NdeI–NdeI GCAP1 fragment was then subcloned in pET30b upstream from the 36 amino acid long linker–437–1054GC1.
Strep-tag, a short peptide (8 amino acids long) with highly selective binding properties for Strep-Tactin (an engineered streptavidin), was also fused to the N-terminus of GCAP1–linker–437–1054GC1 (21). Annealed oligonucleotides (5′-GATCCAATATGGCTAGCAACTGGAGCCACCCGCAATTTGAAAAAGGCGGGCA-′3 and 5′-TATGCCCGCCTTTTTCAAATTGCGGGTGGCTCCAGTTGCTAGCCATATTG-3′) encoding the “MASN-WSHPQFEKGG” Strep-tag peptide, that introduced BamHI and NdeI protruding ends, were cloned with the NdeI–XhoI fragment encoding linker–437–1054GC1 or GCAP1–linker–437–1054GC1 in pFastBac-opened BamHI–SalI.
GC Activity Assay and Protein Purification. Washed rod outer segment (ROS) membranes (22) were prepared from fresh bovine retinas (Schenk Packing Co., Stanwood, WA) reconstituted with recombinant GCAPs and assayed as described previously (23). [Ca2+] was calculated using the computer program Chelator 1.00 (24). All assays were repeated at least twice.
All purification procedures were carried out at 5 °C or on ice. Recombinant GCs and GCAPs were expressed in SF9 insect cells. After homogenization, cell suspensions were centrifuged at 100000g for 20 min at 4 °C. Supernatants were used as a source of recombinant proteins for protein purification. GCAPs and GCAP–GC fusion proteins were purified by Ni2+-nitrilotriacetic acid (Ni2+-NTA) metal-affinity chromatography (Qiagen). The cells were harvested and homogenized with 10 mL (10 plates, 15 cm in diameter) of 10 mM BTP, pH 7.5, containing 2 mM benzamidine. After centrifugation for 10 min at 35000g, the supernatant was incubated for 90 min at 4 °C with 1 mL of Ni-NTA resin. The resin was washed with the same buffer until the absorption at 280 nm was <0.02, and then washed with 10 mM BTP, containing 300 mM NaCl, until A280 nm < 0.02 and eluted with 10 mM Hepes, pH 7.5, containing 100 mM NaCl and 250 mM imidazole. Purified proteins were dialyzed against 10 mM BTP, pH 7.5, containing 50 mM NaCl overnight at 4 °C. The purity of proteins (>90% for GCAPs and >60% for GCAPs–GCs) was estimated by SDS–polyacrylamide gel electrophoresis (PAGE) and Coomassie staining.
PPE–GCAP1–437–1054GC1 protein was purified using antibodies against anti-PPE peptide (PRGPDRPEGIEE) coupled to CNBr-activated Sepharose resin (∼5 mg of IgG per 1 mL of the gel) as described previously (25). Insect cells were homogenized in 10 mM BTP, pH 7.5, and centrifuged (10 min, 100000g, 4 °C), and the supernatant from 10 plates (15 cm) of SF9 cell culture infected with baculovirus-containing GCAP–GC constructs was loaded onto an antibody–Sepharose column (0.8 mg of the gel) equilibrated with 10 mM BTP, pH 7.5, at a flow rate of 6 mL/h. The column was then washed with 10 mM BTP, pH 7.5, containing 200 mM NaCl, and then with 10 mM BTP, pH 7.5. The elution was performed with 1 mM PRGPDRPEGIEE peptide in 10 mM BTP, pH 7.5, containing 50 mM NaCl. Fractions (0.5 mL) were collected and analyzed for GC-stimulating activity and proteins visualized by SDS–PAGE and immunoblotting (Figure 3). Typically, 100 μg of GCAPs and 5–10 μg of GCAP–GC fusion protein were obtained from 10 plates of monolayer SF9 insect cell culture (15 cm in diameter). GCAP–GC proteins were stable for >12 h.
Figure 3.
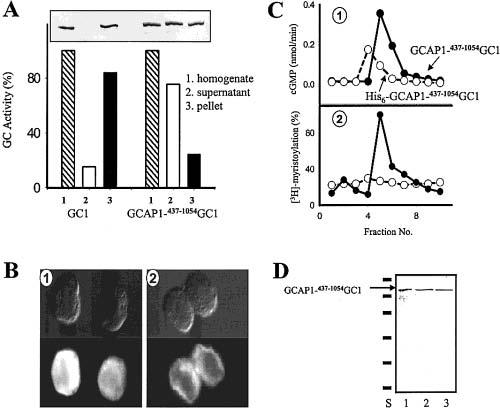
Properties of GCAP1-GC1 fusion proteins in cell culture, biochemical assays, and protein purification. (A) Solubility of GC1 constructs. GC was prepared as described under Materials and Methods. Activity (as a percent of maximal activity) was measured in membrane and soluble cell fractions. Similar results were obtained in four independent measurements. Inset: Immunoblot analysis of GC1 solubility using polyclonal anti-GC1 antibodies. (B) Localization of GCAP1–437–1054GC1 fusion protein in cytoplasm (1) and full-length GC1 in the membrane fraction (2) (upper panels corresponded to image in bright light, bottom panels show fluorescent image). (C) Gel filtration chromatography of GCAP1–437–1054GC1 and His6-GCAP1–437–1054GC1 (Materials and Methods) and correlation with GC stimulation activity. Panel 1, GC activity profile; panel 2, incorporation of the myristoyl group into GCAP1–GC1 mutants. (D) Purification of PPE–GCAP1–437–1054GC1. The GC1 fusion protein was purified as described under Materials and Methods. The purity was assessed by SDS–PAGE (lane 1) and by immunoblotting using anti-GC1 antibody (lane 2) and GCAP1 antibody (lane 3). The standards are (in kDa): phosphorylase B, 116; BSA, 80; ovalbumin, 52.5; carbonic anhydrase, 34.9; soybean trypsin inhibitor, 29.9; lysosyme, 21.8.
For the dilution experiments, the amount of cGMP was determined by standardized Mono Q column chromatography (Pharmacia) using unlabeled GTP as a substrate. Samples (50 μL in 950 μL of 10 mM BTP, pH 7.5) were loaded on the Mono Q column (5 × 50 mm), and nucleotides were separated by a linear gradient from 0% solvent B (300 mM NaCl in 10 mM BTP, pH 7.5)/100% solvent A (10 mM BTP, pH 7.5) to 100% solvent B in 30 min at a flow rate of 0.75 mL/min, monitored at 260 nm. To obtain radiolabeled myristoylated GCAP1–437–1054GC1, the cells were grown in the presence of [9, 10(n)-3H]myristic acid (0.5 mCi/15 mL of cell culture).
Gel Filtration, SDS–PAGE, and Immunoblotting. The supernatant containing soluble recombinant GC (0.25 mL) was loaded on a Superose-6 gel filtration column (10 × 300 mm, Pharmacia), equilibrated with 10 mM BTP, pH 7.5, containing 150 mM NaCl and 0.05% BSA at a flow rate of 0.5 mL/min. Fractions (0.5 mL) were collected, and aliquots of 40 μL were tested for GC activity. The proteins were identified by SDS–PAGE using 15% polyacrylamide gels. For immunoblotting, membranes were blocked with 2% BSA in 20 mM Tris, pH 8.0, containing 150 mM NaCl and 0.05% Tween 20, and incubated for 1 h with primary antibody at a dilution of 1:1000. Cross-reacting anti-GC1 and -GC2 antibody was raised against the conserved dimerization domain (26), while GCAP1- and GCAP2-specific antibodies were generated as described previously (27).
Immunocytochemistry. Insect cells were immersed in chilled 4% formaldehyde in PBS for 2 h. Antibody labeling was assessed by indirect immunofluorescence. Fixed cells were incubated in blocking buffer (PBS containing 3% BSA and 0.2% Triton X-100, pH 7.0) for 30 min at room temperature to reduce nonspecific labeling, washed with PBS, incubated for 1.5 h with rabbit polyclonal antibody (UW28, anti-dimerization domain of GC1 at 1:100 dilution)(26), washed 3 times with PBS, incubated for 30 min with indocarbocyanine (Cy3)-conjugated goat anti-rabbit antibody (1:200 in PBS), extensively washed with PBS, mounted in 5% n-propyl gallate in glycerol, and examined under fluorescence microscopy at 568 nm.
RESULTS AND DISCUSSION
Studies of photoreceptor GC are hindered by several biochemical properties that are difficult to eliminate or control. In general, photoreceptor GCs are poorly extractable with detergents; they are expressed at low levels, sensitive to inactivation during biochemical manipulations, and in ROS, difficult to assay as a consequence of the opposing phosphodiesterase activity. GCs are stimulated by small Ca2+-binding proteins, GCAPs, in a reaction that is highly sensitive to various factors, such as detergents or salts. In particular, the sensitivity to detergent does not allow for a clear determination of whether GCAP activation involves dimerization of GC, as documented to other sTMRs. Inability to reconstitute GC modulation with GCAPs using purified components could also indicate that other proteins, factors of structural elements of ROS, or heterologous expression cell systems are needed for this regulation. Therefore, our approach was to generate a soluble fusion protein that would circumvent these problems (Figure 1).
Figure 1.
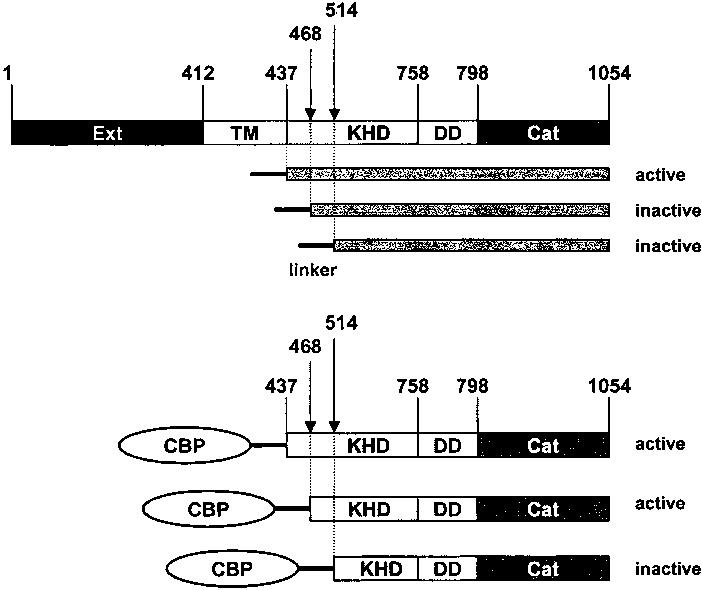
Schematic representation of truncated GCs and CBP–GC fusion proteins. The scheme represents Ext (extracellular), TM (transmembrane), KHD (kinase-homology), DD (dimerization), and Cat (catalytic) domains of GCs. The GC activity was assayed in the whole cell homogenate and in the detergent-free extracts in the presence of GCAP1 at 46 nM [Ca2+]free.
Active Truncated Forms of GCs and Constitutively Active Fusion Proteins between GCs and GCAPs. Recombinant GC1 is associated with the membranes and requires detergent for solubilization. The basal activity of recombinant GC1 expressed in insect cells was 0.02 ± 0.005 nmol/min, and was stimulated 10–20-fold by addition of purified GCAP1 and GCAP2 (Figure 2A) [reviewed also in ref (3)]. The intracellular fragment of GC1 (437–1054) was active, suggesting that the extracellular domain is nonessential for GCAP stimulation as was previously postulated (28, 29). Full-length GC1 and 437–1054GC1 activities (IC50 equal to 225–230 nM) stimulated by soluble GCAP1 had comparable activity versus [Ca2+] relationships (Figure 2A), and similar to the activation of ROS GCs by GCAP1(25). Further systematic truncation of the kinase-homology domain (KHD) led to a 468–1054 product that was stimulated by exogenous GCAP1, but not GCAP2 (data not shown). Additional truncation rendered the GC fragment inactive, indicating that the KHD either is needed for the proper folding of active GC1, is important for GCAP binding (Figure 1), or is required for GC activity, as suggested previously (28). In total, 51 different constructs were generated and tested in this study. All mutations at the C-terminus of GC, such as the addition of a linker (see below), GFP, His6-tag, truncation of the last 6–10 amino acids, led to inactivation of the cyclases and were not useful for further analysis. Shortening the linker also led to inactive cyclase constructs. Therefore, mutants that provided additional information on the properties of GCs and GCAPs are only discussed in this report.
Figure 2.
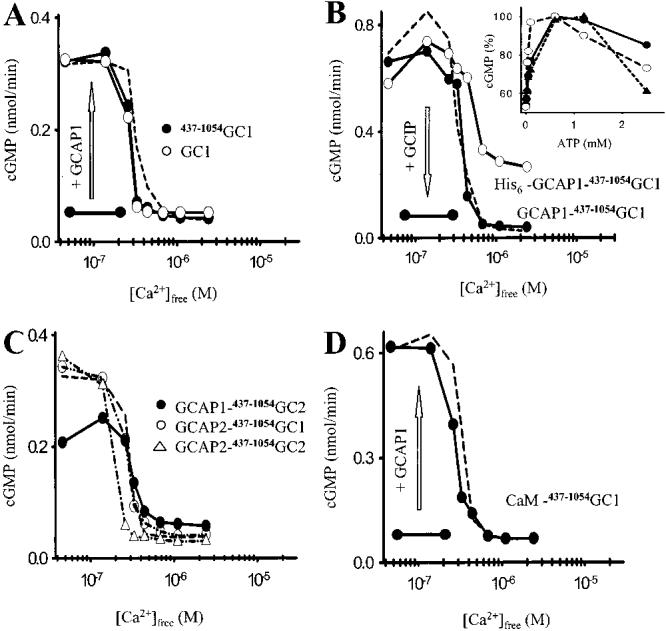
Activity of GC constructs as a function of [Ca2+]free. (A) Activity of GC for full-length GC1 and truncated 437–1054GC1 (enzyme was stimulated by addition of exogenous, recombinant GCAP1). IC50 for GC1 was 230 ± 40 and 225 ± 34 nM for 437–1054GC1 (n = 3). (B) Activity of GC for GCAP1–437–1054GC1 and His6-GCAP1–437–1054GC1 (arrow shows competition of GCIP with GCAP1 in GCAP1–437–1054GC1 fusion protein at low [Ca2+]free). IC50 for His6-GCAP1–437–1054GC1 was 220 ± 34 nM. Inset: ATP titration of GC constructs (solid circles, GCAP1–437–1054GC1; open circles, 437–1054GC1 + GCAP1; open triangles, GC1 + GCAP1) in low [Ca2+]free. Similar results were obtained in three independent measurements. (C) Activity of GC for GCAP1–437–1054GC2, GCAP2–437–1054GC1, and GCAP2–437–1054GC2. IC50 values for GCAP2–437–1054GC1 and GCAP2–437–1054GC1 were 245 ± 74 and 255 ± 30 nM, respectively. (D) Activity of GC for CaM–437–1054GC1 after stimulation by addition of GCAP1. Arrow shows recovery of GC1 activity by reconstitution with GCAP1 in low [Ca2+]free. IC50 for CaM–437–1054GC1 was 195 ± 15 nM. The dashed line represents stimulation of ROS GCs by GCAP1. In all panels, the dashed line represents stimulation of ROS GCs by GCAP1.
Based on these deletion experiments, fusion proteins were generated between selected CBPs and GCs (Figure 1). The fusion proteins were active when CBPs were added to the N-terminus of GC, while the addition of a 35 amino acid long linker (see Materials and Methods) to the GC1 and GC2 C-termini led to inactivation of the enzymes, without significant changes in the expression levels of the constructs (data not shown). Shortening of the GC construct by 44 amino acids from position 468 (Figure 1) and longer systematic deletions (data not shown), as observed for truncated GC1, led to cyclase inactivation.
Although GCAP1/2–GC1/2 constructs were constitutively active at 46 nM [Ca2+]free with IC50 = 220–255 nM [Ca2+]-(Figure 2B,C), further addition of exogenous GCAP1 or GCAP2 increased the GC activity by 10–20%, suggesting that not all molecules in the GCAP–GC complexes are fully enzymatically productive and some are in a dissociated form with the regulatory site of GCs accessible for soluble free GCAPs. Moreover, at low [Ca2+], guanylyl cyclase-inhibitor protein (GCIP), a CBP related to GCAP, competes with GCAP1 in the GCAP1–437–1054GC1 complex (Figure 2B). This observation confirms that the binding of GCAP1 to GC1 in the fusion protein is of low affinity, similar to stimulation in ROS membranes. Both binding sites for GCAP1 and GCIP, therefore, must overlap at least partially.
The inhibition of GC1 at high [Ca2+] required myristoylated GCAP1 (23), and when the recognition site for myristoyl-transferase was disabled by the addition of His6-tag, enhanced cyclase activity was observed in high [Ca2+], which is typically reduced to basal levels by the myristoylated Ca2+-free form of GCAP1 (Figure 2B). These results indicate that the functional role of the N-terminal domain of GCAP1 in the inhibition of GC1 is independent of membrane anchoring.
Photoreceptor GC1 has been reported to be stimulated ∼2-fold by 1–2 mM ATP, while higher concentrations of ATP decreased GC activity, most likely by competing with the substrate, GTP (30). To assess if the fusion protein still responds to ATP, increasing concentrations of this nucleotide were included in the assays. GC responsiveness to ATP was preserved both for the truncated 437-1054GC1 and for the fusion GCAP1–437–1054GC1. At 0.5–1.5 mM ATP, the GC constructs were activated and inhibited at higher concentrations (Figure 2B, inset). As a result, the truncated GC1 fully preserved the responsiveness to ATP, indicating that the membrane component is not needed for this regulation as well.
The GCAP1–437–1054GC1 properties in response to an increase in [Ca2+] were also comparable to those of GCAP2–437–1054GC1, GCAP1–437–1054GC2, and GCAP2–437–1054GC2 (Figure 2C). These results indicate compatibility between GC1 and GC2/GCAP1 and GCAP2, suggesting that underlying mechanisms of activation are similar for both GCAPs and GCs. This finding is in contrast to other observations in reconstituted systems using the mutated and native GCs and GCAPs [reviewed in ref (31)]. These differences between membrane-bound and free systems could result from the alteration in affinities between these two nonlinked components and changes in effective concentrations in a milieu of other proteins on the surface of the membranes as compared to soluble proteins. As expected, not all CBPs have the stimulatory effect on GC1. When CaM was fused to 437–1054GC1, only a basal activity was observed at all tested [Ca2+], and the cyclase activity was enhanced in low [Ca2+] upon addition of exogenous GCAP1 (Figure 2D). Together, these data show that the fusion proteins between GCs and GCAPs display all major enzymatic characteristics of the native system in ROS. Therefore, these constructs were further evaluated to understand the mechanism of cyclase modulation by GCAPs.
Soluble Forms of GC Fusion Proteins. The fraction of active and soluble GCAP1–437–1054GC1 was estimated by the enzymatic assays in membrane suspensions and soluble fractions, and fusion proteins were visualized by immunoblotting with anit-GC1 antibodies. A major fraction of all fusion proteins or truncated GCs was soluble, in contrast to full-length GC1 (Figure 3A). The production of the soluble active products in insect cells is different from the expression of insoluble truncated GC1 obtained in HEK293 cells (29). These differences suggest that the insolubility of the intracellular domain of GC1 may be due to either differences in the expression system (insect cells vs HEK293 cells) or differences in GC1 intracellular sequences (human vs bovine), rather than a general feature of this GC domain. It is worth noting that the intracellular domain of GC type C is also soluble in insect cells (32). The cellular localization of GCAP1–437–1054GC1 showed cytosolic distribution in contrast to plasma membrane association of GC1 (Figure 3B). To determine if GCAP1–437–1054GC1 is myristoylated, the fusion protein was produced in the presence of [3H]-myristic acid. Because [3H-myristoyl]-GCAP1–437–1054GC1 was eluted in fractions that corresponded to the GC-stimulated activity at low [Ca2+], and no radioactivity was detected in the fraction for the control mutant with the disabled myristoylation site, these results and inhibition of GC1 at high [Ca2+] (Figure 2B) suggest that the fusion protein is myristoylated (Figure 3C).
The GCAP1–437–1054GC1 fusion proteins were subjected to various purification schemes. For example, His6-GCAP1–437–1054GC1 was partially purified by Ni2+ metal-affinity chromatography (data not shown). Enzymatic properties (such as [Ca2+] sensitivity) of purified fractions of GCAP–GC fusion proteins were indistinguishable from regulation of membrane-associated GC in ROS or HEK293 membranes. Strep-GCAP1–GC1, containing the MASN-WSHPQFEK-GG-tag sequence, can be developed using the streptavidin affinity purification method (21). Another construct with an antibody recognition tag, PPE–GCAP1–437–1054GC1, containing the PRGPDRPEGIEE sequence, was purified to apparent homogeneity by immunoaffinity chromatography (Figure 3D). It appears, therefore, that no other membrane component is needed for GC1 stimulation by GCAP1 and that these proteins interact directly, rather than through adapter molecules such as cytoskeletal proteins, and that no membrane component is needed for the activation process.
The expression of fusion proteins has been extensively studied in E. coli without success. The vast majority of the expressed protein formed insoluble aggregates, and both the soluble fraction and the insoluble fraction, renatured from urea, were without GC activity. Further improvements, such as simplifying the purification procedures, are required for increasing protein levels in various heterologous expression systems, including insect cells, before structural studies could be embarked on.
Mechanism of GC Stimulation by GCAPs. To probe the mechanism of GC stimulation, we employed GCAP1–437–1054GC1 protein; however, other constructs yielded similar results. To further investigate the affinity of GCAP1 for GCAP1–437–1054GC1 in a mixture of free components and as a fusion protein, the GC activity was measured in the presence of increasing concentrations of the constitutively active GCAP1 mutant at high [Ca2+](33) (Figure 4A). In these conditions, without the constitutively active mutant, native GCAP1 inhibits GC activity (Figure 2A). For GCAP1 to GCAP1–437–1054GC1, the EC50 for stimulation with the GCAP1 mutant was comparable (∼0.8 μM), and similar in our conditions to the EC50 for membrane-bound GC and GCAP1(∼1.5 μM) (33), supporting again the hypothesis that GCAP1 in GCAP1–437–1054GC1 forms a freely dissociable complex and that GCAP1 in the fusion protein can be out-competed by soluble GCAPs. Although the activation of GCAP1–437–1054GC1 could be intermolecular (Figure 2B,D), in diluted conditions, most of the activation occurs through an intramolecular mechanism (Figure 4B) because dilution of GCAP1–437–1054GC1 had only a minor effect on the activity as compared to strong inhibition by the dilution of GCs in ROS in the presence of exogenous GCAP1.
Figure 4.
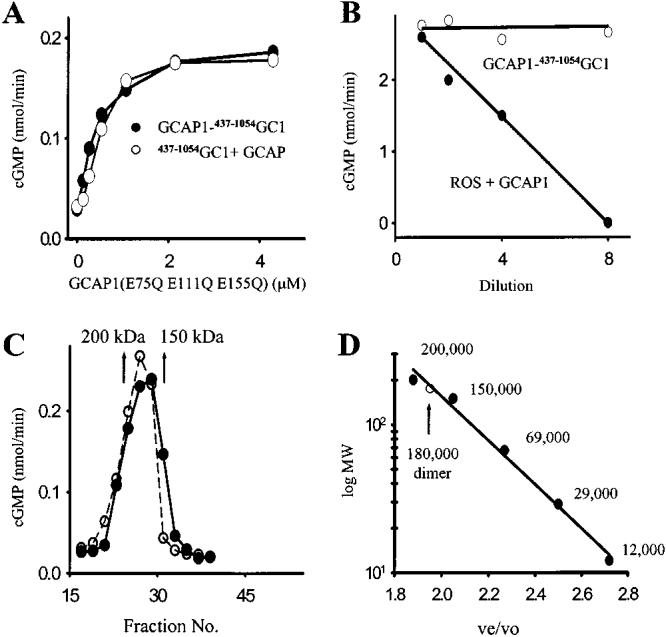
Intramolecular and dimeric nature of GCAP1–GC1-stimulated activity at low [Ca2+]free. (A) Competition of GCAP1–437–1054GC1 and 437–1054GC1 with GCAP1(E75Q E111Q E155Q) at 2 μM [Ca2+]free. The EC50 values were 0.8 ± 0.1 and 0.85 ± 0.1 μM for GCAP1–437–1054GC1 and 437–1054GC1, respectively. (B) Dilution effect on GCAP1–437–1054GC1 and GCAP1/ROS GC1 activities. Samples were diluted 0, 2, 4, and 8 times with increasing concentrations of all reagents in the assay at constant amounts of protein. Results are an average of two measurements. (C) Gel filtration chromatography. GCAP1–437–1054GC1 was loaded at 5 μM [Ca2+]free or with (gray circles and dashed line) addition of 1 mM EDTA (solid circles) on a Superose-6 column as described under Materials and Methods. Arrows show standards: α-amylase (200 kDa) and alcohol dehydrogenase (150 kDa). (D) The calibration curve for the gel filtration column. The standard proteins and compounds used were NaN3, cytochrome c (12 kDa), carbonic anhydrase (29 kDa), bovine serum albumin (67 kDa), alcohol dehydrogenase (150 kDa), α-amylase (200 kDa), and blue dextran (void volume, 2000 kDa) as the high molecular mass standard. The protein standards were detected at 280 nm, blue dextran at 450 nm, and azide at 260 nm. ve/vo is a ratio of the elution volume to void volume. The GC fusion protein eluted at a volume that corresponded to 180 kDa. Similar relationships for the retention time and molecular mass were obtained in three independent experiments.
GCAP1–437–1054GC1 formed a homodimer of intracellular domains as judged by the gel filtration method in both low and high [Ca2+] (Figure 4C). These results are consistent with the observations made using other methods for photoreceptor GCs in membranes (14, 34, 35) and other membrane-associated GCs (12, 32). X-ray structure determination of the soluble extracellular domain also suggests a dimeric form of this part of GC (36), and that the extracellular and intracellular domains permit a set of interactions that brings the two subunits together. The regulatory interactions of GCAPs with the effector GC molecules cause conformational changes that produce the most efficient enhancement in the catalysis, rather than physical dimerization. Recently, we postulated an activation model that assumes changes within the flexible central helix of GCAPs upon Ca2+ dissociation, causing relative reorientation of two structural domains containing a pair of EF-hand motifs, and thus switching its partner, GC, from an inactive to an active conformation (37). The essential components required to transform an inhibitory to an activating protein are contained within the N-terminal region of GCAP1 (residues 1–43) (23, 38, 39). These results deviate from the mechanism proposed for the photoreceptor GC regulation, in which GCAP2 promotes a dimerization of GCs in the Ca2+-free form (35).
In summary, active and soluble GCAP–GC fusion proteins were generated and characterized in biochemical assays. The interaction of GCs with GCAPs did not require any membrane components or adapter proteins. The soluble GCAP–GC fusion proteins are dimers in all tested conditions, and the GC activity showed typical Ca2+-dependent stimulation that was further enhanced by ATP. The myristoyl group of GCAP1 appeared to be critical for the inhibition of GCs at high [Ca2+]. Expression of the active soluble intracellular GC domain in a complex with GCAPs opens new possibilities for using these constructs in structural studies. Thus, the sensitivity of GCs and GCAPs to various reagents, such as salt and detergents, and the relatively low affinities compared to CaM and its target proteins could be overcome by fusing these two components. This raises the possibility to determine the high-resolution structures of the fusion proteins in both Ca2+-free and Ca2+-bound forms. Their critical role in the physiology of photoreceptors and vision makes GC–GCAP systems an attractive target for further studies. These studies may also influence further progress on other sTMRs and their interactions with modulatory proteins.
ACKNOWLEDGMENT
We thank Dr. Ning Li for the constitutively active GCAP1 construct, Dr. Daniel D. Oprian for anti-PPE antibody, and Dr. Geeng-Fu Jang, J. Preston Van Hooser, Naomi Wilson, and Joshua McBee for critical review of the manuscript.
Footnotes
This research was supported by NIH Grants EY08123 (W.B.) and EY08061 (K.P.), a grant from Research to Prevent Blindness, Inc. (RPB), to the Department of Ophthalmology at the University of Washington and the University of Utah, a Center Grant from the Foundation Fighting Blindness, Inc., to the University of Utah, and the Ruth and Milton Steinbach Fund, an Alcon Research Institute award, and the E. K. Bishop Foundation.
REFERENCES
- 1.Pawson T, Gish GD. Cell. 1992;71:359–362. doi: 10.1016/0092-8674(92)90504-6. [DOI] [PubMed] [Google Scholar]
- 2.Polans A, Baehr W, Palczewski K. Trends Neurosci. 1996;19:547–554. doi: 10.1016/s0166-2236(96)10059-x. [DOI] [PubMed] [Google Scholar]
- 3.Palczewski K, Polans AS, Baehr W, Ames JB. Bioessays. 2000;22:337–350. doi: 10.1002/(SICI)1521-1878(200004)22:4<337::AID-BIES4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 4.Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, Chepenik KP, Waldman SA. Pharmacol. Rev. 2000;52:375–414. [PubMed] [Google Scholar]
- 5.Kelsell RE, Gregory Evans K, Payne AM, Perrault I, Kaplan J, Yang RB. Hum. Mol. Genet. 1998;7:1179–1184. doi: 10.1093/hmg/7.7.1179. [DOI] [PubMed] [Google Scholar]
- 6.Sokal I, Li N, Surgucheva I, Warren MJ, Payne AM, Bhattacharya SS, Baehr W, Palczewski K. Mol. Cells. 1998;2:129–133. doi: 10.1016/s1097-2765(00)80121-5. [DOI] [PubMed] [Google Scholar]
- 7.Dizhoor AM, Boikov SG, Olshevskaya EV. J. Biol. Chem. 1998;273:17311–17314. doi: 10.1074/jbc.273.28.17311. [DOI] [PubMed] [Google Scholar]
- 8.Payne AM, Downes SM, Bessant DA, Taylor R, Holder GE, Warren MJ, Bird AC, Bhattacharya SS. Hum. Mol. Genet. 1998;7:273–277. doi: 10.1093/hmg/7.2.273. [DOI] [PubMed] [Google Scholar]
- 9.Duda T, Venkataraman V, Jankowska A, Lange C, Koch KW, Sharma RK. Biochemistry. 2000;39:12522–12533. doi: 10.1021/bi001514d. [DOI] [PubMed] [Google Scholar]
- 10.Koch KW. J. Biol. Chem. 1991;266:8634–8637. [PubMed] [Google Scholar]
- 11.Aparicio JG, Applebury ML. Protein Expression Purif. 1995;6:501–511. doi: 10.1006/prep.1995.1067. [DOI] [PubMed] [Google Scholar]
- 12.Garbers DL. Methods. 1999;19:477–484. doi: 10.1006/meth.1999.0890. [DOI] [PubMed] [Google Scholar]
- 13.Bieger B, Essen LO. EMBO J. 2001;20:433–445. doi: 10.1093/emboj/20.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramamurthy V, Tucker CL, Wilkie SE, Daggett V, Hunt DM, Hurley JB. J. Biol. Chem. 2001;16:16. doi: 10.1074/jbc.M010495200. [DOI] [PubMed] [Google Scholar]
- 15.Sunahara RK, Beuve A, Tesmer JJ, Sprang SR, Garbers DL, Gilman AG. J. Biol. Chem. 1998;273:16332–16338. doi: 10.1074/jbc.273.26.16332. [DOI] [PubMed] [Google Scholar]
- 16.Tesmer JJ, Sunahara RK, Johnson RA, Gosselin G, Gilman AG, Sprang SR. Science. 1999;285:756–760. doi: 10.1126/science.285.5428.756. [DOI] [PubMed] [Google Scholar]
- 17.Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. Science. 1997;278:1907–1916. doi: 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- 18.Tang WJ, Gilman AG. Cell. 1992;70:869–872. doi: 10.1016/0092-8674(92)90236-6. [DOI] [PubMed] [Google Scholar]
- 19.Braunewell KH, Spilker C, Behnisch T, Gundelfinger ED. J. Neurochem. 1997;68:2129–2139. doi: 10.1046/j.1471-4159.1997.68052129.x. [DOI] [PubMed] [Google Scholar]
- 20.Boekhoff I, Braunewell KH, Andreini I, Breer H, Gundelfinger E. Eur. J. Cell Biol. 1997;72:151–158. [PubMed] [Google Scholar]
- 21.Skerra A, Schmidt TG. Methods Enzymol. 2000;326:271–304. doi: 10.1016/s0076-6879(00)26060-6. [DOI] [PubMed] [Google Scholar]
- 22.Papermaster DS. Methods Enzymol. 1982;81:48–52. doi: 10.1016/s0076-6879(82)81010-0. [DOI] [PubMed] [Google Scholar]
- 23.Otto-Bruc A, Buczylko J, Surgucheva I, Subbaraya I, Rudnicka-Nawrot M, Crabb JW, Arendt A, Hargrave PA, Baehr W, Palczewski K. Biochemistry. 1997;36:4295–4302. doi: 10.1021/bi963000d. [DOI] [PubMed] [Google Scholar]
- 24.Schoenmakers TJ, Visser GJ, Flik G, Theuvenet AP. BioTechniques. 1992;12:870–874. [PubMed] [Google Scholar]
- 25.Gorczyca WA, Polans AS, Surgucheva IG, Subbaraya I, Baehr W, Palczewski K. J. Biol. Chem. 1995;270:22029–22036. doi: 10.1074/jbc.270.37.22029. [DOI] [PubMed] [Google Scholar]
- 26.Semple-Rowland SL, Lee NR, Van Hooser JP, Palczewski K, Baehr W. Proc. Natl. Acad. Sci. U.S.A. 1998;95:1271–1276. doi: 10.1073/pnas.95.3.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otto-Bruc A, Fariss RN, Haeseleer F, Huang J, Buczylko J, Surgucheva I, Baehr W, Milam AH, Palczewski K. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4727–4732. doi: 10.1073/pnas.94.9.4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duda T, Goraczniak R, Surgucheva I, Rudnicka-Nawrot M, Gorczyca WA, Palczewski K, Sitaramayya A, Baehr W, Sharma RK. Biochemistry. 1996;35:8478–8482. doi: 10.1021/bi960752z. [DOI] [PubMed] [Google Scholar]
- 29.Laura RP, Dizhoor AM, Hurley JB. J. Biol. Chem. 1996;271:11646–11651. doi: 10.1074/jbc.271.20.11646. [DOI] [PubMed] [Google Scholar]
- 30.Gorczyca WA, Van Hooser JP, Palczewski K. Biochemistry. 1994;33:3217–3222. doi: 10.1021/bi00177a011. [DOI] [PubMed] [Google Scholar]
- 31.Dizhoor AM. Cell. Signalling. 2000;12:711–719. doi: 10.1016/s0898-6568(00)00134-0. [DOI] [PubMed] [Google Scholar]
- 32.Vijayachandra K, Guruprasad M, Bhandari R, Manjunath UH, Somesh BP, Srinivasan N, Suguna K, Visweswariah SS. Biochemistry. 2000;39:16075–16083. doi: 10.1021/bi0013849. [DOI] [PubMed] [Google Scholar]
- 33.Rudnicka-Nawrot M, Surgucheva I, Hulmes JD, Haeseleer F, Sokal I, Crabb JW, Baehr W, Palczewski K. Biochemistry. 1998;37:248–257. doi: 10.1021/bi972306x. [DOI] [PubMed] [Google Scholar]
- 34.Yang RB, Garbers DL. J. Biol. Chem. 1997;272:13738–13742. doi: 10.1074/jbc.272.21.13738. [DOI] [PubMed] [Google Scholar]
- 35.Olshevskaya EV, Ermilov AN, Dizhoor AM. J. Biol. Chem. 1999;274:25583–25587. doi: 10.1074/jbc.274.36.25583. [DOI] [PubMed] [Google Scholar]
- 36.van den Akker F, Zhang X, Miyagi M, Huo X, Misono KS, Yee VC. Nature. 2000;406:101–104. doi: 10.1038/35017602. [DOI] [PubMed] [Google Scholar]
- 37.Sokal I, Li N, Klug CS, Filipek S, Hubbell WL, Baehr W, Palczewski K. J. Biol. Chem. 2001;276(27):43361–43373. doi: 10.1074/jbc.M103614200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li N, Sokal I, Bronson JD, Palczewski K, Baehr W. Biol. Chem. 2001;38:1179–88. doi: 10.1515/BC.2001.148. [DOI] [PubMed] [Google Scholar]
- 39.Ermilov AN, Olshevskaya EV, Dizhoor AM. J. Biol. Chem. 2001 doi: 10.1074/jbc.M107539200. in press. [DOI] [PubMed] [Google Scholar]