

Abstract
This review focuses on two gaseous cellular messenger molecules, CO and H2S, that are involved in cerebrovascular flow regulation. CO is a dilatory mediator in active hyperemia, autoregulation, hypoxic dilation, and counteracting vasoconstriction. It is produced from heme by a constitutively expressed enzyme (heme oxygenase (HO)-2) expressed highly in the brain and by an inducible enzyme (HO-1). CO production is regulated by controlling substrate availability, HO-2 catalytic activity, and HO-1 expression. CO dilates arterioles by binding to heme that is bound to large-conductance Ca2+ activated K+ channels (BKCa channels), which elevates channel Ca2+ sensitivity, increases coupling of Ca2+ sparks to BKCa channel openings and, thereby, hyperpolarizes the vascular smooth muscle. In addition to dilating blood vessels, CO can either inhibit or accentuate vascular cell proliferation and apoptosis, depending on conditions. H2S may also function as a cerebrovascular dilator. It is produced in vascular smooth muscle cells by hydrolysis of L-cysteine catalyzed by cystathione gamma-lyase (CSE). H2S dilates arterioles at physiologically relevant concentrations via activation of KATP channels. In addition to dilating blood vessels, H2S promotes apoptosis of vascular smooth muscle cells and inhibits proliferation-associated vascular remodeling. Thus, both CO and H2S modulate the function and the structure of circulatory system. Both the HO/CO and CSE/H2S systems have potential to interact with NO and prostanoids in the cerebral circulation. Much of the physiology and biochemistry of HO/CO and CSE/H2S in the cerebral circulation remains open for exploration.
Introduction
The group of gaseous autocrine/paracrine messengers continues to expand and now includes nitric oxide (NO), carbon monoxide (CO), hydrogen sulfide (H2S), and several molecules from the broad category of reactive oxygen species (ROS). Diverse functions of the messengers are being detected at an astounding rate, demonstrating that the gaseous autocrine/paracrine messengers are critical to vertebrate, and possibly all living organism, physiology; and extend far beyond just NO. Gaseous messengers, or gasotransmitters (145), are employed for intra- and intercellular communication with high specificity in many, if not all, organ systems. While gaseous messengers function analogously to hormones, neurotransmitters, and lipid mediators, they have distinct attributes. Because they are lipid soluble gases, gasotransmitters are not constrained by cellular membranes. Thus, storage in vesicles for later release is not possible. Although it has been suggested that CO and NO could be bound to heme containing proteins that may be induced to release the messenger (27,53,141), clearly the predominant determination of the strength of signal of gasotransmitters is de novo synthesis. Because gasotransmitters diffuse down partial pressure gradients and are unconstrained by cell membranes, specific uptake or metabolizing processes to terminate the signals need not exist. Signals are terminated by falling concentrations upon reduction of production that are caused by reacting with cellular components (particularly ROS and NO), binding to cellular components (e.g. CO to ferrous heme), or diffusing away. The natures of the interactions of the gasotransmitters with their receptors are, with the exception of NO and guanylyl cyclase, only beginning to be uncovered.
While the overall category of gasotransmitters encompasses NO, CO, H2S, various ROS, and certainly others not yet realized, the present mini-review will be limited to CO and H2S in regulation of cerebrovascular circulation, because roles of NO have been extensively discussed and reviewed before (35,78, 121) and the understanding of signaling roles of ROS, as opposed to the destructive actions, is limited (see reviews 32,44,114).
Carbon monoxide (CO)
Most information on contributions of endogenously produced CO to control of cerebrovascular circulation derives from studies on newborn pigs. Thus, the review below is biased toward newborn pig cerebrovascular circulation. Far fewer data are available on CO in the cerebrovascular circulation from newborns of other species or, for the most part, adults of any species.
The below discussion is summarized pictorially in Fig. 1.
Figure 1.
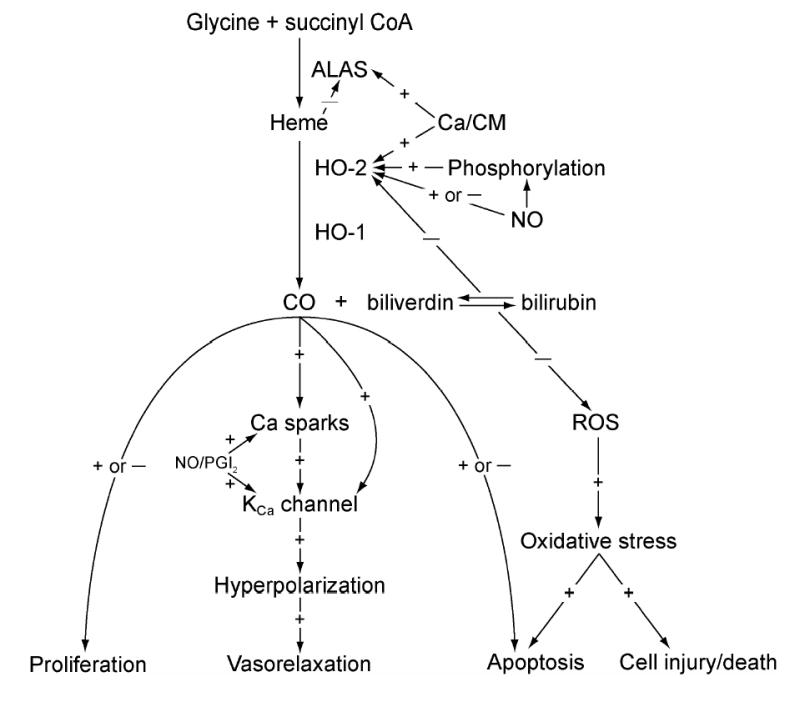
Flow chart summarizing the section of this review on CO.
Control of CO production
The gas, CO, is produced physiologically by catabolism of heme to CO, iron, and biliverdin (94). This reaction is catalyzed by heme oxygenase (HO) with reduction of NADPH. HO is expressed as three known isoforms: HO-1, HO-2 and a third isoform (HO-3) with much lower heme degrading activity (101) and low expression in the brain (129). Recent reports indicate that HO-3 genes are processed pseudogenes derived from HO-2 transcripts that have no known functional significance (47). In freshly isolated cerebral microvessels, as in the intact brain, in vivo, only HO-2 expression was detected on Western blots (117). The constitutive nature of HO-2 in the brain and brain vasculature requires mechanisms for regulation of CO production either by control of enzyme activity or substrate delivery. CO production by HO-2 could be controlled by delivery of electrons from NADPH via cytochrome P450 reductase, by O2 availability, by delivery of heme to the enzyme, and by the catalytic activity of the enzyme.
While evidence to indicate NADPH concentration or PO2 regulates CO production in the cerebral circulation has not been produced, data suggesting O2 availability can affect CO production are available from other tissues. In microsomal fractions of of human placenta chorionic villi, CO production is directly related to PO2 from 18–166 mmHg (5). Also, electrophysiological results from membrane patches of carotid body glomus cells suggest hypoxia may decrease CO production (77). However, hypoxia dilates cerebral arteries, as does CO, so the functional significance of hypoxic inhibition of CO production in the cerebral circulation is unknown.
Endogenous CO production by brain and brain vessels is substrate limited because provision of exogenous heme increases CO production (80). Therefore, mechanisms that regulate cellular heme production will also regulate CO production. Cellular heme manufacture is a multi-step process that includes both mitochondrial and cytoplasmic elements, but the rate-limiting step is production in mitochondria of delta-aminolevulinic acid from succinyl CoA and glycine catalyzed by delta-aminolevulinic acid synthase (ALAS) (66,100). ALAS activity is tightly regulated, being inhibited in a negative feedback manner by heme and hemin (oxidized heme). Control conceivably could also occur by regulation of ferrochelatase or porphobilinogen deaminase, but such control mechanisms have not been demonstrated. In intact cerebral microvessels, increasing cytosolic Ca2+ with ionomycin or activation of protein kinases C (PKC) with phorbol ester increased CO production by increasing heme availability (80). However, in neither case are the cellular mechanisms involved known.
HO-2 catalytic activity, CO production per amount of enzyme from exogenous heme, includes HO catalytic efficiency and activation by intracellular relocation. Negative feedback control of HO-2 catalytic activity by bilirubin has been described (93). Other control mechanisms of HO-2 catalytic activity may be cell type and tissue specific. For example, in neurons HO-2 activity can be stimulated by CK2 catalyzed phosphorylation of serine 79 (10). Glutamatergic activation of HO-2 results from metabotropic glutamate receptor-induced Ca2+ release, activation of PKC, and CK2 phosphorylation (12). Conversely, in freshly isolated piglet cerebral microvessels and microvascular endothelial cells in culture, CO production is increased by ionotropic, but not metabotropic, glutamate receptor stimulation (116) (see below in section on seizures for more extensive discussion of glutamate receptors and CO). In addition, protein tyrosine kinase inhibition decreased and tyrosine phosphatase inhibition increased basal and glutamate-stimulated HO-2 catalytic activity and CO production (79). Furthermore, inhibition of neither PKC nor CK2 altered HO-2 catalytic activity (80,81).
In cerebral microvessels and cortical neurons, calmidizolium, that inhibits calmodulin (CaM), decreased HO-2 catalytic activity and blocked glutamate stimulation of CO production (11,81). Boehning et al (11) demonstrated Ca2+ - dependent CaM binding to HO-2 expressed in yeast. Ionomycin increased HO-2 activity and calmidizolium blocked the response to ionomycin in HEK 293 cells transfected with rat HO-2 gene. Interestingly, in cerebral microvessels we found that the Ca ionophore, ionomycin, in Ca2+-replete media increased CO production but did not detectably increase HO-2 catalytic activity, suggesting elevation of free heme as the major mechanism behind ionomycin-induced stimulation of CO production (81). Nevertheless, the results in both neurons and microvessels are consistent with HO-2 expressed in yeast showing Ca2+/CaM regulation of HO-2 catalytic activity. The reasons for the apparent discrepancy between effects of cytosolic Ca2+ manipulation and those of CaM inhibition are not known.
Another gaseous mediator, NO, can affect HO-2 catalytic activity. HO-2 expressed in Escherichia coli is inhibited by NO donors via binding of NO to a heme regulatory motif on HO-2 (27). Also, a direct inhibitory effect of NO has been reported in HO-1 rich aortic endothelial cell microsomes where nitrosylation of heme prevented catabolism by HO (62). Conversely, in cerebral microvessels we found NO increases HO-2 catalytic activity and thus CO production via a cGMP dependent mechanism (80). The mechanism by which cGMP increases HO-2 activity is unknown, but it appears reasonable to suspect protein kinase G activation of tyrosine kinases or phosphatase inhibition (79,81). Also, in isolated heart (99) and porcine aortic endothelial cells (108) NO increased CO production. It is possible that NO can have a direct inhibitory effect on HO-2 that is masked in the intact system by cGMP-induced stimulation.
Extravascular sources of CO in vivo may contribute to pial arteriolar dilation to glutamate. Of particular note, data on isolated vessels and those from intact cerebrovascular circulation in vivo are not entirely consistent. Thus, in isolated microvessels NOS inhibition totally abolished glutamate –induced CO production (80). However, in vivo, while NOS inhibition blocked glutamate-induced dilation, a constant background amount of NO completely restored dilation to glutamate (71). If glutamate increases NO that increases CO, that is the final mediator of dilation, glutamate should not cause dilation if NO is held constant, but it does. In vivo, cerebral microvessels are accompanied by astrocytes and neurons that also have glutamate receptors (46,54,89), HO-2 (129), and nNOS (157). In fact, involvement of nNOS in glutamate-induced cerebrovascular dilation in mouse cerebellum has been demonstrated (156). Furthermore, because both astrocytes and neurons have HO-2, glutamate could stimulate CO production in these cells. The dilator response to CO would still be endothelial dependent because the endothelium is necessary to provide the obligatory permissive signal of NO.
Localization of HO isoforms and regulation of HO-1 and HO-2 expression
HO-1 and HO-2 isoforms are products of distinct genes located on different chromosomes. The amino acid sequences of HO-1 and HO-2 have only 40% similarity (13). Human HO-1 is a single polypeptide of 288 residues and approximately 32 kDa, while human HO-2 is a 316-residue protein (36 kDa) due to an addition at the N-terminus. A common 24 amino acid domain that forms the heme catalytic pocket is evolutionally conserved in HO-1 and HO-2 except for a single amino acid residue (95). Catalytic mechanisms of heme degradation by the isoforms are similar.
Both HO-1 and HO-2 are membrane-bound proteins anchored to the endoplasmic reticulum membrane via a C-terminal hydrophobic tail (130). In porcine cerebral vascular endothelial cells, HO-1 and HO-2 have similar intracellular localization with strong preference for the nuclear envelope, perinuclear area of the cytoplasm, and endoplasmic reticulum (117). Electron immunocytochemistry data in rat kidney epithelial cells also demonstrated association of HO-2 with the nuclear outer membrane and the endoplasmic reticulum (52). Intracellular localization of HO corresponds to localization of nitric oxide synthase (NOS) and prostaglandin cyclooxygenase (COX) that may suggest functional cross-talk among CO, NO, and prostanoids. In the carotid body, intracellular HO-2 co-localizes with BKCa channels (150). Localization with BKCa channels may be important for cerebral circulatory control because HO-2-derived CO activates BKCa channels to produce vascular smooth muscle relaxation (see below). In pulmonary artery endothelial cells, HO-1 was detectable in plasma membrane caveolae where caveolin binds to HO-1 and regulates its enzymatic activity (68). Under certain conditions, HO-1, but not HO-2, is localized to the nucleus in differentiated astrogial cells (85) and pulmonary artery endothelial cells (68) indicating a possibility of nuclear functions of HO-1.
The highest HO-2 expression is found in the brain, cerebral vasculature, and testes (85,93,95). Steroid hormones are the only known regulators of HO-2 expression (4,92,94,116,147). The presence of glucocorticoid response element (GRE) in the promoter region of the HO-2 gene accounts for upregulation of HO-2 expression (88). High expression in the brain, including selected neuronal populations (29,33,34,57,158), glia (129), and cerebral vasculature (84,117) indicates an important role of HO-2 in brain physiological functions that include regulation of cerebral blood flow and mediation of neuronal activity.
Under basal conditions, HO-1 is expressed strongly only in reticuloendothelial cell-rich tissues, such as spleen and liver were it functions to eliminate potentially toxic heme released from degraded red blood cells (94). In the brain and in cerebral vasculature, no HO-1 expression has been detected under basal conditions (34,84,117,119). HO-1 is an early response gene (heat shock protein Hsp 32) that can be induced by a variety of stress factors including heme, metalloporphyrins, heavy metals, cytokines, oxidative stress and oxidized lipids (2,18,31,38,56,65,75,77,93,95,119,125). Induction of HO-1 is regulated at the level of gene transcription via cell-specific multiple regulatory elements in the promoter region of HO-1 gene, including but not limited to stress response elements (StREs) and antioxidant response elements (AREs) in conjunction with the redox-sensitive transcription factor Nrf2 (98,111), nuclear factor kappa B (NFκB) (76), and cAMP responsive element CRE (75). Enhanced HO-1 gene transcription may also occur via binding of the basic helix-loop-helix-leucine zipper family of transcription factors USF1 and USF2 to the class B E-box located in the proximal promoter of the human HO-1 gene (49). In addition, HO-1 protein induction via translation-independent mRNA stabilization has been described (13).
Mechanism of CO-induced cerebrovascular dilation
It has been proposed that CO-induced cell signaling may be via activation of guanylyl cyclase (51,97). Indeed, treatment of platelets (16) or aorta (41) with CO or a CO releasing molecule (37), as well as overexpression of HO-1 in pulmonary artery or aorta (3,107,127), increase cGMP. CO is much less effective at stimulating guanylyl cyclase than is NO (69,70,71), but involvement of an endogenous substance that increases GC sensitivity to CO has been suggested (24,142). Nevertheless, cGMP as a direct mediator of CO dilation in cerebrovascular circulation under physiological conditions appears unlikely. An increase in cGMP production coincident with CO-induced dilation has not been demonstrated and normal dose-dependent dilation to CO occurs with cGMP held constant (72,82). Furthermore, the ability of guanylyl cyclase inhibition to attenuate vasodilation to CO (37,51,79,144), in the cerebrovascular circulation at least, can be entirely explained by a role of cGMP as a necessary permissive enabling factor (see below).
In contrast to cGMP, extensive evidence suggests CO-induced dilation is mediated by BKCa channel activation. BKCa channel inhibition blocks CO-induced vasodilation (84,110,159). HO inhibitors reduce BKCa channel activity in renal and tail artery smooth muscle cells, suggesting that HO-derived products activate BKCa channels (63,152). O2 can regulate BKCa channels indirectly via CO, because O2 is necessary for heme metabolism by HO (150). Conversely, inhibitors of soluble guanylyl cyclase do not attenuate CO-induced BKCa channel activation in smooth muscle cells isolated from cerebral and renal arteries (63,153). Further, CO activates BKCa channels in excised arterial smooth muscle cell membrane patches that are removed from the intracellular milieu (150,152,153). Chemical modification of histidine residues blocks CO-induced BKCa channel activation, suggesting an important role for this amino acid (147). CO activates BKCa channel α-subunits expressed in the absence of auxiliary β-subunits in mammalian cells, suggesting CO acts on the pore forming α-subunit (152,153). Antisense downregulation of the β-subunit abolished NO-induced BKCa channel activation, but did not alter CO-induced BKCa channel activation in rat-tail artery smooth muscle cells, indicating that CO and NO activate BKCa channels by different mechanisms (152). Taken together, these findings show CO activates BKCa channels by interacting with the α-subunit or an associated regulatory element.
The α-subunit of the BKCa channel contains a heme-binding pocket and binding of heme to the BKCa channel inhibits BKCa channel activity (140). CO, by binding to channel-bound ferrous heme, changes the association of the heme with the channel leading to channel activation (60). Therefore, the BKCa channel is functionally a heme-protein. BKCa channel-bound heme is the receptor for CO, and CO binding increases BKCa channel Ca2+ sensitivity (see below).
In smooth muscle cells, BKCa channels are activated by local intracellular Ca2+ transients termed “Ca2+ sparks” that elevate the local Ca2+ concentration into the micromolar range (61). A single Ca2+ spark activates several BKCa channels, leading to an outward BKCa transient. In the arterial wall, summation of transient KCa currents induces a membrane hyperpolarization that reduces voltage-dependent Ca2+ channel (VDCC) activity, and thus, intracellular Ca2+ concentration. In cerebral artery smooth muscle cells, CO elevates BKCa channel Ca2+ sensitivity, particularly within the micromolar Ca2+ concentration range (153). Accordingly, endogenous HO-derived CO and exogenous CO elevate BKCa channel-transient frequency and amplitude by enhancing the effective coupling of Ca2+ sparks to BKCa channels (59). CO also elevates Ca2+ spark frequency that contributes to the CO-induced transient KCa current frequency and amplitude elevation. Since inhibitors of Ca2+ sparks and BKCa channels block CO-induced cerebrovascular dilation in vivo, Ca2+ spark to BKCa channel coupling is essential for vasodilatory actions of CO (59,84).
In the piglet cerebrovascular circulation, CO interacts with two other prominent endothelial derived vasodilators. Inhibition of either NOS or COX blocks dilation to CO (83). However, these blockades are not the consequence of CO increasing NO and prostacyclin that cause the dilation. The contributions of NO and prostacyclin are permissive in that only sufficient background levels are necessary before CO will produce dilation of piglet cerebral arterioles. Thus, if the concentration of NO and/or prostacyclin is held constant by blocking synthesis, but providing exogenous NO and/or prostacyclin, CO produces dose-dependent pial arteriolar dilation that is indistinguishable from the responses when NOS and COX are not inhibited (83). Furthermore, the final mechanism behind the permissive actions of NO and prostacyclin appear to be the same because either the prostacyclin mimic, iloprost, or the NO generating molecule, sodium nitroprusside (SNP), can return dilation to CO when both COX and NOS are inhibited (82). The source of the permissive mediators may be endothelial because endothelial denudation blocks CO-induced dilation and SNP or 8-br-cGMP restores dilation to CO (8). The actions of both NO and prostacyclin are mediated by protein kinase G, but only the NO action is via cGMP (82). Whether the action of protein kinase G is on the BKCa channel, ryanodine receptor, both, or another mechanism has not as yet been determined.
Functional significance of CO in control of cerebrovascular circulation
Functional significance of purported messengers and processes is usually suggested by actions of pharmacological inhibitors. Pharmacological inhibition of HO with substituted metalloporphyrins is a major source of data on the functional significance of endogenous CO, in addition to measurements of CO production, over expression of endogenous HO, and application of exogenous CO and CO releasing molecules (see below).
Available data suggest the predominant effects of HO inhibitory metalloporphyrins on cerebrovascular circulation in vivo result from HO inhibition. For example, photooxidized protoporphyrins do not block HO (42) and photooxidized CrMP does not alter cerebrovascular responses (123). Furthermore, CuMP, a metal porphyrin that does not inhibit HO, does not affect dilatory responses to AMPA, ATPA or ACPD (123).
Actions of metalloporphyrins unrelated to HO inhibition have been reported. Micromolar concentrations of metal porphyrins can both inhibit (55,131) and activate guanylyl cyclase (131). However, SnPP does not affect dilation to SNP, a cGMP dependent dilator (12). In hippocampal slices, effects of CrMP and ZnPP on NOS activity were greater than those of SnMP and ZnDP, even though all inhibited HO (102). These data suggest CrMP and ZnPP might inhibit NOS independently of HO inhibition. Conversely, ZnPP stimulated NO production by rabbit internal anal sphincter, an action apparently related to HO inhibition (19). ZnPP, but not other protoporphrins may inhibit VDCC in pituitary cells (87). However, the consequence of inhibition of VDCC would be dilation not constriction, so involvement of inhibition of VDCC in metalloporphryrin blockade of putative CO dependent responses in cerebrovascular circulation is unlikely. Metalloporphryrin inhibition of G-protein coupling to second messenger systems has been reported (112, 143). However, in piglet cerebral circulation metalloporphyrins do not inhibit dilation to isoproterenol, suggesting no interference with G-protein function (84). Finally, inhibitory activity toward another necessary component of heme metabolism, NADPH-cytochrome P-450 reductase and possible effects on the expression as well as activity of HO-2 in rat brain have been reported for SnPP, but not for ZnPP (96).
Alternative strategies to pharmacological inhibitors have limitations as well. Antisense oligonucleotide approaches suffer from cell toxicity and ineffectiveness (25,26,128) and siRNA, that can be more efficient than antisense oligonucleotides, also can have unintended actions (48,126,128). Loss of specific functions in KO mice without genes of interest can be very convincing, but no in vivo data are available for HO in cerebrovascular circulation. Also, this approach can be limited by biological compensation for missing proteins and questions of removal of processes and pathways associated with the intended target during development.
Overall, pharmacological inhibition of enzymes and receptors remains the most rapid and effective approach for removal of messages and processes of interest. While there are potential non-HO-mediated actions of metalloporphyrin HO inhibitors, none appear to account for the actions of these compounds on cerebrovascular circulation.
CO is a gasotransmitter related to neural function in the brain (10). Neurally released CO could be a regulator of cerebral blood flow, but no data supporting this concept have been collected to date. In vivo, topical CO (<1nM) dilates pial arterioles (84). Brain production of CO results in accumulation in CSF placed under cranial windows that is contiguous with brain extracellular fluid. Dilator concentrations of CO in this CSF (88±20nM) have been measured under control conditions and 10 fold increases have been measured with strong stimulation (12). Thus, the level of CO production by brain and vessels is sufficient to provide dilator effects on the cerebral circulation.
Indications of functional significance of CO are further supported by alterations of responses to important physiological cerebrovascular regulatory stimuli upon inhibition of HO with substituted metalloporphyrins. Pial arteriolar dilations to both hypoxia and topical glutamate are selectively inhibited following topical treatment with CrMP that blocks CO production (84,123). Furthermore, CO appears to be involved in pial arteriolar dilation to hypotension (64). Mediation of autoregulatory vasodilation could contribute to the protective effects of HO following brain ischemia, but most evidence suggests that the antioxidant properties of bilirubin are largely responsible (71). CO can also attenuate vascular responses to constrictor stimuli (151). For example, inhibition of HO accentuates the constriction of pial arterioles produced by hypertension but has minimal effects when piglets are normotensive. In addition, vasoconstriction to topical application of platelet activating factor is accentuated following inhibition of HO. In rat hypothalamus, CO has been shown to contribute to regulation of vascular tone that is particularly evident in the absence of NO (50). This finding in adult rats appears to be different from results of newborn pigs where NO plays an obligatory permissive role in CO-induced dilation and NOS inhibition blocks dilation to CO (see above).
While reported actions of CO on cerebrovascular tone are uniformly dilator, it should be mentioned that in skeletal muscle vasculature CO has been shown to be capable of producing vasoconstriction, apparently by inhibiting NOS. Thus, in isolated, pressurized, phenylephrine treated, rat gracilis muscle arterioles, either exogenous CO or stimulation of endogenous CO production produced constriction that was prevented by endothelium removal (40). That this constriction involves reduction of endothelial-derived NO is indicated by the ability of L-arginine to prevent the constriction and of NOS inhibition to convert the CO-induced constriction to dilation. Of interest, results from rat gracilis muscle arterioles are opposite of those from small, rat, renal arteries where CO increased NO production that contributed to CO-induced dilation (140).
Protective effects of HO/CO and regulation of CBF during seizures
CO produced by the brain contributes to regulation of cerebral blood flow during seizures (17,105,122). In bicuculline-induced epileptic seizures in newborn pigs, pial arteriolar dilation that occurs simultaneously with neuronal activation correlated with a massive increase in CO concentration in cortical CSF that was sustained for the duration of seizures (17,115). HO inhibitors, CrMP and SnPP, inhibited CO production by the brain and reduced cerebral dilation in response to seizures (17,115,122). The rapid increase in CO production by the brain during epileptic seizures is attributable exclusively to constitutive HO-2, because no induction of HO-1 or HO-2 protein was observed (17,119). The rapid increase in HO-2 enzymatic activity in the cerebral vasculature occurs via a glutamate receptor-mediated mechanism.
The major excitatory neurotransmitter, glutamate, is massively released from neurons during seizures (103). Cerebral microvascular endothelial cells from rat, human and pig, in addition to neurons and glia, have receptors for glutamate (14,23,74,116,133,134). Both ionotropic and metabotropic subtypes have been identified in cerebral vascular endothelial cells (23,116,133,134). Conversely, other investigators were unable to detect glutamate receptors on cerebral microvascular endothelial cells from sheep, rat, or human (9,108). The reason for these opposing results is not apparent. Consistent with the presence of receptors, glutamate receptor agonists cause functional responses in cerebral vascular endothelial cells that include increases in HO activity (116), changes in endothelial monolayer permeability (134), and increases in formation of ROS (14,134). Of significance to the present review, piglet cerebral microvessels respond directly to glutamate and selective NMDA- and AMPA/Kainate receptor agonists by increasing CO production (79,116). We found pressurized pial arteries respond to glutamate by endothelium-dependent vasodilation (11% at 10−5M) (36), although less strongly than in vivo (20% dilation at 10−5M)(123). Others have failed to detect any glutamate-receptor-mediated responses in pressurized bovine middle cerebral arteries (149) or middle cerebral arteries from newborn pigs (135). While the difference in species, age and artery examined could account for divergent results in the first instance, the reason for the inconsistent results in piglets is not evident. The choice of anesthetic, ketamine versus pentobarbital, is different but appears to be an unlikely contributor in an isolated vessel experiment. While the diameters of the vessels in the two studies are similar, the arteries selected may not be the same, since Simandle et al (135) could pressurize to 100 mmHg which was not tolerated by arteries used by Fiumana et al (unpublished observation). Heterogeneity of responses even in pial arteries and arterioles within a cm diameter circle of surface parietal cortex has been described (118). Regardless, it is clear that cerebral arterioles respond more strongly to glutamate in vivo than in vitro, suggesting extravascular cells contribute to glutamatergic dilation.
Epileptic seizures result in prolonged postictal cerebral vascular dysfunction characterized by reduced vasoreactivity to physiologically relevant dilators, including hypercapnia and bradykinin (119). When HO-2 activity is inhibited before seizures, cerebral vascular dysfunction is observed immediately after the ictal episode and is extended for at least 2 days of the postictal period (17,119). In contrast, in animals with intact HO-2 activity, no immediate reduction of cerebral vascular reactivity is detected, but cerebral vascular reactivity is greatly reduced 2 days later (12, 86). Therefore, HO-2 is necessary for a short-term protection but not sufficient for a long-term protection of the cerebral vasculature from detrimental effects of epileptic seizures. However, up-regulation of cerebral HO-1 expression can completely protect the cerebral vasculature during the delayed postictal period (119). Thus it appears that HO-1 can provide long-term protection against postictal cerebral vascular dysfunction. HO-derived CO is important in increasing blood flow to the brain to match excessive neuronal activity during the ictal episode, thus protecting neurons and preventing cerebral vascular injury. HO activity also reduces the amount of pro-oxidant heme, and results in production of biliverdin/bilirubin, which have powerful antioxidant properties as a redox cycling pair that scavenges ROS (7,29,95,104). Also, CO itself may have anti-apoptotic effects in vascular endothelial and smooth muscle cells (see below).
CO, apoptosis, and cell proliferation
In addition to its role as a neuronal and vascular messenger, CO can suppress apoptosis (15,136,160). The molecular mechanism of anti-apoptotic protection by CO is not clear. CO may inhibit generation of free radicals by mitochondria via a direct interaction with the heme protein of the mitochondrial electron transport chain, or interact with the p38 MAPK- or cGMP- signaling pathways that can modulate apoptosis in a cell-and signal- specific manner (136). In addition, CO inhibits the activity of caspases that play a major role in executing apoptosis (138,160). A possible interaction of CO with NFkB-mediated apoptosis signaling also has been proposed (15,90,136). Pro-apoptotic effects of CO have been also reported (139). It appears that the ability of CO to suppress or promote apoptosis depends on the specific apoptotic signal and cell type.
In addition to effects on apoptosis, CO may affect cerebrovascular circulation via regulation of vascular cell proliferation. In rat aortic smooth muscle cells, increasing CO inhibited and scavenging CO increased cell proliferation, actions apparently mediated by increasing cGMP that increases E2F-1 expression (106). Furthermore, CO can reduce endothelial cell proliferation caused by hypoxia by inhibiting VEGF production by adjacent vascular smooth muscle (91). This action as well appears to be mediated via cGMP, by decreasing binding of a hypoxic enhancer to hypoxia-inducible factor-1. Following vascular injury as well as hypoxia, CO suppresses vascular smooth muscle proliferation by increasing cellular cGMP that activates p38 mitogen-activated protein kinase, up-regulating caveolin-1 that prevents proliferation (69). Conversely, CO has been shown to promote proliferation of microvascular endothelial cells (64). Thus, depending upon the conditions, background stimuli, and specific cell type CO can either increase or decrease apoptosis and cell proliferation.
Hydrogen sulfide (H2S)
The below discussion is summarized pictorially in Fig. 2.
Figure 2.
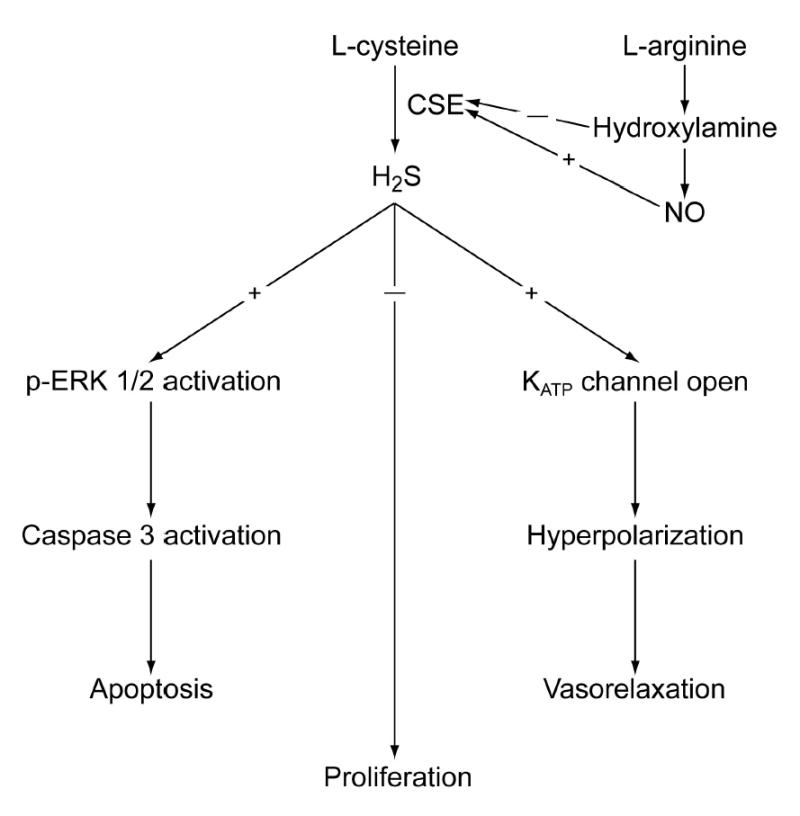
Flow chart summarizing the section of this review on H2S.
Control of H2S production
Production and utilization of H2S have been demonstrated in tissues from different life forms, including bacteria and archae (113), non-mammalian vertebrates (28), and mammals (145,146) with physiological concentrations in the range of 20–160 μM (1,163).
Two pyridoxal-5′-phosphate-dependent enzymes, cystathionine beta-synthase (CBS) and cystathionine gamma-lyase (CSE), are responsible for the endogenous production of H2S in mammalian tissues (1,145,146,163). While expression of CBS is more abundant in liver and neuronal tissues, CSE is the dominant H2S-generating enzyme in cardiovascular system. CSE and CBS catalyze the hydrolysis of cysteine via beta-elimination to generate H2S. A beta-replacement reaction in which cysteine is condensed in the presence of CSE with homocysteine to form cystathionine and H2S has been recently reported in vitro (21). The condensation of cysteine is believed to be 50 times more efficient than hydrolysis of cysteine in terms of H2S production (21).
The synthetic pathway for H2S production is intermingled with the synthetic pathway for NO. The expression and activity of CSE are up-regulated by NO, but hydroxylamine (a precursor of NO) inhibits the activity of CBS (114). S-adenosylmethionine and pyridoxal-5′-phosphate stimulate CSE activity to increase H2S production (70,132). Kimura reported that CBS-mediated hydrolysis of cysteine was regulated by Ca2+ and CaM in mouse brain (70). This observation, however, could not be repeated by Chen et al. in mouse brain, mouse liver, or purified recombinant human CBS expressed in E. coli or yeast (21).
Mechanism of H2S vascular actions
Compelling evidence indicates that KATP channels are major targets of H2S. Opening of KATP channels hyperpolarizes cells and closes VDCC. This provides an important mechanism linking cellular metabolism to excitability and contractility of vascular smooth muscle cells. Patch-clamp studies show that H2S increases whole-cell KATP channel currents in single smooth muscle cells isolated from both aorta and mesenteric arteries (22,163). This stimulatory effect of H2S is concentration dependent and reversible. The KATP inhibitor, glibenclamide blocks and the activator, pinacidil, mimics the effect of H2S on KATP channels. H2S hyperpolarizes smooth muscle cells and the action is antagonized by glibenclamide (163). The molecular mechanisms underlying the effect of H2S on KATP channels are still largely unknown. Alteration of intracellular ATP concentration cannot explain the interaction between H2S and KATP channels since clamping intracellular ATP at different levels did not alter the relative stimulatory effect of H2S on the whole-cell KATP channels (163). Because H2S is a reductant (68), it is possible that H2S directly interacts with KATP channel proteins by reducing the cysteine residues. A mutagenesis approach that replaces cysteine residues with structurally similar serine residues would support this hypothesis if H2S fails to stimulate KATP channels after selective point mutation of cysteine residues.
The pro-apoptotic effect of H2S (see below) is related to increased activity of extracellular signal-regulated kinase (ERK), but not p38 MAPK or c-Jun N-terminal kinase activity. Activation of caspase-3 by ERK could be one downstream target of H2S-ERK interaction (154).
Functional significance of H2S in circulatory regulation
Vascular contractility is regulated by endogenous and exogenous H2S at physiologically relevant concentrations. Intravenous injection of H2S decreases mean arterial blood pressure of anesthetized rats by decreasing vascular resistance (163). Daily intraperitoneal injections of D,L-propargylglycine (PPG), a specific blocker of CSE, for 2–3 weeks elevates of systolic blood pressure (162). Since this PPG treatment suppresses H2S production in vascular and other tissues, PPG-induced hypertension may result from reduced endogenous H2S production in vascular tissues (162). H2S concentration-dependently relaxes phenylephrine-precontracted rat aorta, a conduit artery (161). The isolated and perfused rat mesenteric vascular bed, a model of peripheral resistance arteries, is also relaxed by H2S (16). Although rat aortic and mesenteric artery tissues generate comparable levels of H2S (15,161,162,163), rat mesenteric arteries are much more sensitive to H2S (EC50 of 25.2±3.6 μM) than rat aorta (EC50 of 125±14 μM). L-cysteine, a substrate of CSE and CBS, increases endogenous H2S production and decreases contractility of mesenteric arteries. In contrast, PPG abolishes the L-cysteine-dependent increase in H2S production and relaxation of mesenteric arteries. These findings indicate the importance of endogenous H2S in regulating vascular contractility (22).
H2S can be involved in the control of both proliferation and apoptosis in vascular smooth muscle cells. H2S at physiologically relevant concentrations did not induce necrosis of human aortic smooth muscle cells (154). However, H2S (200–500 μM) increased the numbers of condensed apoptotic nuclei, TUNEL-positive cells, and oligonucleosomal DNA fragmentation. The cells showed the morphological changes typical of apoptosis within 2 h of H2S application. Interestingly, baseline endogenous H2S modulates exogenous H2S-induced apoptosis of human aortic smooth muscle cells. Treatment of these cells with the CSE inhibitor, PPG, alone for 1 h did not induce any apoptotic changes. This preconditioning treatment, however, significantly enhanced the pro-apoptotic effect of exogenously applied H2S. The threshold concentration of H2S to induce apoptosis was reduced from 200 μM to 100 μM under this condition. It is hypothesized that basal endogenous H2S may be high enough to desensitize apoptotic signaling pathways in vascular smooth muscle cells. As such, the basal H2S level may serve as a set point for the basal apoptotic status of smooth muscle cells. Increases or decreases in endogenous H2S levels may consequently alter homeostatic control of smooth muscle cell apoptosis.
H2S also can inhibit cell proliferation. Stable over-expression of CSE in HEK-293 cells increased endogenous H2S production and inhibited cell proliferation and DNA synthesis (154). These effects were significantly reversed in the presence of the H2S scavenger, methemoglobin. Exogenous H2S at the physiologically relevant concentration of 100 μM also inhibited cell proliferation. However, neither over-expression of CSE nor application of exogenous H2S induced apoptosis of HEK-293 cells. The difference between the endogenous H2S basal levels in vascular smooth muscle cells and HEK-293 cells may underlie the lack of pro-apoptotic response of HEK-293 cells to H2S. It has been reported that CSE expression level and its enzymatic activity were higher in kidney (HEK) than in smooth muscle (58).
The function of H2S in cerebral circulation remains poorly understood. H2S is produced in brain tissues, reaching an endogenous level of 50–160 μM (1). The release of corticotropin-releasing hormone from hypothalamus and facilitation of hippocampal long-term potentiation appear to be influenced by H2S metabolism (70). Environmental exposure to H2S at low concentrations for 2 weeks activates protein synthesis in nerve cells and myelinated fibers in the cerebral cortex (137), but the physiological meaning of this study is unclear. It appears likely that endogenously produced H2S exerts similar vasorelaxant effects on the cerebral circulation as in the systemic circulation. Cerebral vascular smooth muscle cells would be exposed to significant amounts of H2S and cerebral vascular smooth muscle cells possess the KATP channels (144), the target of H2S on other vascular smooth muscle cells (145). A recent study showed that NMDA induced dilation of pial arteries in newborn pigs was partially due to activation of KATP channels (121). In glutamatergic neurons, endogenous H2S has been shown to enhance NMDA receptor-mediated transmembrane currents (70).
Conclusions
CO and H2S are gaseous cellular messenger molecules that are involved in cerebrovascular flow regulation. Thus, they join the group with the most studied gaseous mediator, NO. The collective differences between the gasotransmitters and the classical neurotransmitters, hormones, and lipid and peptide autocrine/paracrine messengers in intercellular movement, cellular action mechanisms, and signal termination are already revolutionizing our concepts of cellular communication. This review has focused on two of these molecules, CO and H2S, the physiological significance of which have begun to be appreciated in the last decade or less.
CO exerts a dilator influence on the cerebral circulation and is involved in active hyperemia, autoregulation, hypoxic dilation, and counteracting vasoconstriction. CO is produced by metabolism of cellular heme by a constitutive enzyme expressed highly in the cerebral microcirculation and by an inducible enzyme that is readily up-regulated in brain by potentially injurious conditions. CO production is regulated by controlling substrate, HO-2 catalytic activity, and HO-1 expression. CO causes cerebrovascular dilation by elevating smooth muscle BKCa channel Ca2+ sensitivity, leading to increased coupling to Ca2+ sparks, thereby, hyperpolarizing the cell. The HO/CO vasodilatory system interacts at both the level of messenger production and action with other important mediators of cerebral circulatory control, including NO and prostanoids. While much is becoming known, much more remains unknown about CO and cerebrovascular circulatory control. Even though CO is involved in several cerebral circulatory control mechanisms, many other situations where CO may be a key mediator remain to be explored, and very little is known about pathological alterations of HO/CO system in the cerebral circulation. The cellular origins of CO mediating distinct responses in the intact cerebral circulation are unknown. Knowledge of the mechanisms regulating CO production is only beginning to become available, and those data appear to contrast at times. While mechanisms by which CO can affect the activity of BKCa channels are being revealed, these mechanisms are still not completely understood. In addition, CO can contribute to vascular remodeling by enhancing or depressing both apoptosis and cellular proliferation depending upon cell type and particularly other impinging signals. Thus, much of the physiology and biochemistry of HO/CO in the cerebral circulation remains open for exploration.
H2S is a very recently identified gasotransmitter that is produced by many mammalian cells, including vascular smooth muscle cells. By relaxing vascular smooth muscle cells to dilate blood vessels, promoting apoptosis of vascular smooth muscle cells, and inhibiting proliferation-associated vascular remodeling, H2S modulates both the function and structure of the circulatory system. The mechanisms behind H2S-induced activations of KATP channels on the cell membrane and the ERK signaling pathway inside cells are not yet understood. Genetic approaches to manipulate CSE expression and endogenous production of H2S may help to decisively establish physiological and pathophysiological importance of H2S in regulation of cerebrovascular tone in particular and cardiovascular function in general. Future studies should examine vasorelaxant and pro-apoptotic, antiproliferative effects of H2S on cerebral vascular tissues. Advance in this understanding also has the potential to provide a novel therapeutic avenue for treatment of many vascular diseases linked to H2S-related abnormal cellular contractility and proliferation.
Acknowledgments
C.W. Leffler, J. H. Jaggar, and H Parfenova are supported by NHLBI/NIH. H. Parfenova is also supported by NINDS/NIH. R. Wang is supported by CIHR. We thank G. Short for construction of the figures and M. Lester for clerical assistance.
References
- 1.Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abraham NG, Botros FT, Rezzani R, Rodella L, Bianchi R, Goodman AI. Differential effect of cobalt protoporphyrin on distributions of heme oxygenase in renal structure and on blood pressure in SHR. Cell Mol Biol (Noisy-le-grand) 2002;48:895–902. [PubMed] [Google Scholar]
- 3.Abraham NG, Quan S, Mieyal PA, Yang L, Burke-Wolin T, Mingone CJ, Goodman AI, Nasjletti A, Wolin MS. Modulation of cGMP by human HO-1 retrovirus gene transfer in pulmonary microvessel endothelial cells. Am J Physiol. 2002;283:L1117–24. doi: 10.1152/ajplung.00365.2001. [DOI] [PubMed] [Google Scholar]
- 4.Acevedo CH, Ahmed A. Heme oxygenase-1 inhibits human myometrial contractility via carbon monoxide and is upregulated by progesterone during pregnancy. J Clin Invest. 1998;101:949–955. doi: 10.1172/JCI927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Appleton SD, Marks GS, Nakatsu K, Brien JF, Smith GN, Graham CH. Heme oxygenase activity in placenta: direct dependence on oxygen availability. Am J Physiol. 2002;282:H2055–9. doi: 10.1152/ajpheart.01084.2001. [DOI] [PubMed] [Google Scholar]
- 6.Baram TZ, Snead OC. Bicuculline-induced seizures in infant rats: ontogeny of behavioral and electrocortical phenomena. Dev Brain Res. 1990;57:291–295. doi: 10.1016/0165-3806(90)90055-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baranano DE, Snyder SH. Neural roles for heme oxygenase: contrasts to nitric oxide synthase. Proc Natl Acad Sci U S A. 2001;98:10996–11002. doi: 10.1073/pnas.191351298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barkoudah E, Jaggar JH, Leffler CW. The permissive role of endothelial NO in CO-induced cerebrovascular dilation. Am J Physiol. 2004;287:H1459–H1465. doi: 10.1152/ajpheart.00369.2004. [DOI] [PubMed] [Google Scholar]
- 9.Beart PM, Sheehan KA, Manallack DT. Absence of N-methyl-D-aspartate receptors on ovine cerebral microvessels. J Cereb Blood Flow Metab. 1988;8:879–821. doi: 10.1038/jcbfm.1988.146. [DOI] [PubMed] [Google Scholar]
- 10.Boehning D, Moon C, Sharma S, Hurt KJ, Hester LD, Ronnett GV, Shugar D, Snyder SS. Carbon monoxide neurotransmission activated by CK2 phosphorylation of heme oxygenase-2. Neuron. 2003;40:129–137. doi: 10.1016/s0896-6273(03)00596-8. [DOI] [PubMed] [Google Scholar]
- 11.Boehning D, Sedaghat L, Sedlak TW, Snyder SS. Heme oxygenase-2 is activated by calcium-calmodulin. J Biol Chem. 2004;279:30927–30930. doi: 10.1074/jbc.C400222200. [DOI] [PubMed] [Google Scholar]
- 12.Boehning D, Snyder SH. Novel neural modulators. Ann Rev Neurosci. 2003;26:105–131. doi: 10.1146/annurev.neuro.26.041002.131047. [DOI] [PubMed] [Google Scholar]
- 13.Bouton C, Demple B. Nitric oxide-inducible expression of heme oxygenase-1 in human cells. Translation-independent stabilization of the mRNA and evidence for direct action of nitric oxide. J Biol Chem. 2000 Oct 20;275(42):32688–93. doi: 10.1074/jbc.275.42.32688. [DOI] [PubMed] [Google Scholar]; Brain Res. 2002;954:51–59. doi: 10.1016/s0006-8993(02)03338-3. [DOI] [PubMed] [Google Scholar]
- 14.Bresgen N, Karlhuber G, Krizbai I, Bauer H, Bauer HC, Eckl PM. Oxidative stress in cultured cerebral endothelial cells induced chromosomal aberrations, micronuclei, and apoptosis. J Neurosci Res. 2003;72:327–333. doi: 10.1002/jnr.10582. [DOI] [PubMed] [Google Scholar]
- 15.Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AM, Soares MP. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J Exp Med. 2000;192:1015–1026. doi: 10.1084/jem.192.7.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brune B, Ullrich V. Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol Pharmacol. 1987;32:497–504. [PubMed] [Google Scholar]
- 17.Carratu P, Pourcyrous M, Fedinec A, Leffler CW, Parfenova H. Endogenous heme oxygenase prevents impairment of cerebral vascular functions caused by seizures. Am J Physiol Heart Circ Physiol. 2003;285:H1148–H1157. doi: 10.1152/ajpheart.00091.2003. [DOI] [PubMed] [Google Scholar]
- 18.Caudill TK, Resta TC, Kanagy NL, Walker BR. Role of endothelial carbon monoxide in attenuated vasoreactivity following chronic hypoxia. Am J Physiol. 1998;275:R1025–R1030. doi: 10.1152/ajpregu.1998.275.4.R1025. [DOI] [PubMed] [Google Scholar]
- 19.Chakder S, Rathi S, Ma XL, Rattan S. Heme oxygenase inhibitor zinc protoporphyrin IX causes an activation of nitric oxide synthase in the rabbit internal anal sphincter. J Pharmacol Exp Ther. 1996;277:1376–82. [PubMed] [Google Scholar]
- 20.Chang EF, Wong RJ, Vreman HJ, Igarashi T, Galo E, Sharp F, Stevenson DK, Noble Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene. 2004;336:241–250. doi: 10.1016/j.gene.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Chen X, Jhee KH, Kruger WD. Production of the neuromodulator H2S by cystathionine beta-synthase via the condensation of cysteine and homocysteine. J Biol Chem. 2004;279:52082–52086. doi: 10.1074/jbc.C400481200. [DOI] [PubMed] [Google Scholar]
- 22.Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Sensitive relaxant response of rat mesenteric artery beds to hydrogen sulfide. Am J Physiol. 2004;287:H2316–H2323. doi: 10.1152/ajpheart.00331.2004. [DOI] [PubMed] [Google Scholar]
- 23.Collard CD, Park KA, Montalto MC, Alapati S, Buras JA, Stahl GL, Colgan SP. Neutrophil-derived glutamate regulates vascular endothelial barrier function. J Biol Chem. 2002;277:14801–14811. doi: 10.1074/jbc.M110557200. [DOI] [PubMed] [Google Scholar]
- 24.Colpaert EE, Timmermans JP, Lefebvre RA. Investigation of the potential modulatory effect of biliverdin, carbon monoxideand bilirubin on nitrergic neurotransmission in the pig gastric fundanus. Eur j Pharmacol. 2002;457:177–186. doi: 10.1016/s0014-2999(02)02691-2. [DOI] [PubMed] [Google Scholar]
- 25.Crooke ST, Bennett CF. Progress in antisense oligonucleotide therapeutics. Annu Rev Pharmacol Toxicol. 1996;36:107–29. doi: 10.1146/annurev.pa.36.040196.000543. [DOI] [PubMed] [Google Scholar]
- 26.Crooke ST. Antisense strategies. Curr Mol Med. 2004;4(5):465–87. doi: 10.2174/1566524043360375. [DOI] [PubMed] [Google Scholar]
- 27.Ding Y, McCoubrey WK, Jr, Maines MD. Interaction of heme oxygenase-2 with nitric oxide donors. Is the oxygenase an intracellular ‘sink’ for NO? Eur J Biochem. 1999;264:854–861. doi: 10.1046/j.1432-1327.1999.00677.x. [DOI] [PubMed] [Google Scholar]
- 28.Dombkowski RA, Russell MJ, Schulman AA, Doellman MM, Olson KR. Vertebrate phylogeny of hydrogen sulfide vasoactivity. Am J Physiol. 2005;288:R243–R252. doi: 10.1152/ajpregu.00324.2004. [DOI] [PubMed] [Google Scholar]
- 29.Dore S, Takahashi M, Ferris CD, Zakhary R, Hester LD, Guastella D, Snyder SH. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci U S A. 1999;96:2445–2450. doi: 10.1073/pnas.96.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drummond GS, Greenbaum NL, Kappas A. Tin(Sn+4)-diiododeuteroporphyrin; an in vitro and in vivo inhibitor of heme oxygenase with substantially reduced photoactive properties. J Pharmacol Exp Ther. 1991;257(3):1109–13. [PubMed] [Google Scholar]
- 31.Dwyer BE, Nishimura RN, Lu SY. Differential expression of heme oxygenase-1 in cultured cortical neurons and astrocytes determined by the aid of a new heme oxygenase antibody. Response to oxidative stress. Brain Res Mol Brain Res. 1995;30:37–47. doi: 10.1016/0169-328x(94)00273-h. [DOI] [PubMed] [Google Scholar]
- 32.Ellis A, Tiggle CR. Endothelium-derived activated oxygen species: their relationship to endothelial-dependent hyperpolarization and vascular tone. Can J Physiol Pharmacol. 2003;81:1013–1028. doi: 10.1139/y03-106. [DOI] [PubMed] [Google Scholar]
- 33.Ewing JF, Maines MD. In situ hybridization and immunohistochemical localization of heme oxygenase-2 mRNA and protein in normal rat brain: differential distribution of isozyme 1 and 2. Mol Cell Neurosci. 1992;3:559–570. doi: 10.1016/1044-7431(92)90068-d. [DOI] [PubMed] [Google Scholar]
- 34.Ewing JF, Weber CM, Maines MD. Biliverdin reductase is heat resistant and coexpressed with constitutive and heat shock forms of heme oxygenase in brain. J Neurochem. 1993;61:1015–23. doi: 10.1111/j.1471-4159.1993.tb03615.x. [DOI] [PubMed] [Google Scholar]
- 35.Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- 36.Fiumana E, Parfenova H, Jaggar JH, Leffler CW. Carbon monoxide mediates vasodilator effects of glutamate in isolated pressurized cerebral arterioles of newborn pigs. Am J Physiol. 2003;284:H1073–1079. doi: 10.1152/ajpheart.00881.2002. [DOI] [PubMed] [Google Scholar]
- 37.Foresti R, Hammad J, Clark JE, Johnson TR, Mann BE, Friebe A, Green CJ, Motterlini R. Vasoactive properties of CORM-3, a novel water-soluble carbon monoxide-releasing molecule. Br J Pharmacol. 2004;142:453–60. doi: 10.1038/sj.bjp.0705825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foresti R, Sarathchandra P, Clark JE, Green CJ, Motterlini R. Peroxinitrite induces haem oxygenase-1 in vascular endothelial cells: a link to apoptosis. Biochem J. 1999;339:729–736. [PMC free article] [PubMed] [Google Scholar]
- 39.Friebe A, Schultz G, Koesling D. Sensitizing soluble guanylate cyclase to become a highly CO-sensitive enzyme. EMBO J. 1996;15:6863–6868. [PMC free article] [PubMed] [Google Scholar]
- 40.Fruzsina K, Johnson RA. Carbon monoxide promotes endothelium-dependent constriction of isolated gracilis muscle arterioles. Am J Physiol. 2003;285:R536–R541. doi: 10.1152/ajpregu.00624.2002. [DOI] [PubMed] [Google Scholar]
- 41.Furchgott RF, Jothianandan D. Endothelium-dependent and –independent vasodilation involving cyclic GMP: relation induced by nitric oxide, carbon monoxide and light. Blood Vessels. 1991;28:52–61. doi: 10.1159/000158843. [DOI] [PubMed] [Google Scholar]
- 42.Greenbaum NL, Kappas A. Comparative photoactivity of tin and zinc porphyrin inhibitors heme oxygenase: pronounced photolability of the zinc compounds. Photochem Photobiol. 1991;54(2):183–92. doi: 10.1111/j.1751-1097.1991.tb02005.x. [DOI] [PubMed] [Google Scholar]
- 43.Grundemar L, Ny L. Pitfalls using metalloporphyrins in carbon monoxide research. TiPS. 1997;18:193–195. doi: 10.1016/s0165-6147(97)01065-1. [DOI] [PubMed] [Google Scholar]
- 44.Gutterman DD, Miura H, Liu Y. Redox modulation of vascular tone. Focus of potassium channel mechanisms of dilation. Arterioscler Thromb Vasc Biol. 2005;25:671–678. doi: 10.1161/01.ATV.0000158497.09626.3b. [DOI] [PubMed] [Google Scholar]
- 45.Haeusslein LJ. Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J Neurosci. 2003;23:3689–3966. doi: 10.1523/JNEUROSCI.23-09-03689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harder DR, Zhang C, Gebremedhin Astrocytes function in matching blood flow to metabolic activity. News Physiol Sci. 2002;16:27–31. doi: 10.1152/physiologyonline.2002.17.1.27. [DOI] [PubMed] [Google Scholar]
- 47.Hayashi S, Omata Y, Sakamoto H, Higashimoto Y, Hara T, Sagara Y, Noguchi M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene. 2004;336 :241–50. doi: 10.1016/j.gene.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 48.Heidenreich O. Oncogene suppression by small interfering RNAs. Curr Pharm Biotechnol. 2004;5:349–54. doi: 10.2174/1389201043376733. [DOI] [PubMed] [Google Scholar]
- 49.Hock TD, Nick HS, Agarwal A. Upstream stimulatory factors, USF1 and USF2, bind to the human haem oxygenase-1 proximal promoter in vivo and regulate its transcription. Biochem J. 2004;383:209–18. doi: 10.1042/BJ20040794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horvath B, Hrabak A, Kaldi K, Sandor P, Benyo Z. Contribution of the heme oxygenase pathway to the maintenance of the hypothalamic blood flow during diminished nitric oxide synthesis. J Cereb Blood Flow Metab. 2003;23:653–657. doi: 10.1097/01.WCB.0000071890.63724.C9. [DOI] [PubMed] [Google Scholar]
- 51.Hosein S, Marks GS, Brien JF, McGlaughlin BE, Nakatsu K. An extracellular source of heme can induce a significant heme oxygenase mediated relaxation in the rat aorta. Can J Physiol Pharmacol. 2002;80:761–5. doi: 10.1139/y02-086. [DOI] [PubMed] [Google Scholar]
- 52.Hu Y, Ma N, Yang M, Semba R. Expression and distribution of heme oxygenase-2 mRNA and protein in rat kidney. J Histochem Cytochem. 1998;46:249–256. doi: 10.1177/002215549804600214. [DOI] [PubMed] [Google Scholar]
- 53.Huang TJ, McCoubrey WK, Jr, Maines MD. Heme oxygenase-2 interaction with metalloporphyrins: function of heme regulatory motifs. Antioxid Redox Signal. 2001;3:685–96. doi: 10.1089/15230860152543023. [DOI] [PubMed] [Google Scholar]
- 54.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nature Rev Neuroscience. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 55.Ignarro LJ, Ballot B, Wood KS. Regulation of soluble guanylate cyclase activity by porphyrins and metalloporphyrins. J Biol Chem. 1984;259:6201–7. [PubMed] [Google Scholar]
- 56.Immenschuh S, Ramadori Gene regulation of heme oxygenase-1 as a therapeutic target. Biochem Pharmacol. 2000;60:1121–1128. doi: 10.1016/s0006-2952(00)00443-3. [DOI] [PubMed] [Google Scholar]
- 57.Ingi T, Chiang G, Ronnett GV. The regulation of heme turnover and carbon monoxide biosynthesis in cultured primary rat olfactory receptor neurons. J Neurosci. 1996;16:5621–5628. doi: 10.1523/JNEUROSCI.16-18-05621.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ishii I, Akahoshi N, Yu XN, Kobayashi Y, Namekata K, Komaki G, Kimura H. Murine cystathionine gamma-lyase: complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem J. 2004;381:113–123. doi: 10.1042/BJ20040243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaggar JH, Leffler CW, Cheranov SY, Tcheranova D, E S, Cheng X. Carbon monoxide dilates cerebral arterioles by enhancing the coupling of Ca2+ sparks to Ca2+-activated K+ channels. Circ Res. 2002;91:610–617. doi: 10.1161/01.res.0000036900.76780.95. [DOI] [PubMed] [Google Scholar]
- 60.Jaggar, JH, A. Li, H. Parfenova, J. Liu, E. S. Umstot, A. M. Dopico, and C. W. Leffler. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ. Res. (in press) [DOI] [PMC free article] [PubMed]
- 61.Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol. 2000;278:C235–C256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- 62.Juckett M, Zheng Y, Yuan H, Pastor T, Antholine W, Wber M, Vercellotti G. Heme and the endothelium, Effects of nitric oxide on catalytic iron and heme degradation by heme oxygenase. J Biol Chem. 1998;273:23388–23397. doi: 10.1074/jbc.273.36.23388. [DOI] [PubMed] [Google Scholar]
- 63.Kaide JI, Zhang F, Wei Y, Jiang H, Yu C, Wang WH, Balazy M, Abraham NG, Nasjletti A. Carbon monoxide of vascular origin attenuates the sensitivity of renal arterial vessels to vasoconstrictors. J Clin Invest. 2001;107:1163–1171. doi: 10.1172/JCI11218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanu A, Whitfield J, Leffler CW. Carbon monoxide contributes to cerebrovascular vasodilation in hypotensive piglets. FASEB J. 2005;19:A1249. doi: 10.1152/ajpheart.01368.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kietzmann T, Samoylenko A, Immenschuh S. Transcriptional regulation of heme oxygenase-1 gene expression by MAP kinases of the JNK and p38 pathways in primary cultures of rat hepatocytes. J Biol Chem. 2003;278:17927–17936. doi: 10.1074/jbc.M203929200. [DOI] [PubMed] [Google Scholar]
- 66.Kikuchi G, Hayashi N. Regulation by heme of synthesis and intracellular translocation of delta-aminolevinulinate synthase in the liver. Mol Cell Biochem. 1981;37:27–41. doi: 10.1007/BF02355885. [DOI] [PubMed] [Google Scholar]
- 67.Kim C, Zhou Q, Deng B, Thornton EC, Xu H. Chromium(VI) reduction by hydrogen sulfide in aqueous media: stoichiometry and kinetics. Environ Sci Technol. 2001;35:2219–2225. doi: 10.1021/es0017007. [DOI] [PubMed] [Google Scholar]
- 68.Kim HP, Wang X, Galbiati F. Ryter SW, and Choi AMK. Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J. 2004;18:1080–1089. doi: 10.1096/fj.03-1391com. [DOI] [PubMed] [Google Scholar]
- 69.Kim HP, Wang X, Nakao A, Kim SI, Murase N, Choi ME, Ryter SW, Choi AM. Caveolin-1 expression by means of p38beta mitogen-activated protein kinase mediates the antiproliferative effect of carbon monoxide. Proc Natl Acad Sci U S A. 2005;102:11319–24. doi: 10.1073/pnas.0501345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kimura H. Hydrogen sulfide as a neuromodulator. Mol Neurobiol. 2002;26:13–19. doi: 10.1385/MN:26:1:013. [DOI] [PubMed] [Google Scholar]
- 71.Koehler R, Traystman RJ. Cerebrovascular effects of carbon monoxide. Antiox Redo Signal. 2002;4:279–290. doi: 10.1089/152308602753666334. [DOI] [PubMed] [Google Scholar]
- 72.Koneru P, Leffler CW. Role of cyclic GMP in carbon monoxide induced vasodilation in piglets. Am J Physiol. 2004;286:H304–H309. doi: 10.1152/ajpheart.00810.2003. [DOI] [PubMed] [Google Scholar]
- 73.Kourembanas S, Morita T, Liu Y, Christou H. Mechanisms by which oxygen regulates gene expression and cell-cell interaction in the vasculature. Kidney Int. 1997;51:438–443. doi: 10.1038/ki.1997.58. [DOI] [PubMed] [Google Scholar]
- 74.Krizbai IA, Deli MA, Pestenacz A, Siklos L, Szabo CA, Andras I, Joo F. Expression of glutamate receptors on cultured cerebral endothelial cells. J Neurosci Res. 1998;54:814–819. doi: 10.1002/(SICI)1097-4547(19981215)54:6<814::AID-JNR9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 75.Kronke G, Bochkov VN, Huber J, Gruber F, Bluml S, Furnkranz A, Kadl A, Binder BR, Leitinger N. Oxidized phospholipids induce expression of human heme oxygenase-1 involving activation of cAMP-responsive element-binding protein. J Biol Chem. 2003;278:51006–51014. doi: 10.1074/jbc.M304103200. [DOI] [PubMed] [Google Scholar]
- 76.Lavrovsky Y, Schwartzman ML, Abraham NG. Novel regulatory sites of the human heme oxygenase-1 promoter region. Biochem Biophys Res Commun. 1993;196:336–41. doi: 10.1006/bbrc.1993.2253. [DOI] [PubMed] [Google Scholar]
- 77.Lee PJ, Hiang B, Chin B, Iyer N, Alam J, Semenza G, Choi A. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase gene in response to hypoxia. J Biol Chem. 1997;272:5373–5381. [PubMed] [Google Scholar]
- 78.Lee TJ. Nitric oxide and the cerebral vascular function. J Biomed Sci. 2000;7:16–26. doi: 10.1007/BF02255914. [DOI] [PubMed] [Google Scholar]
- 79.Leffler CW, Balabanova L, Fedinec AL, Waters CM, Parfenova H. Mechanism of glutamate stimulation of CO production in cerebral microvessels. Am J Physiol. 2003;285:H74–H80. doi: 10.1152/ajpheart.01081.2002. [DOI] [PubMed] [Google Scholar]
- 80.Leffler CW, Balabanova L, Fedinec AL, Parfenova H. Nitric oxide increases carbon monoxide production by piglet cerebral microvessels. Am J Physiol (In press) [DOI] [PMC free article] [PubMed]
- 81.Leffler CW, Balabanova L, Sullivan CD, Wang X, Fedinec AL, Parfenova H. Regulation of CO production in cerebral microvessels of newborn pigs. Am J Physiol. 2003;285:H292–H297. doi: 10.1152/ajpheart.01059.2002. [DOI] [PubMed] [Google Scholar]
- 82.Leffler CW, Fedinec AL, Parfenova H, Jaggar JH. Permissive contributions of NO and prostacyclin in CO-induced cerebrovascular dilation in piglets. Am J Physiol. 2005;289:H432–H438. doi: 10.1152/ajpheart.01195.2004. [DOI] [PubMed] [Google Scholar]
- 83.Leffler CW, Nasjletti A, Johnson RA, Fedinec AL. Contributions of prostacyclin and nitric oxide to carbon monoxide induced cerebrovascular dilation in newborn pigs. Am J Physiol. 2001;280:H1490–H1495. doi: 10.1152/ajpheart.2001.280.4.H1490. [DOI] [PubMed] [Google Scholar]
- 84.Leffler CW, Nasjletti A, Yu C, Johnson RA, Fedinec AL, Walker N. Carbon monoxide and cerebral microvascular tone in newborn pigs. Am J Physiol Heart Circ Physiol. 1999;276:H1641–H1646. doi: 10.1152/ajpheart.1999.276.5.H1641. [DOI] [PubMed] [Google Scholar]
- 85.Li Volti G, Ientile R, Abraham NG, Vanella A, Cannavo G, Mazza F, Curro M, Raciti G, Avola R, Campisi A. Immunocytochemical localization and expression of heme oxygenase-1 in primary astroglial cell cultures during differentiation: effect of glutamate. Biochem Biophys Res Commun. 2004;315:517–524. doi: 10.1016/j.bbrc.2004.01.090. [DOI] [PubMed] [Google Scholar]
- 86.Li Volti G, Sacerdoti D, Sangras B, Vanella A, Mezentsev A, Scapagnini G, Falck JR, Abraham NG. Carbon monoxide signaling in promoting angiogenesis in human microvessel endothelial cells. Antiox&Redox Signal. 2005;7:704–710. doi: 10.1089/ars.2005.7.704. [DOI] [PubMed] [Google Scholar]
- 87.Linden DJ, Narasimhan K, Gurfel D. Protoporphyrins modulate voltage-gated Ca current in AtT-20 pituitary cells. J Neurophysiol. 1993;70(6):2673–7. doi: 10.1152/jn.1993.70.6.2673. [DOI] [PubMed] [Google Scholar]
- 88.Liu N, Wang X, McCoubrey WK, Maines MD. Developmentally regulated expression of two transcripts for heme oxygenase-2 with a first exon specific to rat testis; control by corticosterone of the oxygenase protein expression. Gene. 2000;241:175–183. doi: 10.1016/s0378-1119(99)00439-4. [DOI] [PubMed] [Google Scholar]
- 89.Liu Q, Xu Q, Arcuino G, Kang J, Nedergaard M. Astrocyte-mediated activationm of neuronal kainite receptors. Proc Nat Acad Sci USA. 2004;101:3172–3177. doi: 10.1073/pnas.0306731101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu XM, Chapman GB, Peyton KJ, Schafer AI, Durante W. Carbon monoxide inhibits apoptosis in vascular smooth muscle cells. Cardiovasc Res. 2002;55:396–405. doi: 10.1016/s0008-6363(02)00410-8. [DOI] [PubMed] [Google Scholar]
- 91.Liu Y, Christou H, Morita T, Laughner E, Semenza GL, Kourembanas S. Carbon monoxide and nitric oxide suppress the hypoxic induction of vascular endothelial growth factor gene via the 5′ enhancer. J Biol Chem. 1998;273:15257–62. doi: 10.1074/jbc.273.24.15257. [DOI] [PubMed] [Google Scholar]
- 92.Maines MD, Eke BC, Zhao X. Corticosterone promotes increase heme oxygenase-2 protein and transcript expression in newborn rat. Brain Res. 1996;722:83–94. doi: 10.1016/0006-8993(96)00184-9. [DOI] [PubMed] [Google Scholar]
- 93.Maines MD. Carbon monoxide: an emerging regulator of cGMP in the brain. Mol Cell Neurosciences. 1993;4:389–397. doi: 10.1006/mcne.1993.1049. [DOI] [PubMed] [Google Scholar]
- 94.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 95.Maines MD. The heme oxygenase system and its functions in the brain. Cell Mol Biol. 2000;46:573–585. [PubMed] [Google Scholar]
- 96.Mark JA, Maines MD. Tin-protopoorphyrin-mediated disruption in vivo of heme-oxygenase-2 protein integrity and activity in the rat brain. Pediatr Res. 1992;32:324–329. doi: 10.1203/00006450-199209000-00016. [DOI] [PubMed] [Google Scholar]
- 97.Marks GS, Brien JF, Nakatsu K, McGlaughlin BE. Does carbon monoxide have a biological function? Trends Pharmacol Sci. 1991;12:185–8. doi: 10.1016/0165-6147(91)90544-3. [DOI] [PubMed] [Google Scholar]
- 98.Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, De Galarreta CM, Cuadrado A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem. 2004;279:8919–8929. doi: 10.1074/jbc.M309660200. [DOI] [PubMed] [Google Scholar]
- 99.Maulik N, Engelman DT, Watanabe M, Engelman RM, Das DK. Nitric oxide-a retrograde messenger for carbon monoxide signaling in ischemic heart. Mol Cell Biochem. 1996;157:75–86. doi: 10.1007/BF00227883. [DOI] [PubMed] [Google Scholar]
- 100.May BK, Bhasker CR, Bawden MJ, Cox TC. Molecular regulation of 5-aminolevulinate synthase. Diseases related to heme biosynthesis. Mol Biol Med. 1990;7:405–421. [PubMed] [Google Scholar]
- 101.McCoubrey WK, Huang TJ, Maines MD. Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur J Biochem. 1997;247:725–732. doi: 10.1111/j.1432-1033.1997.00725.x. [DOI] [PubMed] [Google Scholar]
- 102.Meffert MK, Haley JE, Schuman EM, Schulman H, Madison DV. Inhibition of hippocampal heme oxygenase, nitric oxide synthase, and long-term potentiation by metalloporphyrins. Neuron. 1994;13(5):1225–33. doi: 10.1016/0896-6273(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 103.Meldrum BS. Concept of activity-induced cell death in epilepsy: historical and contemporary perspectives. Prog Brain Res. 2002;135:3–11. doi: 10.1016/S0079-6123(02)35003-9. [DOI] [PubMed] [Google Scholar]
- 104.Minetti M, Mallozzi C, Di Stasi AM, Pietraforte D. Bilirubin is an effective antioxidant of peroxynitrite-mediated protein oxidation in human blood plasma. Arch Biochem Biophys. 1998;352:165–174. doi: 10.1006/abbi.1998.0584. [DOI] [PubMed] [Google Scholar]
- 105.Montecot C, Seylaz J, Pinard E. Carbon monoxide regulates cerebral blood flow in epileptic seizures but not in hypercapnia. Neuroreport. 1998;9:2341–2346. doi: 10.1097/00001756-199807130-00035. [DOI] [PubMed] [Google Scholar]
- 106.Morita T, Mitsialis SA, Koike H, Liu Y, Kourembanas S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J Biol Chem. 1997;272:32804–9. doi: 10.1074/jbc.272.52.32804. [DOI] [PubMed] [Google Scholar]
- 107.Morita T, Perrella MA, Lee ME, Kourembanas S. Smooth muscle cell-derived carbon monoxide is a regulator of vascular cGMP. Proc Natl Acad Sci U S A. 1995;92:1475–9. doi: 10.1073/pnas.92.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Morley P, Small DL, Murray CL, Mealing GA, Poulter MO, Durkin JP, Stanimirovic DB. Evidence that functional glutamate receptors are not expressed on rat or human cerebromicrovascular endothelial cells. J Cereb Blood Flow Metab. 1998;18:396–406. doi: 10.1097/00004647-199804000-00008. [DOI] [PubMed] [Google Scholar]
- 109.Motterlini R, Foresti R, Intaglietta M, Winslow RW. NO-mediated activation of heme oxygenase: endogenous cytoprotection against oxidative stress to endothelium. Am J Physiol. 1996;270:H107–H114. doi: 10.1152/ajpheart.1996.270.1.H107. [DOI] [PubMed] [Google Scholar]
- 110.Naik JS, Walker BR. Heme oxygenase-mediated vasodilation involves vascular smooth muscle cell hyperpolarization. Am J Physiol. 2003;285:H220–H228. doi: 10.1152/ajpheart.01131.2002. [DOI] [PubMed] [Google Scholar]
- 111.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 112.Ny L, Andersson KE, Grundemar L. Inhibition by zinc protoporphyrin-IX of receptor-mediated relaxation of the rat aorta in a manner distinct from inhibition of haem oxygenase. Br J Pharmacol. 1995;115:186–90. doi: 10.1111/j.1476-5381.1995.tb16337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276:734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- 114.Paravicini TM, Soby CG. Cerebral vascular effects of reactive oxygen species: recent evidence for a role of NADPH-oxidase. Clin Exp Pharmacol Physiol. 2003;30:855–859. doi: 10.1046/j.1440-1681.2003.03920.x. [DOI] [PubMed] [Google Scholar]
- 115.Parfenova H, Daley ML, Carratu P, Leffler CW. Heme Oxygenase Inhibition Reduces Neuronal Activation Evoked by Bicuculline in Newborn Pigs. Brain Res. 2004;1014:87–96. doi: 10.1016/j.brainres.2004.03.052. [DOI] [PubMed] [Google Scholar]
- 116.Parfenova H, Fedinec A, Leffler CW. Ionotropic glutamate receptors in cerebral microvascular endothelium are functionally linked to heme oxygenase. J Cereb Blood Flow Metab. 2003;23:190–197. doi: 10.1097/01.WCB.000004823561824.C4. [DOI] [PubMed] [Google Scholar]
- 117.Parfenova H, Neff RA, III, Alonso JS, Shlopov BV, Jamal CN, Sarkasova SA, Leffler CW. Cerebrovascular endothelial heme oxygenase: expression, intracellular compartmentalization, and activation by glutamate. Am J Physiol. 2001;281:C1954–C1963. doi: 10.1152/ajpcell.2001.281.6.C1954. [DOI] [PubMed] [Google Scholar]
- 118.Parfenova H, Zuckerman Z, Leffler CW. Inhibitory effect of indomethacin on prostacyclin receptor-mediated cerebral vascular responses. Am J Physiol. 1995;268:H1184–H1890. doi: 10.1152/ajpheart.1995.268.5.H1884. [DOI] [PubMed] [Google Scholar]
- 119.Parfenova H, Carratu P, Tcheranova D, Fedinec A, Pourcyrous M, Leffler CW. Epileptic seizures cause extended cerebral vascular dysfunction that is prevented by HO-1 overexpression. Am J Physiol. 2005;288:H2843–H2850. doi: 10.1152/ajpheart.01274.2004. [DOI] [PubMed] [Google Scholar]
- 120.Pelligrino DA, Baughman VL, Koenig HM. Nitric oxide and the brain. Int Anesthesiol Clin. 1996;34:113–32. doi: 10.1097/00004311-199603440-00009. [DOI] [PubMed] [Google Scholar]
- 121.Philip S, Armstead WM. NMDA dilates pial arteries by KATP and KCa channel activation. Brain Res Bull. 2004;63:127–131. doi: 10.1016/j.brainresbull.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 122.Pourcyrous M, Bada H, Parfenova H, Daley M, Korones S, Leffler CW. Cerebrovasodilatory contribution of endogenous carbon monoxide during seizures in newborn pigs. Pediatr Res. 2002;51:579–585. doi: 10.1203/00006450-200205000-00006. [DOI] [PubMed] [Google Scholar]
- 123.Robinson JS, Fedinec AL, Leffler CW. Role of CO in glutamate receptor-induced dilation of newborn pig pial arterioles. Am J Physiol. 2002;282:H2371–H2376. doi: 10.1152/ajpheart.00911.2001. [DOI] [PubMed] [Google Scholar]
- 124.Rosenblum WI. ATP-sensitive potassium channels in the cerebral circulation. Stroke. 2003;34:1547–1552. doi: 10.1161/01.STR.0000070425.98202.B5. [DOI] [PubMed] [Google Scholar]
- 125.Ryter SW, Choi AMK. Heme oxygenase-1: molecular mechanisms of gene expression in oxygen-related stress. Antiox Redux Signal. 2002;4 :625–632. doi: 10.1089/15230860260220120. [DOI] [PubMed] [Google Scholar]
- 126.Sachse C, Echeverri CJ. Oncology studies using siRNA libraries: the dawn of RNAi-based genomics. Oncogene. 2004;23(51):8384–91. doi: 10.1038/sj.onc.1208072. [DOI] [PubMed] [Google Scholar]
- 127.Sammut IA, Foresti R, Clark JE, Exon DJ, Vesely MJ, Sarathchandra P, Green CJ, Motterlini R. Carbon monoxide is a major contributor to the regulation of vascular tone in aortas expressing high levels of haeme oxygenase-1. Br J Pharmacol. 1998;125:1437–44. doi: 10.1038/sj.bjp.0702212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Scanlon KJ. Anti-genes: siRNA, ribozymes and antisense. Curr Pharm Biotechnol. 2004;5:415–20. doi: 10.2174/1389201043376689. [DOI] [PubMed] [Google Scholar]
- 129.Scapagnini G, D’Agata V, Calabrese V, Pascale A, Colombrita C, Alkon D, Cavallaro S. Gene expression profiles of heme oxygenase isoforms in the rat brain. Brain Res. 2002;954:51–59. doi: 10.1016/s0006-8993(02)03338-3. [DOI] [PubMed] [Google Scholar]
- 130.Schuller DJ, Wilks A, Ortiz de Montellano PR, Poulos TL. Crystal structure of human heme oxygenase-1. Nat Struct Biol. 1999;6:860–867. doi: 10.1038/12319. [DOI] [PubMed] [Google Scholar]
- 131.Serfass L, Burstyn JN. Effect of heme oxygenase inhibitors on soluble guanylyl cyclase activity. Arch Biochem Biophys. 1998;359:8–16. doi: 10.1006/abbi.1998.0887. [DOI] [PubMed] [Google Scholar]
- 132.Shan X, Dunbrack RL, Jr, Christopher SA, Kruger WD. Mutations in the regulatory domain of cystathionine beta synthase can functionally suppress patient-derived mutations in cis. Hum Mol Genet. 2001;10:635–643. doi: 10.1093/hmg/10.6.635. [DOI] [PubMed] [Google Scholar]
- 133.Sharp CD, Hines I, Houghton J, Warren A, Jackson TH, 4th, Jawahar A, Nanda A, Elrod JW, Long A, Chi A, Minagar A, Alexander JS. Glutamate causes a loss in human cerebral endothelial barrier integrity through activation of NMDA receptor. Am J Physiol Heart Circ Physiol. 2003;285:H2592–H2598. doi: 10.1152/ajpheart.00520.2003. [DOI] [PubMed] [Google Scholar]
- 134.Sharp CD, Houghton J, Elrod JW, Warren A, Jackson TH, 4th, Jawahar A, Nanda A, Minagar A, Alexander JS. N-methyl-D-aspartate receptor activation in human cerebral endothelium promotes intracellular oxidant stress. Am J Physiol. 2005;288:H1893–9. doi: 10.1152/ajpheart.01110.2003. [DOI] [PubMed] [Google Scholar]
- 135.Simandle SA, Kerr BA, Lacza Z, Eckman DM, Busija DW, Bari F. Piglet pial arteries respond to N-methyl-d-aspartate in vivo but not in vitro. Microvasc Res. 2005 doi: 10.1016/j.mvr.2005.04.007. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 136.Soares MP, Usheva A, Brouard S, Berberat PO, Gunther L, Tobiash E, Bach FH. Modulation of endothelial cell apoptosis by heme oxygenase-1-derived carbon monoxide. Antiox Redox Signaling. 2002;4:321–328. doi: 10.1089/152308602753666370. [DOI] [PubMed] [Google Scholar]
- 137.Solnyshkova TG, Shakhlamov VA. Ultrastructural and morphometric characteristics of nerve cells and myelinated fibers in the cerebral cortex after chronic exposure to natural gas containing hydrogen sulfide in low concentrations. Bull Exp Biol Med. 2002;134:411–413. doi: 10.1023/a:1021937104829. [DOI] [PubMed] [Google Scholar]
- 138.Song R, Mahidhara RS, Zhou Z, Hoffman RA, Seol DW, Flavell RA, Billiar TR, Otterbein LE, Choi AM. Carbon monoxide inhibits T lymphocyte proliferation via caspase-dependent pathway. J Immunol. 2004;172:1220–1226. doi: 10.4049/jimmunol.172.2.1220. [DOI] [PubMed] [Google Scholar]
- 139.Song R, Zhou Z, Kim PK, Shapiro RA, Liu F, Ferran C, Choi AM, Otterbein LE. Carbon monoxide promotes Fas/CD95-induced apoptosis in Jurkat cells. J Biol Chem. 2004;279:44327–44334. doi: 10.1074/jbc.M406105200. [DOI] [PubMed] [Google Scholar]
- 140.Tang XD, Rong X, Reynolds MF, Garcia ML, Heinemann SM, Hoshi T. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature. 2003;425:531–535. doi: 10.1038/nature02003. [DOI] [PubMed] [Google Scholar]
- 141.Thorup C, Jones CL, Gross SS, Moore LC, Goligorsky MS. Carbon monoxide induces vasodilation and nitric oxide release but suppresses endothelial NOS. Am J Physiol. 1999;277:F882–889. doi: 10.1152/ajprenal.1999.277.6.F882. [DOI] [PubMed] [Google Scholar]
- 142.Tulis DA. Salutary properties of YC-1 in the cardiovascular and hematological systems. Curr Med Chem Cardiovasc Hematol Agents. 2004;2:343–59. doi: 10.2174/1568016043356200. [DOI] [PubMed] [Google Scholar]
- 143.Undem BJ, Ellis JL, Meeker S, Fischer A, Canning BJ. Inhibition by zinc protoporphyrin-IX of vasoactive intestinal peptide-induced relaxations of guinea pig isolated trachea. J Pharmacol Exp Ther. 1996;278:964–70. [PubMed] [Google Scholar]
- 144.Wang R, Wang Z, Wu L. Carbon monoxide-induced vasorelaxation and the underlying mechanisms. Br J Pharmacol. 1997;121:927–34. doi: 10.1038/sj.bjp.0701222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang R. The gasotransmitter role of hydrogen sulfide. Antioxid Redox Signal. 2003;5:493–501. doi: 10.1089/152308603768295249. [DOI] [PubMed] [Google Scholar]
- 146.Wang R. Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 147.Wang R, Wu L. The chemical modification of KCa channels by carbon monoxide in vascular smooth muscle cells. J Biol Chem. 1997;272:8222–8226. doi: 10.1074/jbc.272.13.8222. [DOI] [PubMed] [Google Scholar]
- 148.Weber CM, Eke BC, Maines MD. Corticosterone regulates heme oxygenase-2 and NO synthase transcription and protein expression in rat brain. J Neurochemistry. 1994;63:953–962. doi: 10.1046/j.1471-4159.1994.63030953.x. [DOI] [PubMed] [Google Scholar]
- 149.Wendling WW, Chen D, Daniels FB, Monteforte MR, Fischer MB, Harakal C, Carlsson C. The effects of N-methyl-D-aspartate agonists and antagonists on isolated bovine cerebral arteries. Anesth Analg. 1996;82:264–268. doi: 10.1097/00000539-199602000-00009. [DOI] [PubMed] [Google Scholar]
- 150.Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004;306:2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- 151.Winestone JS, Bonner C, Leffler CW. Carbon monoxide as an attenuator of vasoconstriction in piglet cerebral circulation. Experimental Biology and Medicine. 2003;228:46–50. doi: 10.1177/153537020322800106. [DOI] [PubMed] [Google Scholar]
- 152.Wu L, Cao K, Lu Y, Wang R. Different mechanisms underlying the stimulation of K(Ca) channels by nitric oxide and carbon monoxide. J Clin Invest. 2002;110:691–700. doi: 10.1172/JCI15316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Xi Q, Tcheranova D, Parfenova H, Horowitz B, Leffler CW, Jaggar JH. Carbon monoxide activates KCa channels in newborn arteriole smooth muscle cells by increasing apparent Ca2+ sensitivity of alpha-subunits. Am J Physiol. 2004;286:H610–H618. doi: 10.1152/ajpheart.00782.2003. [DOI] [PubMed] [Google Scholar]
- 154.Yang G, Cao K, Wang R. Cystathionine γ-lyase overexpression inhibits cell proliferation via a H2S-dependent modulation of ERK1/2 phosphorylation and p21Cip/WAK-1. J Biol Chem. 2004;279:49199–49205. doi: 10.1074/jbc.M408997200. [DOI] [PubMed] [Google Scholar]
- 155.Yang G, Sun X, Wang R. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of MAP kinases and caspase-3. FASEB J. 2004;18:1782–1784. doi: 10.1096/fj.04-2279fje. [DOI] [PubMed] [Google Scholar]
- 156.Yang G, Zhang Y, Ross ME, Iadecola C. Attenuation of activity –induced increases in cerebellar blood flow in mice lacking neuronal nitric oxide synthase. Am J Physiol. 2003;285:H298–H304. doi: 10.1152/ajpheart.00043.2003. [DOI] [PubMed] [Google Scholar]
- 157.Yuan ZR, Liu B, Zhang Y, Yuan L, Muteliefu G, Lu J. Upregulated expression of neuronal nitric oxide synthase by insulin in both neurons and astrocytes. Brain Res. 2004;1008:1–10. doi: 10.1016/j.brainres.2004.01.076. [DOI] [PubMed] [Google Scholar]
- 158.Zakhary R, Gaine SP, Dinerman JL, Ruat M, Flavahan NA, Snyder SH. Heme oxygenase 2: endothelial and neuronal localization and role in endothelium-dependent relaxation. Proc Natl Acad Sci U S A. 1996;93:795–798. doi: 10.1073/pnas.93.2.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang F, Kaide J, Wei Y, Jiang H, Yu C, Balazy M, Abraham NG, Wang W, Nasjletti A. Carbon monoxide produced by isolated arterioles attenuates pressure-induced vasoconstriction. Am J Physiol. 2001;281:H350–H358. doi: 10.1152/ajpheart.2001.281.1.H350. [DOI] [PubMed] [Google Scholar]
- 160.Zhang X, Shan P, Otterbein LE, Alam J, Flavell RA, Davis RJ, Choi AM, Lee PJ. Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activated protein kinase pathway and involves caspase 3. J Biol Chem. 2003;278:1248–1258. doi: 10.1074/jbc.M208419200. [DOI] [PubMed] [Google Scholar]
- 161.Zhao W, Wang R. H2S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol. 2002;283:H474–H480. doi: 10.1152/ajpheart.00013.2002. [DOI] [PubMed] [Google Scholar]
- 162.Zhao W, Ndisang JF, Wang R. Modulation of endogenous production of H2S in rat tissues. Can J Physiol Pharmacol. 2003;81:848–853. doi: 10.1139/y03-077. [DOI] [PubMed] [Google Scholar]
- 163.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]