

It is well-known that amino acids exist as zwitterions in aqueous solution within a range of pH. Whether zwitterions exist in the absence of solvent has been extensively discussed in the literature.1–6 In the gas phase, experimental evidence that glycine is not a zwitterion has been obtained by millimeter wave spectroscopy1 and by measurements of substituent effects on gas-phase basicity.2 In addition, the potential energy surface of isolated glycine has been thoroughly investigated by ab initio methods.3,4 At the highest levels of theory, the zwitterion is ~16–20 kcal/mol less stable than the neutral with no barrier for proton transfer between the protonated amino and the deprotonated carboxyl groups.4 Calculations indicate that two water molecules complexed with glycine can stabilize the zwitterion; if the two water molecules are hydrogen bonded only to glycine, a barrier to proton transfer occurs although the zwitterion is still higher in energy than the neutral.4d Results of Rizzo et al.5 indicate that electronically excited tryptophan formed in a supersonic jet is a zwitterion. Recently, evidence for salt-bridge interactions in gas-phase peptide ions has been reported.7 For example, the most stable form of protonated bradykinin (Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg) is one in which both terminal arginine residues are protonated and interact via the deprotonated carboxyl of the C-terminus.7a Salt-bridge interactions can be significantly stabilized by intramolecular hydrogen bonding to polar groups, such as carbonyl oxygens, which provide the near equivalent of a solvent shell around the charges.8 Here, we present experimental evidence that protonated dimers of arginine are bound by a salt-bridge in the gas phase. Further, our calculations indicate that the most stable form of arginine itself is a zwitterion. We believe that this is the first evidence for a stable ground state zwitterion form of an amino acid in the absence of solvent.
The dissociation kinetics of proton-bound dimers of several amino acids were investigated with blackbody infrared radiative dissociation (BIRD) in a 2.7-T Fourier-transform mass spectrometer. These experiments are described elsewhere.9 Proton-bound dimers were formed by electrospray ionization and dissociated at cell temperatures ranging from 37 to 149 °C. Unimolecular rate constants are obtained from the slope of ln-{[D+]/([D+] + [F+])} vs time where [D+] and [F+] refer to the dimer ion abundance and sum of fragment ion abundances, respectively. The resulting Arrhenius plot for dissociation of these dimers is shown in Figure 1. The proton-bound dimer of Gly, (Glyd), dissociates more slowly than both Alad and the methyl ester (ME) of Gly dimer itself (GlyMEd). The increased dissociation rates of the latter two ions are primarily due to their increased molecular size. This results in both an increased rate of blackbody photon absorption and an increased population above the threshold dissociation energy. A small difference in binding energy (<0.05 eV) or relative transition state entropy may also partially contribute to the different dissociation rates. Alad and the GlyMEd are isomers. Their similar dissociation kinetics indicate that these ions have comparable binding energies. In contrast, the dissociation kinetics of Argd and ArgMEd differ much more dramatically than those of Glyd and GlyMEd (Figure 1).
Figure 1.
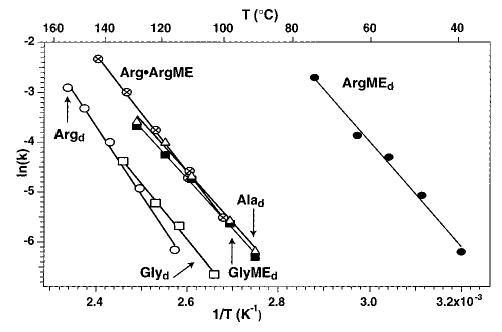
Arrhenius plot for the dissociation of the proton-bound dimer of (○) arginine, (□) glycine, (▪) glycine methyl ester, (▵) alanine, (⊕) arginine·arginine methyl ester, and (•) arginine methyl ester.
Threshold dissociation energies (Eo) can be extracted from these kinetic data by using master equation modeling, the method for which has been described in detail elsewhere.9 The results of this modeling are given in Table 1. Assuming a negligible reverse activation barrier for these dissociation processes, Eo can be equated to the binding energy. The dimers of Gly, Ala, and GlyME all have similar binding energies (1.10–1.15 ± 0.05 eV). The similar binding energy for Glyd and its methyl ester indicates that the hydrogen of the carboxylic acid functional group does not play a role in the binding. This is consistent with the lowest energy ab initio structure in which the protonated N-terminus of one residue interacts with both the N-terminus nitrogen and the carbonyl oxygen of the other.9
Table 1.
Measured Arrhenius Activation Parameters of Proton-Bound Amino Acid Dimers and Methyl Esters (ME) Obtained by BIRD
| dimer | measured Ea (eV) | measured log A | master eq Eo (eV) |
|---|---|---|---|
| Arg | 1.19 ± 0.05 | 12.8 ± 0.6 | 1.33 ± 0.08 |
| ArgME·Arg | 1.01 ± 0.03 | 11.2 ± 0.3 | 1.20 ± 0.05 |
| ArgME | 0.91 ± 0.05 | 12.0 ± 0.8 | 1.05 ± 0.05 |
| Gly | 0.96 ± 0.04 | 10.0 ± 0.5 | 1.15 ± 0.05 |
| GlyME | 0.86 ± 0.03 | 9.3 ± 0.4 | 1.10 ± 0.05 |
| Ala | 0.88 ± 0.04 | 9.5 ± 0.4 | 1.10 ± 0.05 |
The binding energy of ArgMEd is slightly lower (1.05 ± 0.05 eV). The side-chain guanidine group is highly basic and is the expected site of protonation. Charge delocalization should occur to a more significant extent on the guanidine group than, for example, on the N-terminus. This delocalization should reduce the ion–dipole interaction, consistent with the decreased binding energy observed. The binding energies for Argd and Arg·ArgME are significantly higher (1.33 ± 0.08 and 1.20 ± 0.05 eV, respectively). We believe that the higher binding energies indicate that these dimers are bound by salt-bridges in which both guanidine groups are protonated and the carboxyl group of an Arg is deprotonated, i.e., one of the arginines in the dimer is a zwitterion (Figure 2, part a). The zwitterion form is not possible in ArgMEd. The significantly larger dipole moment of a zwitterion10 should increase the binding energy of these complexes.
Figure 2.
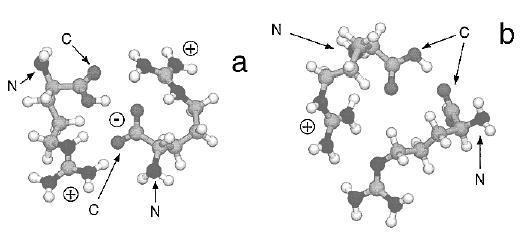
Minimum energy structures calculated at the AM1 semiempirical level of protonated arginine dimers modeled as (a) a salt-bridge in which both guanidine side chains are protonated and the carboxyl group of one is deprotonated, i.e., one arginine is a zwitterion, and (b) a simple proton-bound ion–molecule complex. The C- and N-terminus of each monomer is labeled.
The ~0.15 eV higher binding energy of Arg·ArgME vs ArgMEd is consistent with recent results for protonated complexes of betaine which show an increased binding energy of 0.2 eV when betaine is in its zwitterionic form.11 We attribute the even higher binding energy of Argd to extra stabilization of the carboxylic acid group of protonated Arg which solvates the zwitterion (Figure 2a). This is consistent with results from both AM1 and PM3 semiempirical modeling which indicate only one stable structure for Argd but several different structures of comparable energy for Arg·ArgME (both modeled as salt-bridges). The existence of several structures for Arg·ArgME but not for Argd indicates that the amino and ester groups are less effective in stabilizing the zwitterion.
A zwitterion can be stabilized by a nearby charge, as is the case for the dimer. To determine if arginine by itself is a zwitterion, both the zwitterion and the neutral form were modeled at modest levels of theory.12 At the BLYP/6-31G* level, the zwitterion is a stationary point and is 1.0 kcal/mol more stable than the neutral with the zero-point correction (0.4 kcal/mol without) (Figure 3). A different neutral conformer observed in the molecular modeling appears to be consistent with an intermediate structure for proton transfer (Figure 3). This conformer is a minimum on the potential energy surface and is 5 kcal/mol less stable than the zwitterion at the BLYP/6-31G* level. At the MP2/6-31G* level, the zwitterion is 0.3 kcal/mol more stable than the neutral form.13 We are not currently able to extend the calculations using a higher basis set for this relatively large molecule and have not achieved convergence. However, the excellent agreement between these two different computational methods provides significant support for the comparable stability of the zwitterion and neutral forms.13 A key problem with these calculations is the lowest energy neutral geometry may not have been found. However, the similarity in energy of these structures suggests that the zwitterion is likely to be stable in the gas phase.
Figure 3.
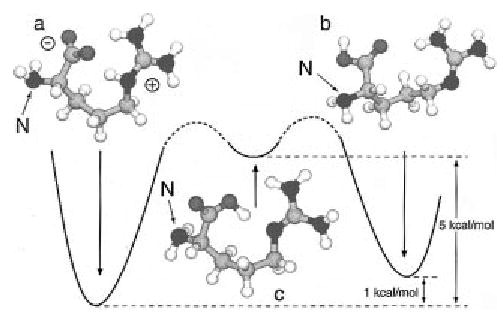
Optimized structures of arginine calculated at the BLYP/6-31G* ab initio level as (a) a zwitterion, (b) the lowest energy neutral conformation found, and (c) a higher energy stable conformer that is consistent with an intermediate to proton transfer from the carboxyl to the guanidine groups. The N-terminus of each structure is labeled.
In conclusion, these experimental results suggest that protonated dimers of arginine are bound by a salt-bridge in the gas phase. This type of interaction appears favored even without intramolecular self-solvation to stabilize the charge separation. For arginine itself, the high basicity of the guanidine side-chain functionality should stabilize the zwitterion vs neutral forms relative to these forms for glycine. Guanidine ((NH2)2C=NH) is ~21 kcal/mol more basic than glycine.14 This difference is larger than the calculated difference in stability between the glycine zwitterion and neutral forms. Thus, from basicity arguments alone, the guanidine side chain should be a competitive site of protonation relative to the carboxylate group making the zwitterion a potentially favored structure for arginine in the gas phase. This conclusion is supported by our ab initio calculations. These results suggest that uncharged arginine is an extremely interesting molecule in the gas phase and that both further measurement, e.g., absorption spectroscopy, and calculations at a higher level of theory are warranted.
Acknowledgments
The authors thank Profs. Martin Head-Gordon and Richard J. Saykally for helpful comments and the National Science Foundation (CHE-9258178) and the National Institutes of Health (IR29GM50336-01A2) for generous financial support.
References
- 1.Suenram RD, Lovas FJ. J Mol Spectrosc. 1980;72:372–382. [Google Scholar]
- 2.Locke MJ, McIver RT., Jr J Am Chem Soc. 1983;105:4226–4232. [Google Scholar]
- 3.a Jensen JH, Gordon MS. J Am Chem Soc. 1991;113:7917–7924. [Google Scholar]; b Hu CH, Shen M, Schaefer HF., III J Am Chem Soc. 1993;115:2923–2928. [Google Scholar]; c Csaszar AG. J Am Chem Soc. 1992;114:9568–9575. [Google Scholar]
- 4.a Gordon MS, Jensen JH. Acc Chem Res. 1996;29:536–543. [Google Scholar]; b Ding YB, Krogh-Jespersen K. Chem Phys Lett. 1992;199:261–266. [Google Scholar]; c Yu D, Armstrong DA, Rauk A. Can J Chem. 1992;70:1762–1772. [Google Scholar]; d Jensen JH, Gordon MS. J Am Chem Soc. 1995;117:8159–8170. [Google Scholar]
- 5.Rizzo TR, Park YD, Levy DH. J Chem Phys. 1986;85:6945–6951. [Google Scholar]
- 6.Grenie Y, Lassegues J, Garrigou-Lagrange C. J Chem Phys. 1970;53:2980–2982. [Google Scholar]
- 7.a Schnier PD, Price WD, Jockusch RA, Williams ER. J Am Chem Soc. 1996;118:7178–7189. doi: 10.1021/ja9609157. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Price WD, Schnier PD, Jockusch RA, Strittmatter EF, Williams ER. J Am Chem Soc. 1996;118:10640–10644. doi: 10.1021/ja961812r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Grese RP, Cerny RL, Gross ML. J Am Chem Soc. 1989;111:2835–2842. [Google Scholar]; d Cambell S, Rodgers MT, Marzluff EM, Beauchamp JL. J Am Chem Soc. 1995;117:12840–12854. [Google Scholar]; e Cox KA, Gaskell SJ, Morris M, Whiting A. J Am Soc Mass Spectrom. 1996;7:522–531. doi: 10.1016/1044-0305(96)00019-0. [DOI] [PubMed] [Google Scholar]
- 8.a Gross DS, Williams ER. J Am Chem Soc. 1995;117:883–890. [Google Scholar]; b Wyttenbach T, von Helden G, Bowers MT. J Am Chem Soc. 1996;118:8355–8364. [Google Scholar]
- 9.Price WD, Schnier PD, Williams ER. J Phys Chem B. 1997;101:664–673. doi: 10.1021/jp9628702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The dipole moment of the arginine zwitterion calculated at the RHF/6-31G* level is 10.3 D.
- 11.Price, W. D.; Jockusch, R. A.; Williams, E. R. J. Am. Chem. Soc. Submitted for publication.
- 12.A total of 120 conformers of each structure was generated by molecular dynamics and simulated annealing. One structure was obtained for the zwitterion; many different conformers were obtained for the neutral. Upon optimization at both the semiempirical AM1 and PM3 levels, the six lowest energy conformers of the neutral reduced to four conformers of similar energy. Three of these conformers were eliminated on the basis of RHF/6-31G* calculations. Full geometry optimizations up to the BLYP/6-31G* (GAUSSIAN 92) and MP2/6-31G* levels were performed on the zwitterion and the remaining neutral conformer.
- 13.Head-Gordon, M., Personal communication. This value does not include a zero point energy correction.
- 14.Gas-phase basicity of glycine and arginine from: Lias SG, Liebman JF, Levin RD.J Phys Chem Ref Data 198413695–808.Amekraz B, Tortajada J, Morizur JP, Gonzalez AI, Mo O, Yanez M, Leito I, Maria PC, Gal JF.New J Chem 199620 1011–1021.respectively. [Google Scholar]