

Abstract
Background
Prefrontal D1 systems have been implicated in the regulation of working memory and in the pathophysiology of schizophrenia. D1 hypofunction might contribute to reduced sensorimotor gating in schizophrenia patients since D1 activity in the medial prefrontal cortex (MPFC) regulates prepulse inhibition of startle (PPI) in animal models. We studied the neurochemical basis for the D1 regulation of PPI in rats.
Methods
PPI to weak (1–5 dB over background) prepulses was measured after systemic or intra-MPFC administration of the D1 antagonist, SCH 23390, in rats pretreated systemically with the D2 antagonist, haloperidol (vehicle or 0.1 mg/kg).
Results
After vehicle pretreatment, systemic and intra-MPFC SCH 23390 disrupted PPI produced by weak prepulses. This effect was not significantly opposed by pretreatment with haloperidol (0.1 mg/kg). In contrast, the PPI-disruptive effects of the DA agonist amphetamine were significantly opposed by this dose of haloperidol.
Conclusions
D1 blockade reduces PPI, but this effect does not appear to be mediated entirely via increased dopamine transmission at D2 receptors.
Keywords: Dopamine, Medial prefrontal cortex, Nucleus accumbens, Prepulse inhibition, Schizophrenia, Sensorimotor gating, Startle
1. Introduction
It has been proposed that dopamine (DA) dysfunction in schizophrenia includes both hyperfunction of subcortical D2 and hypofunction of cortical D1 systems, the latter being related to working memory deficits and perhaps positive as well as negative symptoms of this disorder (Goldman-Rakic, 1998; Lewis et al., 2004; Seamans and Yang, 2004). Specifically, reduced D1 ‘‘tone’’ in the prefrontal cortex has been linked theoretically to ‘‘premature termination of information in working memory prior to the completion of a thought or action’’ that would leave thoughts ‘‘‘contaminated’ by ‘weak’ stimuli that are normally ignored’’ and thereby ‘‘result in distractibility, tangential or intrusive thought patterns’’ (Seamans and Yang, 2004; p. 43). One operational model for the loss of information-protective preattentional mechanisms is prepulse inhibition of the startle reflex (PPI).
PPI is the normal inhibition of a startle response to an intense, abrupt stimulus when it is preceded by a weak ‘‘prepulse’’ (Graham, 1975). PPI is impaired in schizophrenia patients (Braff et al., 1978, 1999, 2005; cf. Braff et al., 2001) and in patients from a number of other disorders characterized by intrusive sensory, cognitive or motor information (e.g. Tourette syndrome (Castellanos et al., 1996; Swerdlow et al., 2001a), obsessive compulsive disorder (Swerdlow et al., 1993; Hoenig et al., 2005), and Huntington’s disease (Swerdlow et al., 1995; Valls-Sole et al., 2004)). PPI can also be studied in laboratory animals to understand its neural and genetic substrates (Swerdlow et al., 2004a). In rats, PPI is disrupted both by manipulations that lead to subcortical D2 overactivity (Swerdlow et al., 1986; cf. Swerdlow et al., 2000) and by those that lead to cortical D1 hypoactivity (Ellenbroek et al., 1996; Zavitsanou et al., 1999; Bubser and Koch, 1994; Koch and Bubser, 1994). While there is a relatively well-developed understanding of the neural basis for reduced PPI after subcortical D2 activation (cf. Koch and Schnitzler, 1997; Swerdlow et al., 2001b), much less is understood about the neural basis for reduced PPI after blockade of D1 receptors or DA depletion in the medial prefrontal cortex (MPFC). A model has been proposed that suggests that reduced MPFC DA tone results in subcortical DA hyperactivity, that might provide one explanation for PPI deficits after MPFC D1 blockade (Koch and Bubser, 1994), consistent with related models for the pathophysiology of schizophrenia (cf. Abi-Dargham, 2004).
We have previously reported that the effects of DAergic manipulations on PPI are highly dependent on two characteristics of discrete prepulses—intensity and interval (time preceding pulse onset). Specifically, DA agonists that disrupt PPI elicited by intense prepulses (e.g. 15 dB over background) or relatively long prepulse intervals (e.g. 100 ms) can actually potentiate PPI elicited by weaker (1–5 dB) prepulses or shorter (e.g. 10–30 ms) prepulse intervals (Swerdlow et al., 2001c, Swerdlow et al., 2004a). In one recent report, we demonstrated that a low systemic dose of the D1 antagonist SCH 23390 (0.05 mg/kg) significantly potentiated PPI elicited with intense prepulses at short (10–30 ms) prepulse intervals (Swerdlow et al., 2004a); others have previously reported PPI-potentiating effects of D1 blockade (Schwarzkopf et al., 1993). In the present study, we assessed PPI elicited by weak (1–5 dB) prepulses after systemic and intra-MPFC administration of the D1 antagonist, SCH 23390, and examined the sensitivity of SCH 23390-induced PPI deficits to pretreatment with the D2 blocker, haloperidol.
2. Methods and materials
2.1. Experimental animals
Adult male Sprague–Dawley rats (225–250 g; Harlan, San Diego) were housed in groups of 2–3 and maintained on a reversed 12:12 h light/dark schedule with ad lib food and water. Testing occurred between 0900 and 1700 hours, during the dark phase. Rats were handled individually within 2 days of arrival. Surgery occurred between 7 and 10 days after arrival. All efforts were made to minimize animal suffering and reduce the number of animals used. All experiments conform to the National Institutes of Health Guide for the care and use of laboratory animals (NIH Publications No. 85–23) and were approved by the Animal Subjects Committee at the University of California, San Diego (protocol #S01221).
2.2. Surgical preparations
Rats were given 0.1 ml atropine sulfate (Vedco, 0.054 mg/ml SC) 15–30 min before they were fully anesthetized with sodium pentobarbital (Abbott, 60.0 mg/kg IP) and placed in a Kopf stereotaxic instrument in a flat skull position (toothbar 3.3 mm below the interaural line). Bilateral 23 gauge cannulae (9 mm) were aimed 1 mm above the MPFC at coordinates: AP 2.2 (Bregma), L ± 0.8, DV − 4.0, based on previous reports (Ellenbroek et al., 1996).
2.3. Behavioral testing
Each of the 4 startle chambers (SR-LAB, San Diego Instruments, San Diego, CA) was housed in a sound-attenuated room with a 60 dB(A) ambient noise level and consisted of a Plexiglas cylinder 8.2 cm in diameter resting on a 12.5 × 25.5 cm Plexiglas frame within a ventilated enclosure. Noise bursts were presented via a speaker mounted 24 cm above the cylinder. A piezoelectric accelerometer mounted below the Plexiglas frame detected and transduced motion within the cylinder. The delivery of stimuli was controlled by the SR-LAB microcomputer and interface assembly, which also digitized (0–4095), rectified and recorded stabilimeter readings, with 100 1-ms readings collected beginning at stimulus onset. Startle amplitude was defined as the average of the 100 readings. Background noise and all acoustic stimuli were delivered through one Radio Shack Supertweeter (frequency response predominantly between 5 and 16 kHz) in each chamber.
To reduce inter-group variability, dose groups were assigned based on matched PPI from a brief pretest session. For studies of intra-MPFC administration, this matching session took place 1 week post-surgery. In this session, rats were exposed to 5 min of 70 dB background noise followed by 17 ‘‘P-ALONE’’ trials of 40 ms 120 dB noise bursts and 3 ‘‘PREPULSE’’ trials consisting of a 20 ms 82 dB (12 dB above background) prepulse followed 100 ms later by a 120 dB pulse (onset to onset).
Testing began approximately 3 days later. SCH 23390 (Sigma Chemical Co., St. Louis, MO) was dissolved in 0.9% saline. For systemic administration (n =34), SCH 23390 (0 or 0.1 mg/kg) was administered SC in saline vehicle (1 ml/kg), 10 min after systemic pretreatment with the D2 antagonist haloperidol (vehicle or 0.1 mg/kg, in saline). For intra-MPFC administration of SCH 23390 (n =49), 10 min after systemic administration of haloperidol (vehicle or 0.1 mg/kg SC), stylets were removed from the cannulae and replaced by a 30-gauge 10 mm needle. Rats received either active dose infusions (3 μg SCH 23390 in 0.5 μl bilaterally) or vehicle infusions over 84 s using a Hamilton microsyringe connected to the needle via polyethylene tubing. Needles remained in place for 30 s after the injection and then were replaced with stylets. Rats were then placed immediately into the startle chambers. One week later, rats were retested in an identical fashion, except that the dose SCH 23390 was reversed, with half of the rats receiving each dose on each week. Data from a subgroup of these vehicle-pretreated rats were reported previously to justify coordinates for MPFC lesions in a separate study (Shoemaker et al., in press). Lastly, to assess the impact of this dose of haloperidol on a DA-mediated disruption of PPI, a final group of rats (n =19) was tested after pretreatment with haloperidol (vehicle or 0.1 mg/kg) followed 10 min later by treatment with d-amphetamine sulfate (AMPH) (saline vehicle vs. 4.5 mg/kg SC). This dose of amphetamine is known to potently disrupt PPI (Mansbach et al., 1988).
For testing, immediately after intra-MPFC SCH 23390 and 15 min after SC administration of SCH 23390 or 10 min after SC administration of AMPH, rats were placed in the startle chambers for a 5-min acclimation period with a 70 dB(A) background noise. After this period, rats were exposed to six trial types: (1) 40 ms– 120 dB noise burst (P-ALONE); (2) P-ALONE preceded 100 ms by 20 ms noise burst that was either 1, 2, 3, 4 or 5 dB(A) above background (PP1dB, PP2dB, PP3dB, PP4dB, and PP5dB; (3) NOSTIM trials (stabilimeter recordings obtained when no stimulus was presented). Trials were presented in pseudorandom order, with 12 repetitions of each trial with a NOSTIM trial between each pulse or prepulse trial. The session began and ended with 3 P-ALONE trials to permit the assessment of SCH 23390 effects on startle habituation. The session had a total of 155 trials (78 active trials and 77 NOSTIM trials). NOSTIM trials were used to assess gross motor activity during the test session but were not included in the calculation of intertrial intervals, which were variable and averaged 15 s. One rat from the intra-MPFC infusion study and two rats from the haloperidol × AMPH study were excluded from analysis due to startle magnitudes<5 units on P-ALONE trials.
2.4. Anatomy/histology
After behavioral testing, all MPFC-infused animals were deeply anesthethized with pentobarbital and transcardially perfused with a buffered 10% formalin solution. Brains were removed and kept in 10% formalin solution plus 30% sucrose for 3 days at 4 °C. Frozen sections (40 μm) were cut on a microtome and mounted on gelatin-coated glass slides and Nissl stained. To verify cannulae placement, stained sections were digitally scanned, sized and oriented in Adobe Photoshop (v. 7.0), and then superimposed on corresponding digitized drawings of a rat stereotaxic atlas (Paxinos and Watson, 1998) in Adobe Illustrator (v. 11.0). Injector placements were drawn freehand in Illustrator from these composite section/atlas images. Drawings were completed blind to the behavioral data.
2.5. Statistics
Prepulse inhibition was defined as 100 − [(startle amplitude on prepulse trials /startle amplitude on P-ALONE trials) × 100] and was analyzed by mixed-design analyses of variance (ANOVAs), with specific comparisons noted for each experiment. Separate analyses were performed using raw startle magnitude on P-ALONE and prepulse trials to determine whether changes in %PPI reflected a diminished ability of prepulses to inhibit startle. Alpha was 0.05 for all statistical analyses. For relevant significant interaction effects, all values of p are based on a Greenhouse–Geisser correction.
3. Results
3.1. Experiment 1. Systemic SCH 23390
Systemic administration of SCH 23390 disrupted PPI caused by 3–5 dB prepulses and this effect was not significantly opposed by haloperidol (Fig. 1). ANOVA of %PPI revealed no significant effect of haloperidol (F <1), SCH 23390 (F < 1), trial block (F < 1) or any 2- or 3-way interactions (all ns). There was a significant effect of prepulse intensity (F =59.47, df 4,120, p < 0.0001), a trend-level interaction of haloperidol × intensity (F = 2.27, df 4, 120, p <0.07), and a significant interaction of SCH 23390 × intensity (F =7.22, df 4,120, p <0.0006). The 3-way interaction of haloperidol × SCH 23390 × intensity was not significant (F = 1.54, df 4, 120, ns). The interaction of SCH 23390 × intensity reflected an SCH 23390-induced increase in PPI at the weakest prepulse intensity (F =4.35, df 1,30, p <0.05) and an SCH 23390-induced decrease in PPI for prepulses of 3–5 dB over background (main effect of SCH 23390 dose for PPI elicited by 3–5 dB prepulses: F =5.42, df 1,30, p <0.03; SCH 23390 × intensity interaction: ns; haloperidol × SCH 23390 × intensity interaction: ns).
Fig. 1.
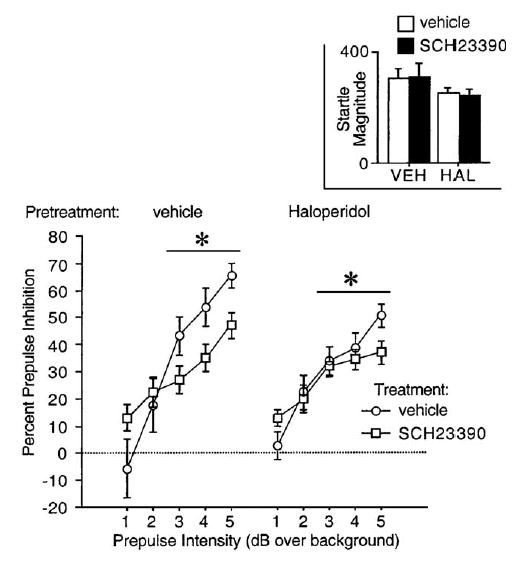
Percent prepulse inhibition (SEM) in rats after pretreatment with haloperidol (vehicle vs. 0.1 mg/kg SC) and treatment with SCH 23390 (vehicle vs. 0.1 mg/kg SC). (*p <0.03, main effect of SCH 23390 dose for PPI elicited by 3–5 dB prepulses after significant interaction of SCH 23390 × intensity).
Analysis of P-ALONE startle magnitude during PPI testing revealed no significant main effects of haloperidol (F =1.68, df 1,30, ns) or SCH 23390 (F <1), or haloperidol × SCH 23390 interaction (F <1) (Fig. 1, inset). ANOVA of NOSTIM activity revealed no significant effect of haloperidol (F < 1), a significant SCH 23390-induced reduction in NOSTIM activity (F =4.80, df 1,30, p <0.04), and no haloperidol × SCH 23390 interaction (F <1) (Table 1).
Table 1.
Signal amplitudes on NOSTIM trials (mean startle units (SEM))
| Treatment | Pretreatment
|
||
|---|---|---|---|
| Vehicle | Haloperidol | ||
| Experiment 1 | |||
| Vehicle | 0.11 (0.04) | 0.06 (0.03) | |
| SCH 23390 | 0.01 (0.01) | 0.00 (0.00) | |
| Experiment 2 | |||
| Vehicle | 0.123 (0.04) | 0.05 (0.03) | |
| SCH 23390 | 0.08 (0.02) | 0.02 (0.01) | |
| Experiment 3 | |||
| Vehicle | 0.15 (0.09) | 0.00 (0.00) | |
| AMPH | 0.11 (0.07 | 0.00 (0.00) | |
3.2. Experiment 2. Intra-MPFC SCH 23390
Histological assessment revealed SCH 23390 infusion sites within cingulate and prelimbic cortices (Fig. 2A). Intra-MPFC administration of SCH 23390 disrupted PPI and this effect was not significantly opposed by systemic administration of haloperidol (Fig. 2B). ANOVA of %PPI revealed no significant effect of haloperidol (F <1), a significant effect of SCH 23390 (F =19.33, df 1,41, p <0.0001) and trial block (F =10.60, df 1,41, p <0.003), but no significant interactions of haloperidol × SCH 23390 (F =1.51, df 1,41, ns) or block × haloperidol × SCH 23390 (F <1). There was a significant effect of prepulse intensity (F =40.31, df 4,164, p <0.0001) and a trend–level interaction of SCH 23390 × intensity (F =2.31, df 4,164, p < 0.065), but no significant interactions of haloperidol × intensity (F <1) or intensity × haloperidol × SCH 23390 (F =1.00, df 4, 164, ns).
Fig. 2.
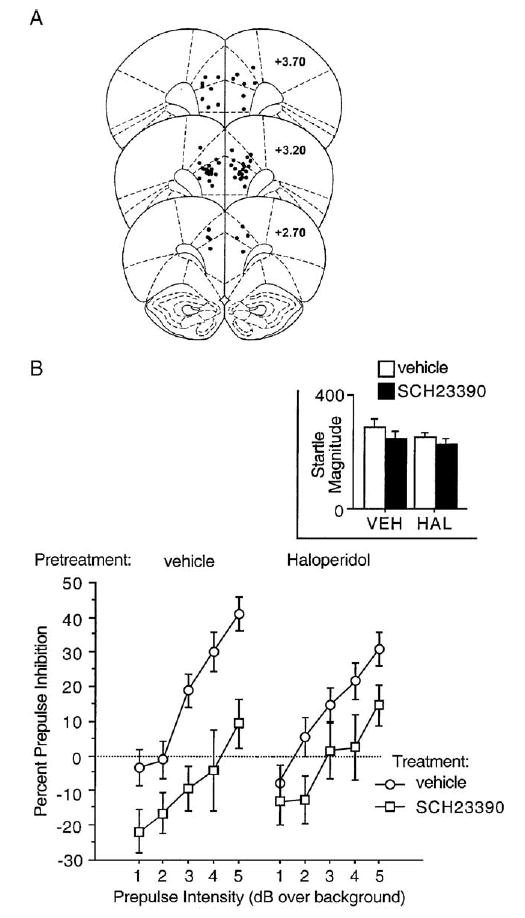
A. SCH 23390 infusion sites within cingulate and prelimbic cortices. B. Percent prepulse inhibition (SEM) in rats after pretreatment with haloperidol (vehicle vs. 0.1 mg/kg SC) and treatment with SCH 23390 (vehicle vs. 3.0 μg/side). Inset shows startle magnitude in response to pulse stimuli.
Analysis of P-ALONE startle magnitude during PPI testing revealed no significant main effects of haloperidol (F <1) or SCH 23390 (F =2.67, df 1,41, ns) or haloperidol × SCH 23390 interaction (F < 1) (Fig. 2B, inset). ANOVA of NOSTIM activity revealed a significant reduction of NOSTIM activity by haloperidol (F =6.11, df 1,41, p <0.02) but not SCH 23390 (F =1.37, df 1,41, ns) and no haloperidol × SCH 23390 interaction (F <1) (Table 1).
3.3. Experiment 3. Haloperidol vs. amphetamine
As expected, AMPH disrupted PPI and this effect was prevented by haloperidol pretreatment. ANOVA revealed no significant main effects of AMPH (F =1.73, df 1,15, ns) or haloperidol (F =2.34, df 1,15, ns), but a significant interaction of AMPH × haloperidol (F =5.09, df 1,15, p <0.04), and a significant interaction of intensity × AMPH × haloperidol (F =5.11, df 4,60, p <0.004). This 3-way interaction reflected the more robust disruption of PPI at higher prepulse intensities and hence the greater range of suppression of this effect by haloperidol at these intensities (Fig. 3).
Fig. 3.
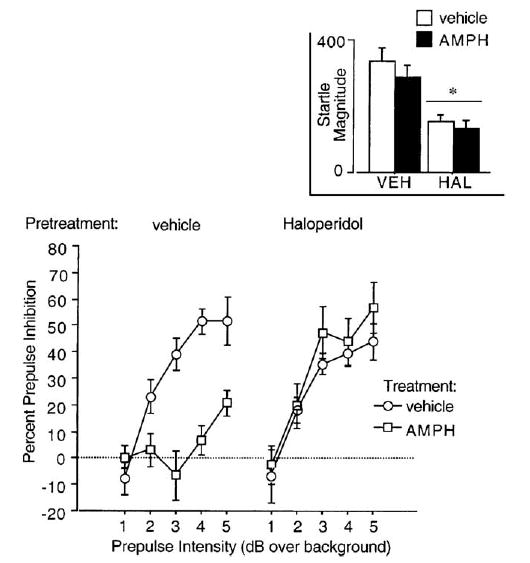
Percent prepulse inhibition (SEM) in rats after pretreatment with haloperidol (vehicle vs. 0.1 mg/kg SC) and treatment with AMPH (vehicle vs. 4.5 mg/kg SC). Inset shows startle magnitude in response to pulse stimuli (*significant main effect of haloperidol on startle magnitude).
Analysis of P-ALONE startle magnitude during PPI testing revealed no significant main effects of AMPH (F <1), a significant reduction of startle by haloperidol (F =14.39, df 1, 15, p < 0.002), but no significant AMPH × haloperidol interaction (F <1) (Fig. 3, inset). ANOVA of NOSTIM activity revealed a significant reduction of NOSTIM activity by haloperidol (F =4.73, df 1,15, p <0.05), but no significant effect of AMPH (F <1) and no haloperidol × AMPH interaction (F <1) (Table 1).
4. Discussion
These findings provide further evidence for the regulation of PPI by D1 receptors. PPI was disrupted by either systemic or intra-MPFC infusion of the D1 antagonist, SCH 23390. Previous reports have demonstrated reduced PPI after D1 blockade in the MPFC (Ellenbroek et al., 1996; Zavitsanou et al., 1999), although Bast et al. (2002) failed to observe reduced PPI after MPFC infusion of the mixed D1/D2 antagonist, cis-flupenthixol dihydrochloride. In this latter report, which involved Wistar rats, data suggested a reduction in PPI by MPFC D1 blockade at the weakest prepulse intensity (4 dB over background; p. 671, Fig. 2), which was the only condition shared by the present studies. In contrast, increased PPI was observed in Long Evans rats after infusion of SCH 23390 into the basolateral amygdala (BLA) (Stevenson and Gratton, 2004); we have previously demonstrated substantial differences in the dopaminergic regulation of PPI between Sprague–Dawley (SD) and Long Evans rats, which may be linked to heritable differences in signal transduction pathways (Swerdlow et al., in press). Nonetheless, changes in PPI after systemic administration of SCH 23390 in SD rats in the present study were presumably accompanied by (and thus perhaps mediated in part via) blockade of D1 receptors in both the MPFC and BLA, in addition to the basal ganglia and other D1-containing regions. D1-mediated reductions in PPI were not accompanied by changes in startle magnitude to P-ALONE trials, suggesting that these drug effects on PPI reflected a diminution of the inhibitory effects of weak prepulses, rather than generalized changes in startle stimulus reactivity (Swerdlow et al., 2000).
Systemic (but not intra-MPFC) administration of SCH 23390 was also associated with an increase in PPI produced by 1 dB prepulses. Certainly, this finding must be viewed with caution because the magnitude of both the stimulus and the resulting inhibition are quite small. However, the rationale for using very weak prepulses in the present study was based on our past observations of increased PPI after D1 blockade under other conditions of ‘‘low inhibitory drive’’, e.g. with short prepulse intervals (Swerdlow et al., 2004a). We have also reported that PPI with 1 dB prepulses is increased by the DA agonist pergolide administered at very low doses (0.005 mg/kg) that theoretically may diminish forebrain DA transmission (Swerdlow et al., 2001c). The fact that SCH 23390-induced increases in 1 dB PPI were not observed after intra-MPFC infusion of SCH 23390 might suggest that, if this is a ‘‘real’’ pharmacological effect, it is likely to be mediated outside of the MPFC.
Systemic but not intra-MPFC administration of SCH 23390 also reduced activity on NOSTIM trials. This observation is of interest because reduced PPI stimulated by direct DA agonists and NMDA antagonists is often (but not always, e.g. see Experiment 3) accompanied by increases in NOSTIM activity (Swerdlow et al., 2004b). The possibility that these processes—reduced PPI and increased NOSTIM activity—are causally linked is not consistent with the present results, in which reduced PPI is associated with reduced NOSTIM activity. The use of weak prepulses in the present study also greatly diminished the possibility that either PPI or its drug sensitivity in the present study reflects changes in prepulse-induced motor responses, as we have previously demonstrated that no detectable motor activity occurs using prepulses of 1–5 dB over a 70 dB(A) background (Swerdlow et al., 2004b).
There are a number of limitations to this study. A single dose of both systemic (0.1 mg/kg) and intra-MPFC SCH 23390 (3 ug/side) was used and pretreatment of haloperidol was also limited to a single dose (0.1 mg/kg). It is certainly possible that a more comprehensive dose–response function might have revealed either different effects on PPI or greater sensitivity to PPI-protective effects of haloperidol. However, we have previously demonstrated orderly, monotonic dose–response effects of systemic and intra-MPFC SCH 23390 on PPI (Swerdlow et al., 2004a; Shoemaker et al., in press), with maximal effects at the doses used in the present study. We and others have also demonstrated that the present dose of haloperidol fully protects PPI from the disruptive effects of DA agonists (Mansbach et al., 1988; Swerdlow et al., 1994; Caine et al., 1995) and Experiment 3 was used as an additional ‘‘control’’ study to demonstrate the effectiveness of this dose vs. AMPH, under the same conditions in which it failed to oppose the PPI-disruptive effects of SCH 23390. Thus, while single-dose effects must always be interpreted with caution, this limitation is mitigated somewhat when viewed in the context of information generated in previous studies and present controls.
Another limitation to this study reflects the lack of specificity of SCH 23390 for D1 receptors. In addition to D1 receptors, SCH 23390 also has affinity for 5HT(2) and 5HT(1C) serotonin receptors (cf. Bourne, 2001) and may also stimulate 5HT(2C) serotonin receptors in the rat MPFC (Ramos et al., in press). It is certainly conceivable that the PPI-disruptive effects of SCH 23390 might reflect, in part or entirely, stimulation of 5HT receptors (cf. Geyer et al., 2001). However, this would not alter the most conservative interpretation of the present findings: that the PPI-disruptive effects of systemic and intra-MPFC SCH 23390 are not mediated via increased transmission at D2 receptors, as neither of these effects are significantly blocked by a dose of haloperidol that completely prevents the PPI-disruptive effects of a rather large dose of the DA releaser, AMPH (Experiment 3). This dose of AMPH is known to produce robust DA release within the ventral striatum, with a time course that closely parallels the reduction in PPI (Zhang et al., 2000). Data shown in Fig. 1 and to a lesser degree in Fig. 2 suggest that the PPI-disruptive effects of SCH 23390 in haloperidol-pretreated rats were less than they were in vehicle-pretreated rats. Thus, despite the lack of a significant interaction of SCH 23390 × haloperidol or SCH 23390 × haloperidol × prepulse intensity, one might argue that there is some participation of D2 receptor activation in the PPI-disruptive effects of SCH 23390. However, close inspection reveals that the relative diminution of the ‘‘SCH 23390 effects’’ in these experiments primarily reflects a haloperidol-induced reduction in PPI in rats treated with vehicle (particularly for 3–4 dB prepulses). While reduced PPI after haloperidol is not the rule, we have reported such effects previously (e.g. Martinez et al., 2002), also under conditions of low levels of PPI.
In contrast, haloperidol completely opposed the PPI-disruptive effects of AMPH in Experiment 3. In Experiment 3, the ability of haloperidol to oppose the PPI-disruptive effects of AMPH was demonstrated by a significant haloperidol-induced increase in PPI among AMPH-treated rats. Clearly, the PPI-disruptive effects of a drug that acts through enhanced subcortical DA release are completely prevented by this dose of haloperidol. Thus, to the degree that intra-MPFC or systemic SCH 23390 enhance subcortical DA release, these effects do not appear to be critical to its PPI-disruptive effects.
More generally, the present findings are consistent with most (Koch and Bubser, 1994; Bubser and Koch, 1994; Ellenbroek et al., 1996; Zavitsanou et al., 1999) but not all (Bast et al., 2002) reports linking reduced MPFC DA function and, particularly, D1 function to reduced PPI in rats. These findings are at least conceptually consistent with the notion that prefrontal D1 hypofunction in schizophrenia might lead to a reduction in normal information-protective mechanisms and contribute to the intrusion of irrelevant stimuli into consciousness. Our findings further suggest that such deficits—like most cognitive deficits in schizophrenia—would not be fully sensitive to acute D2 receptor blockade. Studies of the effects of atypical and chronic antipsychotic exposure in this model and of the effects of D1 antagonists on PPI in normal human subjects would be important next steps in understanding the relationship between D1 hypofunction, PPI deficits and clinical symptoms in schizophrenia.
Acknowledgments
Supported by MH01436 and MH53484.
References
- Abi-Dargham A. Do we still believe in the dopamine hypothesis? New data bring new evidence. Int J Neuropsychopharmacol. 2004;7(Suppl 1):S1–5. doi: 10.1017/S1461145704004110. [DOI] [PubMed] [Google Scholar]
- Bast T, Pezze MA, Feldon J. Dopamine receptor blockade in the rat medial prefrontal cortex reduces spontaneous and amphetamine-induced activity and does not affect prepulse inhibition. Behav Pharmacol. 2002;13:669–73. doi: 10.1097/00008877-200212000-00010. [DOI] [PubMed] [Google Scholar]
- Bourne JA. SCH 23390: the first selective dopamine D1-like receptor antagonist. CNS Drug Rev. 2001;7:399–414. doi: 10.1111/j.1527-3458.2001.tb00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L. Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology. 1978;14:339–43. doi: 10.1111/j.1469-8986.1978.tb01390.x. [DOI] [PubMed] [Google Scholar]
- Braff DL, Swerdlow NR, Geyer MA. Symptom correlates of prepulse inhibition deficits in male schizophrenia patients. Am J Psychiatry. 1999;156:596–602. doi: 10.1176/ajp.156.4.596. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology. 2001;156:234–58. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Braff DL, Light GA, Ellwanger J, Sprock J, Swerdlow NR. Female schizophrenia patients have prepulse inhibition deficits. Biol Psychiatry. 2005;57:817–20. doi: 10.1016/j.biopsych.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Bubser M, Koch M. Prepulse inhibition of the acoustic startle response of rats is reduced by 6-hydroxydopamine lesions of the medial prefrontal cortex. Psychopharmacology. 1994;113:487–92. doi: 10.1007/BF02245228. [DOI] [PubMed] [Google Scholar]
- Caine SB, Geyer MA, Swerdlow NR. Effects of D3/D2 dopamine receptor agonists and antagonists on prepulse inhibition of acoustic startle in the rat. Neuropsychopharmacology. 1995;12:139–45. doi: 10.1016/0893-133X(94)00071-7. [DOI] [PubMed] [Google Scholar]
- Castellanos FX, Fine EJ, Kaysen DL, Marsh WL, Rapoport JL, Hallett M. Sensorimotor gating in boys with Tourette’s syndrome and ADHD: preliminary results. Biol Psychiatry. 1996;39:33–41. doi: 10.1016/0006-3223(95)00101-8. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, Budde S, Cools AR. Prepulse inhibition and latent inhibition: the role of dopamine in the medial prefrontal cortex. Neuroscience. 1996;75:535–42. doi: 10.1016/0306-4522(96)00307-7. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. The cortical dopamine system: role in memory and cognition. Adv Pharmacol. 1998;42:707–11. doi: 10.1016/s1054-3589(08)60846-7. [DOI] [PubMed] [Google Scholar]
- Graham F. The more or less startling effects of weak prestimuli. Psychophysiology. 1975;12:38–48. doi: 10.1111/j.1469-8986.1975.tb01284.x. [DOI] [PubMed] [Google Scholar]
- Hoenig K, Hochrein A, Quednow BB, Maier W, Wagner M. Impaired prepulse inhibition of acoustic startle in obsessive–compulsive disorder. Biol Psychiatry. 2005;57:1153–8. doi: 10.1016/j.biopsych.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Koch M, Bubser M. Deficient sensorimotor gating after 6-hydroxydopamine lesion of the rat medial prefrontal cortex is reversed by haloperidol. Eur J Neurosci. 1994;6:1837–45. doi: 10.1111/j.1460-9568.1994.tb00576.x. [DOI] [PubMed] [Google Scholar]
- Koch M, Schnitzler HU. The acoustic startle response in rats—circuits mediating evocation, inhibition and potentiation. Behav Brain Res. 1997;89:35–49. doi: 10.1016/s0166-4328(97)02296-1. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Cruz D, Eggan S, Erickson S. Postnatal development of prefrontal inhibitory circuits and the pathophysiology of cognitive dysfunction in schizophrenia. Ann N Y Acad Sci. 2004;1021:64–76. doi: 10.1196/annals.1308.008. [DOI] [PubMed] [Google Scholar]
- Mansbach RS, Geyer MA, Braff DL. Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology. 1988;94:507–14. doi: 10.1007/BF00212846. [DOI] [PubMed] [Google Scholar]
- Martinez ZA, Platten A, Pollack E, Shoemaker J, Ro H, Pitcher L, et al. ‘‘Typical’’ but not ‘‘atypical’’ antipsychotic effects on startle gating deficits in prepubertal rats. Psychopharmacology. 2002;161:38–46. doi: 10.1007/s00213-001-0977-y. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 4th ed. San Diego, CA: Academic Press Inc; 1998.
- Ramos M, Goni-Allo B, Aguirre N. Administration of SCH 23390 into the medial prefrontal cortex blocks the expression of MDMA-induced behavioral sensitization in rats: an effect mediated by 5-HT(2C) receptor stimulation and not by D(1) receptor blockade. Neuropsychopharmacology (in press). [DOI] [PubMed]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–57. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Schwarzkopf SB, Bruno JP, Mitra T. Effects of haloperidol and SCH 23390 on acoustic startle and prepulse inhibition under basal and stimulated conditions. Prog Neuropsychopharmacol Biol Psychiatry. 1993;17:1023–36. doi: 10.1016/0278-5846(93)90028-q. [DOI] [PubMed] [Google Scholar]
- Shoemaker, J.M., Saint Marie, R.L., Bongiovanni, M.J., Neary, A.C., Tochen, L.S., Swerdlow, N.R. Prefrontal DA and ventral hippocampal NMDA regulation of startle gating in rats. Neuroscience (in press). [DOI] [PMC free article] [PubMed]
- Stevenson CW, Gratton A. Role of basolateral amygdala dopamine in modulating prepulse inhibition and latent inhibition in the rat. Psychopharmacology. 2004;176:139–45. doi: 10.1007/s00213-004-1879-6. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA, Koob GF. Central dopamine hyperactivity in rats mimics abnormal acoustic startle response in schizophrenics. Biol Psychiatry. 1986;21:23–33. doi: 10.1016/0006-3223(86)90005-3. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Benbow CH, Zisook S, Geyer MA, Braff DL. A preliminary assessment of sensorimotor gating in patients with obsessive compulsive Disorder (OCD) Biol Psychiaty. 1993;33:298–301. doi: 10.1016/0006-3223(93)90300-3. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Taaid N, Geyer MA. Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch Gen Psychiatry. 1994;51:139–54. doi: 10.1001/archpsyc.1994.03950020063007. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Paulsen J, Braff DL, Butters N, Geyer MA, Swenson MR. Impaired prepulse inhibition of acoustic and tactile startle in patients with Huntington’s disease. J Neurol Neurosurg Psychiatry. 1995;58:192–200. doi: 10.1136/jnnp.58.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA. Animal models of deficient sensorimotor gating: what we know, what we think we know, and what we hope to know soon. Behav Pharmacol. 2000;111:185–204. doi: 10.1097/00008877-200006000-00002. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL. Neural circuitry of prepulse inhibition of startle in the rat: current knowledge and future challenge. Psychopharmacology. 2001a;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Karban B, Ploum Y, Sharp R, Geyer MA, Eastvold A. Tactile pre-puff inhibition of startle in children with Tourette syndrome: in search of an ‘‘fMRI-friendly’’ startle paradigm. Biol Psychiatry. 2001b;50:578–85. doi: 10.1016/s0006-3223(01)01164-7. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Platten A, Shoemaker J, Pitcher L, Auerbach P. Effects of pergolide on sensorimotor gating of the startle reflex in rats. Psychopharmacology. 2001c;158:230–40. doi: 10.1007/s002130100856. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Shoemaker JM, Auerbach PP, Pitcher L, Goins J, Platten A. Heritable differences in the dopaminergic regulation of sensorimotor gating: II Temporal, pharmacologic and generational analyses of apomorphine effects on prepulse inhibition. Psychopharmacology. 2004;174:452–62. doi: 10.1007/s00213-003-1480-4. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Talledo J, Shoemaker JM, Codon K, Goins J, Auerbach PP. Weak prepulses inhibit but do not elicit startle in rats and humans. Biol Psychiatry. 2004;55:1195–8. doi: 10.1016/j.biopsych.2004.02.030. [DOI] [PubMed] [Google Scholar]
- Swerdlow, N.R., Krupin, A.S., Bongiovanni, M.J., Shoemaker, M.J., Goins, J.C., Hammer, Jr. R.P. Heritable differences in the dopaminergic regulation of behavior in rats: relationship to D2-like receptor G protein function. Neuropsychopharmacology (in press). [DOI] [PMC free article] [PubMed]
- Valls-Sole J, Munoz JE, Valldeoriola F. Abnormalities of prepulse inhibition do not depend on blink reflex excitability: a study in Parkinson’s disease and Huntington’s disease. Clin Neurophysiol. 2004;115:1527–36. doi: 10.1016/j.clinph.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Zavitsanou K, Cranney J, Richardson R. Dopamine antagonists in the orbital prefrontal cortex reduce prepulse inhibition of the acoustic startle reflex in the rat. Pharmacol Biochem Behav. 1999;63:55–61. doi: 10.1016/s0091-3057(98)00234-2. [DOI] [PubMed] [Google Scholar]
- Zhang J, Forkstam C, Engel JA, Svensson L. Role of dopamine in prepulse inhibition of acoustic startle. Psychopharmacology. 2000;149:181–8. doi: 10.1007/s002130000369. [DOI] [PubMed] [Google Scholar]