

Abstract
Background
Sensorimotor gating, as measured by prepulse inhibition of the startle reflex, is deficient in schizophrenia patients, and in rats after specific manipulations of limbic cortico–striato–pallido–thalamic circuitry. For example, prepulse inhibition in rats is disrupted after D1 blockade in the medial prefrontal cortex, and after N-methyl-d-aspartate infusion into the ventral hippocampus. In the present study, we examined whether these two substrates form part of an integrated circuit regulating sensorimotor gating, which might contribute to the loss of prepulse inhibition in patient populations.
Methods
Prepulse inhibition was assessed in male Sprague–Dawley rats after systemic or intra-medial prefrontal cortex administration of the D1 antagonist, SCH 23390. Separate rats received intra-medial prefrontal cortex infusion of the retrograde transported label Fluoro-Gold. In rats with sham or electrolytic lesions of the medial prefrontal cortex, pre-pulse inhibition was tested after infusion of N-methyl-d-aspartate or vehicle into ventral hippocampus regions that were determined to send projections to the medial prefrontal cortex.
Results
Prepulse inhibition was disrupted after systemic SCH 23390 treatment and after infusion of SCH 23390 into medial prefrontal cortex sites within the prelimbic and cingulate cortices. Fluoro-Gold infusion into these medial prefrontal cortex sites labeled cells in the ventral hippocampus complex, including regions CA1 and entorhinal cortex. N-methyl-d-aspartate infusions into these ventral hippocampus regions disrupted prepulse inhibition in rats after sham but not electrolytic lesions of the medial prefrontal cortex.
Conclusions
Prepulse inhibition appears to be regulated by interacting substrates within the ventral hippocampus and MPFC. Specifically, NMDA activation of the ventral hippocampus appears to disrupt prepulse inhibition in a manner that is dependent on the integrity of infralimbic or cingulate cortical regions that also support a D1-mediated regulation of pre-pulse inhibition. Conceivably, dysfunction within these hippocampal-frontal circuits may contribute to sensorimotor gating deficits in schizophrenia.
Keywords: dopamine, medial prefrontal cortex, prepulse inhibition, SCH 23390, schizophrenia, ventral hippocampus
Abbreviations: ANOVA, analysis of variance; AP, anterior—posterior; Cg1, cingulate cortex, area 1; DA, dopamine; DV, dorsal—ventral; FG, Fluoro-Gold; IL, infralimbic cortex; L, lateral; MO/VO, medial/ventral orbital cortex; MPFC, medial prefrontal cortex; M2, secondary motor cortex; NAC, nucleus accumbens; NMDA, N-methyl-d-aspartate; PPI, prepulse inhibition; PrL, prelimbic cortex; VH, ventral hippocampus
“Prepulse inhibition” (PPI) is the normal suppression of startle when the startling stimulus is preceded by a weak prestimulus (Graham, 1975). This automatic inhibition of a motor reflex by a weak sensory event is an operational measure of sensorimotor gating, that is impaired in a number of neuropsychiatric disorders, including schizophrenia (cf. Braff et al., 2001). Because PPI can be studied in virtually all mammalian species, substantial effort has been directed toward identifying the neural circuit regulation of PPI in laboratory animals, as a way to understand the neural basis for deficient sensorimotor gating in schizophrenia and other disorders (Swerdlow et al., 1992, 2000a, 2001a; Koch and Schnitzler, 1997).
Hippocampal abnormalities in schizophrenia have been identified by many groups (Benes, 1999; Bogerts et al., 1985; Razi et al., 1999; Seidman et al., 1999; Shenton et al., 1992; Stefanis et al., 1999; Velakoulis et al., 1999). Reduced hippocampal volume and an enlarged hippocampal fissure may even be present at the onset of the illness (Smith et al., 2003). Conceivably, hippocampal abnormalities in schizophrenia might give rise to PPI deficits in this disorder, as substantial evidence suggests that the ventral hippocampus (VH) regulates PPI in rodents (cf. Swerdlow et al., 2001a). For example, we reported PPI deficits in rats after intra-VH infusion of carbachol (Swerdlow et al., 1992) or of N-methyl-d-aspartate (NMDA) (Wan et al., 1996). Within the hippocampal complex, the PPI-disruptive effects of NMDA are limited to the ventral portions, with perhaps the most potent effects seen within the entorhinal cortex (Swerdlow et al., 2001b); they are reversed by co-infusion of the NMDA antagonist amino-5-phosphonopentanoate, though not by systemic pretreatment with the D2 antagonist, haloperidol, or by blockade of non-NMDA glutamatergic receptors in the NAC core or shell (Wan et al., 1996).
Our working hypothesis had been that reduced PPI after NMDA activation of the VH was mediated via efferents leaving the hippocampal complex through the fornix (FX) to the nucleus accumbens (NAC) (Brog et al., 1998; Finch et al., 1995; Groenewegen et al., 1987; Kelley and Domesick, 1982; Totterdell and Meredith, 1997). This model was based on three pieces of evidence: 1) the NAC potently regulates PPI (cf. Swerdlow et al., 2001a); 2) VH NMDA infusion stimulates NAC cFOS activation (Klarner et al., 1998), and 3) locomotor activation after VH NMDA infusion is mediated via VH-NAC projections (Mogensen and Nielsen, 1984). However, contrary to this prediction, fornix transection did not interfere with the PPI-disruptive effects of intra-VH NMDA infusion (Swerdlow et al., 2004b).
The persistent influence of the VH on PPI after fornix transection suggests that this influence is mediated via information exiting the hippocampal complex via a nonfornical route. One such route in primates passes first to the entorhinal cortex, and then rostrally, to the frontal and prefrontal cortices (Friedman et al., 2002); in rodents, frontal projections from the entorhinal cortex traverse via several nonfornical routes, including via tracts lateral to the ventricles, the angular bundle and the cingulum (Sorensen, 1985). Conceivably, changes within this projection might impact PPI via substrates in the medial prefrontal cortex (MPFC), a region that is known to regulate PPI via D1 mechanisms (Ellenbroek et al., 1996; Zavitsanou et al., 1999), in addition to other substrates (Japha and Koch, 1999; Schwabe and Koch, 2004). In the present study, we confirmed this hypothesis by: 1) identifying regions within the MPFC that support a D1-mediated regulation of PPI; 2) demonstrating the innervation of these regions by the VH; and 3) determining that destruction of these MPFC regions blocks the PPI-disruptive effects of intra-VH NMDA infusion.
EXPERIMENTAL PROCEDURES
Experimental animals
Adult male Sprague–Dawley rats (225–250 g; Harlan, San Diego, CA, USA) were housed in groups of two to three and maintained on a reversed 12-h light/dark schedule with ad libitum food and water. Testing occurred between 09:00 and 17:00 h, during the dark phase. Rats were handled individually within 2 days of arrival. All surgery occurred between seven and 10 days after arrival. All efforts were made to minimize animal suffering and reduce the number of animals used. All experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80–23) and were approved by the Animal Subjects Committee at the University of California, San Diego (protocol #S01221). Throughout these studies, all efforts were made to minimize animal suffering and to reduce the number of animals used.
Surgical preparations
Rats were given 0.1 ml atropine sulfate (Vedco, St. Joseph, MO, USA, 0.054 mg/ml s.c.) 15–30 min before they were fully anesthetized with sodium pentobarbital (Abbott, North Chicago, IL, USA, 60.0 mg/kg i.p.) and placed in a Kopf stereotaxic instrument in a flat skull position (toothbar 3.3 mm below the interaural line). In rats used for MPFC infusion of SCH 23390 (n=17), bilateral 23 gauge cannulae (9 mm) were aimed 1 mm above the MPFC at coordinates: anterior–posterior (AP) 2.2 (Bregma), lateral (L)±0.8, dorsal–ventral (DV) −4.0, based on previous reports (Ellenbroek et al., 1996). In rats used for VH NMDA infusions (n=39), bilateral 23 gauge cannulae (10 mm) were aimed 3 mm above the VH at coordinates: AP −6.0 (Bregma), L ±5.4, DV −8.2, consistent with the outcome of retrograde labeling from Fluoro-Gold (FG) infusions into the MPFC (below). Cannulae were anchored to the skull with cement and screws, surrounded by a plastic cap and filled with wire stylets. In 24 rats, bilateral electrolytic lesions of the MPFC were made by passing a 1.2 mA × 15 s current at coordinates AP 3.2 (Bregma), L ±0.7, DV −4.2; another 15 rats received sham lesions, consisting of drilling the skull hole and lowering the electrode to a DV coordinate of −1.5.
Behavioral testing
Each of four startle chambers (SR-LAB, San Diego Instruments, San Diego, CA, USA) was housed in a sound-attenuated room with a 60 dB(A) ambient noise level, and consisted of a Plexiglas cylinder 8.2 cm in diameter resting on a 12.5×25.5 cm Plexiglas frame within a ventilated enclosure. Noise bursts were presented via a speaker mounted 24 cm above the cylinder. A piezoelectric accelerometer mounted below the Plexiglas frame detected and transduced motion within the cylinder. The delivery of stimuli was controlled by the SR-LAB microcomputer and interface assembly which also digitized (0–4095), rectified and recorded stabilimeter readings, with 100 1-ms readings collected beginning at stimulus onset. Startle amplitude was defined as the average of the 100 readings. Background noise and all acoustic stimuli were delivered through one Radio Shack Supertweeter (Ft. Worth, TX, USA, frequency response predominantly between 5 and 16 kHz) in each chamber.
SCH23390 studies
For studies involving intra-MPFC infusion of SCH 23390, preliminary dose- and stimulus-finding studies identified a sensitive dose of 3.0 μg/side, consistent with previous reports (Zavitsanou et al., 1999). Systemic doses were based on our previous findings of PPI-disruptive effects of systemic SCH 23390 in a paradigm utilizing intense prepulses and variable prepulse intervals (Swerdlow et al., 2004a). For subsequent studies involving either intra-MPFC (n=17) or systemic SCH 23390 administration (n=28), to reduce inter-group variability, dose groups were assigned based on matched PPI from a brief pretest session. For studies of intra-MPFC administration, this matching session took place 1 week post-surgery. In this session, rats were exposed to 5 min of 70 dB background noise followed by 17 “P-ALONE” trials of 40 ms 120 dB noise bursts and three “PREPULSE” trials consisting of a 20 ms 82 dB (12 dB above background) prepulse followed 100 ms by a 120 dB pulse (onset to onset).
Testing began approximately 3 days later. For intra-MPFC administration of SCH 23390, stylets were removed from the cannulae and replaced by a 30 gauge 10 mm needle immediately prior to the test session. SCH 23390 (Sigma Chemical Co., St. Louis, MO, USA) was dissolved in 0.9% saline. Rats received either active dose infusions (3 μg SCH23390 in 0.5 μl bilaterally) or saline vehicle infusions, over 84 s using a Hamilton microsyringe connected to the needle via polyethylene tubing. Needles remained in place for 30 s after the injection, and then were replaced with stylets. Testing was repeated one week later, with each rat receiving the opposite dose of SCH 23390 (vehicle or 3 μg/side), to complete a within-subject dose design. Some of these rats later participated in a study examining the effects of haloperidol on PPI, but these data are not presented in this report. For systemic administration, SCH 23390 (0, 0.01, 0.03 or 0.1 mg/kg) was administered s.c. in saline vehicle (1 ml/kg), in a between-subject (single test) design (n=6–8 per dose).
Immediately (intra-MPFC study) or 15 min later (systemic study), rats were placed in the startle chambers for a 5 min acclimation period with a 70 dB(A) background noise. After this period, rats were exposed to six trial types: (1) 40 ms–120 dB noise burst (P-ALONE); (2) P-ALONE preceded 100 ms by a 20 ms noise burst that was either 1, 2, 3, 4 or 5 dB(A) above background (PP1dB, PP2dB, PP3dB, PP4dB, PP5dB); (3) NOS-TIM trials (stabilimeter recordings obtained when no stimulus was presented). Trials were presented in pseudorandom order, with 12 repetitions of each trial with a NOSTIM trial between each pulse or prepulse trial. The session began and ended with three P-ALONE trials to permit the assessment of SCH 23390 effects on startle habituation. The session had a total of 155 trials (78 active trials and 77 NOSTIM trials). NOSTIM trials were used to assess gross motor activity during the test session, but were not included in the calculation of intertrial intervals, which were variable and averaged 15 s.
VH NMDA infusions
A matching procedure was used, identical to that described above. Testing began 3 days later. Immediately prior to the test session, stylets were removed from the cannulae and replaced by a 30 gauge needle (13 mm). NMDA (Sigma Chemical Co.) was dissolved in 0.9% saline. Rats received either active dose infusions (0.8 μg NMDA in 0.5 μl bilaterally) or saline vehicle infusions, over 42 s using a Hamilton microsyringe connected to the needle via polyethylene tubing. Needles remained in place for 30 s after the injection, and then were replaced with a stylet. Rats were then placed in the startle chambers for a 5 min acclimation period with a 70 dB(A) background noise. After this period, rats were exposed to nine trial types: (1) 40 ms–120 dB noise burst (P-ALONE); (2) P-ALONE preceded 100 ms by a 20 ms noise burst that was either 1, 2, 3, 4, 5, 10, 15 dB(A) above background (PP1dB, PP2dB, PP3dB, PP4dB, PP5dB, PP10dB, PP15dB); (3) NOSTIM trials (stabilimeter recordings obtained when no stimulus was presented). The more intense (10 and 15 dB) prepulses were added to this session after data from SCH 23390 studies suggested that greater group separation could be achieved with pre-pulses more intense than 5 dB over background. Trials were presented in pseudorandom order, with eight repetitions of each trial type with a NOSTIM trial between each pulse or prepulse trial for a total of 128 trials (64 active trials and 64 NOSTIM trials). Because our previous studies have detected no effects of intra-VH NMDA infusion on startle habituation (Swerdlow et al., 2001b), extra startle trials were not added to this session. NOSTIM trials were used to assess gross motor activity during the test session, but were not included in the calculation of intertrial intervals, which were variable and averaged 15 s. Testing was repeated one week later, with each rat receiving the opposite dose of NMDA (vehicle or 0.8 μg/side), to complete a within-subject dose design. Cement cap instability precluded a second test in two rats (one lesion, one sham). Of the rats included in the final statistical analyses, none exhibited seizure-like activity during testing, but three of the NMDA infused-rats exhibited some form of seizure activity within the 2 h after testing.
Anatomy/histology
After completion of behavioral testing, all animals were deeply anesthetized with pentobarbital and transcardially perfused with a buffered 10% formalin solution. Brains were removed and kept in the 10% formalin solution plus 30% sucrose for 3 days at 4 °C. Frozen sections (40 μm) were cut on a microtome and mounted on gelatin-coated glass slides and Nissl stained. To verify cannulae and lesion placement, stained sections were digitally scanned, sized and oriented in Adobe Photoshop (San Jose, CA, USA; ver. 7.0), then superimposed on corresponding digitized drawings of a rat stereotaxic atlas (Paxinos and Watson, 1998) in Adobe Illustrator (ver. 11.0). Injector placements and lesion boundaries were drawn free-hand in Illustrator from these composite section/atlas images. Lesion damage in the various subdivisions of the MPFC was calculated using Image-Pro Plus (Media Cybernetics, Silver Springs, MD, USA). Drawings were completed blind to the behavioral data. Data were analyzed from all rats that completed testing, for which bilateral injection sites could be confirmed within the target regions. All MPFC infusion sites were confirmed bilaterally, while in some animals (n=6 sham, n=7 lesion) VH injection sites could not be confirmed bilaterally, or were located outside of the VH complex.
To assess the connectivity of the VH and MPFC, other rats received infusions of the retrograde tracer, hydroxystilbamidine (Molecular Probes, Eugene, OR, USA; 0.25–0.5 μl of 2.5% w/v in sterile saline) into the MPFC. Hydroxystilbamidine is equivalent to FG, a trademark of Fluorochrome (Denver, CO, USA). These rats were maintained for 7–10 days before they were deeply anesthetized with pentobarbital and perfused with a buffered 4% paraformaldehyde solution. Brains were dissected and cryoprotected in 30% sucrose in the buffered fixative for 3–5 days. Alternate series of 50 μm frozen sections were either mounted on glass slides for fluorescent microscopy, Nissl stained for conventional histology, or immunoreacted with a rabbit antibody to Fluorogold (Chemicon, Temecula, CA, USA) and processed with avidin/biotin diaminobenzidine/Ni histochemistry (Vector Laboratories; Burlingame, CA, USA) to stabilize the retrograde tracer for conventional light microscopy. Injection sites of hydroxystilbamidine into the MPFC were confirmed for accurate placement to assure reliable interpretation of the VH labeling.
Statistics
PPI was defined as 100−[(startle amplitude on prepulse trials/startle amplitude on P-ALONE trials)×100], and was analyzed by mixed-design analyses of variance (ANOVAs), with specific comparisons noted for each experiment. For the SCH 23390 dose-response study, significant main effects of dose were followed by pair-wise ANOVAs of vehicle vs. active dose with repeated measures on prepulse intensity. Separate analyses were performed using raw startle magnitude on P-ALONE and prepulse trials, to determine whether changes in percent PPI reflected a diminished ability of prepulses to inhibit startle. A variable representing the “impact” of NMDA was calculated to allow correlations of lesion size and behavioral change; “impact” was defined as the difference between the mean PPI levels (averaged across prepulse conditions) for the vehicle condition minus drug condition. In this manner, a greater difference reflected a larger “impact” of the drug in reducing PPI levels. Alpha was 0.05.
RESULTS
Anatomy/histology
Histological assessment revealed SCH23390 infusion sites within prelimbic and infralimbic subregions of the MPFC (Fig. 2B). FG infusion sites were confirmed within these same regions of the MPFC. NMDA infusion sites were distributed within the VH (Fig. 1A, B), primarily within the ventral CA1, ventral subiculum and entorhinal cortex regions that also demonstrated the most dense retrograde labeling after intra-MPFC infusions of FG (Fig. 1C). Electrolytic lesions of the MPFC were confined in the A/P plane between Bregma levels+4.70 and +2.70 (Paxinos and Watson, 1998) and resulted in damage to secondary motor cortex (M2), cingulate cortex area 1 (Cg1), prelimbic cortex (PrL), and in some rats medial and ventral orbital cortex (MO/VO) and infralimbic cortex (IL) (Fig. 1D, E). Percent destruction of these subregions (mean (range)) was: M2 (10.3% (1.6–31.3)); Cg1 (33.0% (5.4–69.7)); PrL (46.8% (10.8–64.4)); MO/VO (23.7% (0–71.9)); IL (10.7% (0–81.8)).
Fig. 2.
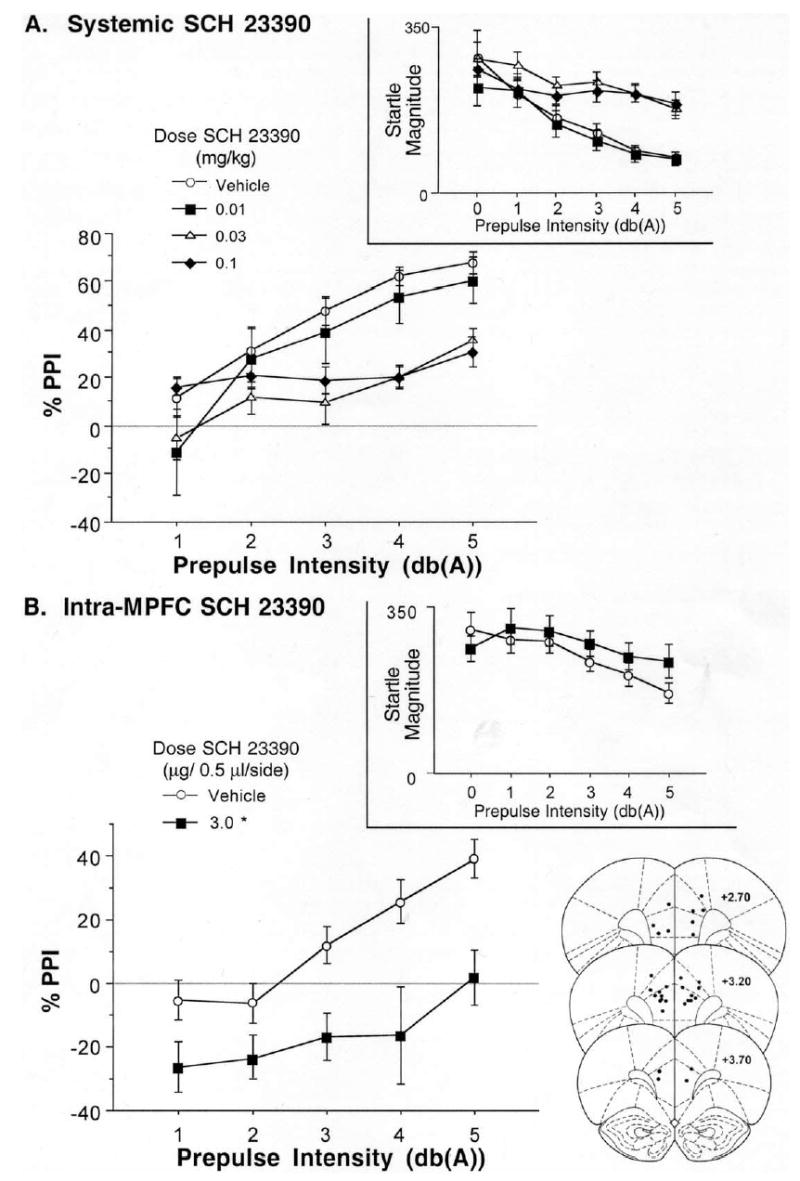
%PPI and raw startle magnitude on pulse alone and prepulse+pulse trials (insets) after: (A) systemic injection of SCH 23390, and (B) intra-MPFC infusion of the D1 antagonist, SCH 23390. PPI was significantly disrupted by SCH 23390 after either systemic or intra-MPFC administration. Also shown are SCH 23390 infusion sites within prelimbic and infralimbic subregions of the MPFC (*) significant reduction in PPI vs. vehicle, P<0.05.
Fig. 1.
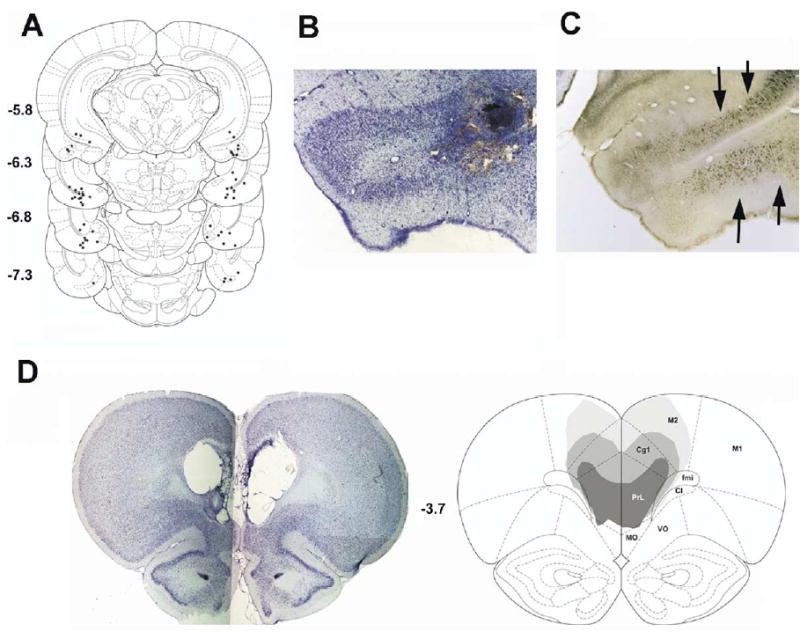
(A) Distribution of NMDA infusion sites within the VH complex, primarily within the ventral CA1, ventral subiculum and entorhinal cortex regions. (B) Example of typical NMDA injection sites within the VH region that also demonstrated (C) the most dense retrograde labeling after intra-MPFC infusions of FG. Additional labeling was found to extend from the subiculum to the pyramidal layer of ventral and caudal portions of CA1; and from the entorhinal cortex to perirhinal and ectorhinal cortices. (D) Nissl stain of a “representative” MPFC lesion, and computer reconstruction of smallest (darkest), largest (lightest), and “average” damage produced by electrolytic lesions of the MPFC.
Behavior
Systemic injection of SCH 23390 resulted in a dose- and intensity-dependent disruption of PPI (Fig. 2A). ANOVA of PPI revealed a significant main effect of dose (F=3.56, df 3, 24, P<0.03), a significant effect of prepulse intensity (F=63.57, df 4, 96, P<0.0001), and a significant dose×intensity interaction (F=6.26, df 12, 96, P<0.0001). Pair-wise ANOVAs revealed that, compared with vehicle, PPI was significantly reduced by the 0.03 and 0.1 mg/kg doses of SCH 23390 (P<0.002 and 0.015, respectively), with effects being most robust for the most intense (5 dB over background) prepulses. Inspection of the raw startle magnitude revealed that reduced PPI at these doses of SCH 23390 reflected the diminished ability of prepulses to reduce startle magnitude on prepulse+pulse trials (Fig. 2A, inset). There was no significant effect of SCH 23390 on startle magnitude on P-ALONE trials (F<1).
SCH 23390 infusion into the MPFC resulted in a significant reduction in PPI (Fig. 2B). ANOVA of PPI revealed significant effects of SCH 23390 dose (F=15.67, df 1, 16, P<0.001) and prepulse intensity (F=11.34, df 4, 64, P<0.0001), with no significant dose×intensity interaction (F=1.94, df 4, 64, ns). As with systemic SCH 23390 (above), inspection of the raw startle magnitude after intra-MPFC infusion of SCH 23390 revealed that reduced PPI reflected the diminished ability of prepulses to reduce startle magnitude on prepulse+pulse trials (Fig. 2B, inset). There was no significant effect of SCH 23390 on startle magnitude on P-ALONE trials (F=1.00, df 1, 16, ns).
Collectively, these anatomical and behavioral data demonstrate that a region of the MPFC that is innervated by the VH potently regulates PPI, and provide an empirical a priori rationale (together with the theoretical rationale discussed earlier) for assessing the role of this MPFC region in mediating the PPI-disruptive effects of VH activation. Intra-VH infusion of NMDA significantly reduced PPI in rats with sham lesions of the MPFC, but not in rats with electrolytic lesions of the MPFC (Fig. 3). ANOVA revealed a main effect of NMDA dose (F=6.49, df 1, 22, P<0.02) and prepulse intensity (F=46.83, df 6, 132, P<0.0001). There was no significant effect of lesion (F<1) and only a trend toward a dose×lesion interaction (F=3.44, df 1, 22, P<0.08). Inspection of the data revealed significant PPI-reducing effects of NMDA in rats with sham lesions (F=13.63, df 1, 7, P<0.008) but not electrolytic lesions of the MPFC (F<1). This same pattern of results was obtained if the analyzed sample included rats that were originally excluded because their VH injector sites could not be confirmed bilaterally in the VH (main effect of NMDA dose, P<0.005; significant effect in sham (F=9.05, df 1, 13, P=0.01) but not lesion group rats (ns)). There was some evidence that MPFC lesions selectively blunted the effects of intra-VH NMDA on PPI, and that MPFC-lesioned rats were not simply insensitive to all behavioral effects of NMDA: intra-VH NMDA infusion stimulated basal (NOS-TIM) motor activity (main effect of NMDA dose: F=8.60, df 1, 22, P<0.008), an effect that was intact in MPFC lesion group rats (dose×lesion interaction: ns; main effect of dose in MPFC lesion group: F=9.72, df 1, 15, P<0.007).
Fig. 3.
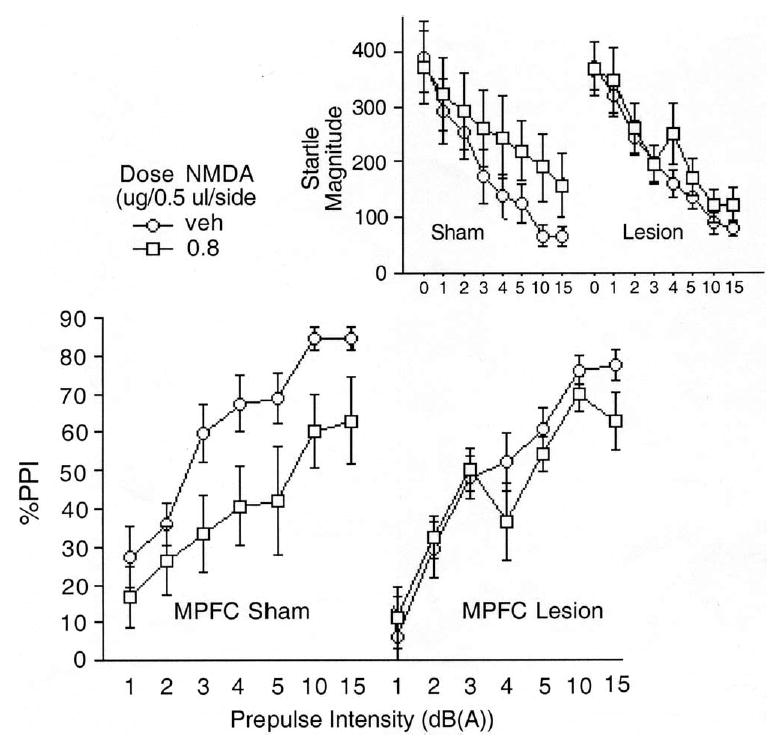
%PPI and startle magnitude on pulse alone and prepulse+pulse trials (inset) after intra-VH infusion of vehicle or NMDA (0.8 μg/side) in rats after sham or electrolytic lesions of the MPFC. Intra-VH NMDA significantly reduced PPI in MPFC sham, but not electrolytic lesion group rats.
The PPI-disruptive effects of intra-VH NMDA infusion were examined in relation to different anatomical characteristics of MPFC lesions. All MPFC lesions resulted in damage to the PrL, and to a more limited degree Cg1, between A/P+4.70 and +2.70. Damage to other cortical areas was inconsistent, and absent among some rats. While primary damage to PrL (and perhaps Cg1) would appear to be responsible for the observed behavioral effects, the magnitude of the NMDA-induced reduction in PPI did not correlate significantly with the percent destruction of any single MPFC subregion, or with the overall lesion size. Other strategies (e.g. comparing regional lesion size among rats with extreme levels of NMDA effects) also did not identify damage to any particular MPFC subregion to be associated with the greatest reductions in NMDA sensitivity.
DISCUSSION
Numerous studies have demonstrated both anatomical connectivity and functional interactions between the VH complex and the MPFC. Efferent projections from the hippocampus (CA1–CA2 fields), subiculum and entorhinal cortex form monosynaptic inputs onto pyramidal neurons and GABAergic interneurons within prelimbic, infralimbic and orbital cortex (Jay and Witter, 1991; Jay et al., 1992; Condé et al., 1995; Carr and Sesack, 1996; Gabbott et al., 2002). Evidence from a variety of sources suggests that these excitatory aspartate/glutamate projections terminate onto AMPA-type receptors, often in apposition with dopamine (DA)-containing terminals arising from midbrain DA nuclei and terminating on D1 receptors (Carr and Sesack, 1996). Hippocampal complex projections may arrive at the MPFC via multiple different pathways (Conde et al., 1995), to converge not only with ascending DAergic fibers but also with excitatory inputs from the mediodorsal thalamus (Sorensen, 1985; Friedman et al., 2002; Delatour and Witter, 2002; Floresco and Grace, 2003). It is speculated that this converging circuitry plays an important role in executive functioning, working memory and other higher order cognitive processes (Compte et al., 2000). Perhaps more importantly, pathology in these connections has been implicated in the pathophysiology of schizophrenia, based on neuroimaging, neuropathological and animal model studies (Lewis et al., 2004).
While no evidence for a direct functional interaction is presented here, the present findings suggest at least a spatial convergence of hippocampal efferents to the MPFC, and D1 substrates within the MPFC, both of which regulate sensorimotor gating of startle. Specifically, we report that: 1) chemical stimulation of the HPC→MPFC projection disrupts PPI; 2) this effect depends on the integrity of the MPFC terminal regions; and 3) and pharmacologic blockade of D1 receptors in these same terminal regions also disrupts PPI.
It has been proposed, by our investigative group and by others, that the hippocampal regulation of PPI is mediated via VH efferents to the NAC (Swerdlow et al., 1992, 2001a; Klarner et al., 1998). At the time, based on existing evidence, this seemed like reasonable conjecture. The VH is known to project to the NAC and this projection is thought to mediate other VH-dependent behaviors (Mogensen and Nielsen, 1984). Intra-VH NMDA infusion, which disrupts PPI, stimulates cFOS activation in the NAC (Klarner et al., 1998). VH lesions in adult rats lead to an enhanced sensitivity to the PPI-disruptive effects of the DA agonist, apomorphine (Swerdlow et al., 1995, 2000, 2004) and amphetamine (Caine et al., 2001), drugs that are thought to regulate PPI at least in part via stimulation of D2 activity in the NAC (cf. Swerdlow et al., 2001a). Thus, the indirect evidence for this VH→NAC PPI model motivated studies in pursuit of more direct evidence and a mechanistic explanation.
But our efforts to understand the neural mechanisms by which such a VH→NAC projection might regulate PPI have yielded a number of findings that have made this hypothesis increasingly untenable. First, we and others reported that the PPI-disruptive effects of intra-VH NMDA infusion are not prevented by D2 blockade, either from the typical antipsychotic haloperidol (Wan et al., 1996; Bast et al., 2001), or from the atypical antipsychotic clozapine (Bast et al., 2001, but also see Zhang et al., 2000). Thus, if a VH→NAC projection mediates this effect, it must do so via non-D2-mediated mechanisms. Second, we reported that these effects of intra-VH NMDA are not opposed by blockade of NAC AMPA receptors (Wan et al., 1996), despite the fact that this VH→NAC projection is believed to terminate on non-NMDA receptors in the NAC (Wan and Swerdlow, 1996a). Most recently, we demonstrated that the PPI-disruptive effects of intra-VH NMDA persisted after physical disconnection of the VH and NAC via lesions of the fornix (Swerdlow et al., 2004b). This last finding forced a reassessment of the role of the VH→NAC projection in the regulation of PPI. Based on these past findings and those of the present study, one working hypothesis is that the PPI-disruptive effects of VH activation are mediated via nonfornical projections to the MPFC.
This is not to say that a VH→NAC projection via the fornix does not contribute to the regulation of PPI, but rather that it is not required for the reduction of PPI observed after acute infusion of NMDA into the VH. In contrast, the enhanced sensitivity to the PPI-disruptive effects of DA agonists, observed 4 weeks after lesions of the VH (Swerdlow et al., 1995, 2000, 2004), might be mediated by mechanisms quite different from those engaged by acute VH stimulation, and might reasonably be hypothesized to reflect changes in response to destruction of the VH→NAC projection. Tests of this model are in progress. Similarly, reduced PPI after excitotoxic lesions of the entorhinal cortex in adult (Goto et al., 2002), but not neonatal rats (Schmadel et al., 2004), have been proposed to reflect an alteration in NAC DAergic activity that might conceivably reflect changes resulting from the loss of entorhinal cortical projections to the NAC (Goto et al., 2004).
Anatomical studies have demonstrated that projections from the ventral hippocampal complex reach the MPFC via multiple pathways (Rosene and Van Hoesene, 1977; Irle and Markowitsch, 1982; Goldman-Rakic et al., 1984; Barbas and Blatt, 1995; Verwer et al., 1997; Delatour and Witter, 2002). Thus, not only do MPFC afferents arise from hippocampal fields CA1 and CA2, ventral subiculum, entorhinal and perirhinal cortex (e.g. Sorensen, 1985; Jay and Witter, 1991; Conde et al., 1995; Insausti et al., 1997; Vann et al., 2000), but these projections may reach their targets via different routes. Our inability to detect significant correlations between damage to a specific MPFC subregion and reduced PPI sensitivity to intra-VH NMDA may reflect the involvement of multiple MPFC subregions in this behavioral response. This would be consistent with our findings that reduced PPI can result from NMDA infusion into any one of several regions within the VH complex (Swerdlow et al., 2001b). Alternatively, it is possible that our lesions lacked adequate variability to permit us to detect the differential involvement of specific MPFC subregions in this behavioral change.
We do not mean to suggest that the hippocampal regulation of PPI resides entirely within the VH. We (Caine et al., 1991) and others have reported that various chemical manipulations of the dorsal hippocampus can impair PPI, although this is not the case for NMDA infusion (Swerdlow et al., 2001b). Nor do we mean to suggest that the MPFC regulation of PPI entirely reflects activity within a VH→MPFC projection. The MPFC is innervated by numerous regions known to contribute to the regulation of PPI, including the basolateral amygdala (Wan and Swerdlow, 1996b) and the midbrain DA nuclei (Zhang et al., 1995). Finally, it is very possible that some of the PPI disruptive effects of intra-VH NMDA are mediated via VH projections via non-fornical, or even fornical routes. The present data suggest only that the full impact of VH activation on PPI requires the integrity of the MPFC.
There may be some basis for speculating about the general types of neural interactions within the MPFC that might account for the changes in PPI observed in the present studies. In the simplest model of our data, reduced PPI followed both: 1) activation of MPFC AMPA-type receptors, a result of NMDA stimulating an excitatory VH→MPFC aspartate/glutamate projection; and 2) a reduction of DAergic activity at MPFC D1 receptors, a result of SCH23390-induced blockade of MPFC D1 receptors. That the PPI-disruptive effects of intra-VH NMDA infusion were not evident after MPFC lesions at least tentatively suggests that activation of MPFC AMPA receptors disrupts PPI by stimulating MPFC efferent projections. Thus, in this simplest model, both AMPA stimulation and D1 blockade in the MPFC disrupt PPI by activating descending MPFC projections into some “next level” of PPI-regulatory circuitry. Presumably, these MPFC efferent projections might involve layer five pyramidal neurons, which project to portions of the striatum and brainstem, likely “next levels” of this PPI circuitry (cf. Swerdlow et al., 1992, 2001a; Koch and Schnitzler, 1997). Of course, activation of these efferents can reflect the impact of AMPA receptor stimulation or D1 receptor blockade on various populations of MPFC interneurons, such as calretinin-positive GABAergic interneurons, that might allow for a downstream “disinhibition” of layer five pyramidal cell projection neurons (cf. Lewis et al., 2004).
How the activation of MPFC efferents leads to a loss of PPI is perhaps not so easily explained. Our findings suggest that such a process would not involve stimulation of subcortical D2 receptors via activation of mesolimbic DA transmission, as neither the effects of intra-VH NMDA (Wan et al., 1996) nor those of intra-MPFC SCH 23390 (Crain et al., 2003) are prevented by systemic D2 blockade with haloperidol. Nor would such a process involve stimulation of NAC AMPA receptors, as the PPI-disruptive effects of intra-VH NMDA are not prevented by intra-NAC infusion of CNQX (Wan et al., 1996). One might argue that the critical MPFC efferents responsible for reduced PPI project not to the ventral tegmental nucleus or NAC, but instead to the thalamus, via MPFC cells in layer 6. However, reduced PPI follows thalamic inhibition (Kodsi and Swerdlow, 1997; Swerdlow et al., 2002), which would be opposite to the effect predicted after activation of excitatory corticothalamic efferents.
The use of electrolytic lesions of the MPFC in the present study does not preclude the possibility that the critical substrate destroyed by these lesions involved fibers of passage through the MPFC, rather than cells residing in this region. Such fibers might subserve cortical–cortical connections, or alternatively, be part of aminergic fibers projecting rostrally to the frontal poles before extending caudally across the cortical hemispheres. To examine this possibility, we are presently assessing whether the PPI-disruptive effects of VH NMDA infusion are maintained or disrupted after fiber-sparing excitotoxic lesions of the MPFC.
While we have no immediate explanation for the neurophysiological basis of reduced PPI after MPFC activation, the psychophysiological basis is somewhat more clear. After both intra-VH NMDA infusion and systemic and intra-MPFC D1 blockade, reduced PPI was accompanied by increases in startle magnitude on prepulse+pulse trials (Figs. 2–3). This pattern of changes reflects a diminished gating effect of the prepulse on the motor response to the startle stimulus, i.e. a true loss of sensorimotor gating (Swerdlow et al., 2000). This is the process that has been speculated to contribute to symptomatology in a number of disorders, including schizophrenia. Recent findings from our group suggest that deficient PPI in schizophrenia patients is associated with impairment in neuropsychological and functional measures mediated by the frontal cortex (Swerdlow et al., 2005). The present findings may provide an initial basis for studying mechanisms by which dysfunction in VH→MPFC circuits in schizophrenia may lead to reduced PPI and associated impairments in frontal function in schizophrenia.
Acknowledgments
Supported by MH 01436 and MH 53484. The authors gratefully acknowledge the assistance of Ms. Pamela Auerbach and Ms. Lillian Ma in the completion of this work.
References
- Barbas H, Blatt GJ. Topographically specific hippocampal projections target functionally distinct prefrontal areas in the rhesus monkey. Hippocampus. 1995;5:511–533. doi: 10.1002/hipo.450050604. [DOI] [PubMed] [Google Scholar]
- Bast T, Zhang W, Heidbreder C, Feldon J. Hyperactivity and disruption of prepulse inhibition induced by N-methyl-D-aspartate stimulation of the ventral hippocampus and the effects of pretreatment with haloperidol and clozapine. Neuroscience. 2001;103:325–335. doi: 10.1016/s0306-4522(00)00589-3. [DOI] [PubMed] [Google Scholar]
- Benes FM. Evidence for altered trisynaptic circuitry in schizophrenic hippocampus. Biol Psychiatry. 1999;46:589–599. doi: 10.1016/s0006-3223(99)00136-5. [DOI] [PubMed] [Google Scholar]
- Bogerts B, Meertz E, Schonfeldt-Bausch R. Basal ganglia and limbic system pathology in schizophrenia. A morphometric study of brain volume and shrinkage. Arch Gen Psychiatry. 1985;42:784–791. doi: 10.1001/archpsyc.1985.01790310046006. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: Normal subjects, patient groups, and pharmacological studies. Psychopharmacology. 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Brog JS, Salyapongse A, Deutch AY, Zahm DS. The patterns of afferent innervation of the core and shell in the “accumbens” part of the rat ventral striatum: immunohistochemical detection of retrogradely transported fluoro-gold. J Comp Neurol. 1998;338:255–278. doi: 10.1002/cne.903380209. [DOI] [PubMed] [Google Scholar]
- Caine SB, Geyer MA, Swerdlow NR. Carbachol infusion into the dentate gyrus disrupts sensorimotor gating of startle in the rat. Psycopharmacology. 1991;105:347–354. doi: 10.1007/BF02244429. [DOI] [PubMed] [Google Scholar]
- Caine SB, Humby T, Robbins TW, Everitt BJ. Behavioral effects of psychomotor stimulants in rats with dorsal or ventral subiculum lesions: locomotion, cocaine self-administration, and prepulse inhibition of startle. Behav Neurosci. 2001;115:880–894. doi: 10.1037//0735-7044.115.4.880. [DOI] [PubMed] [Google Scholar]
- Carr DB, Sesack SR. Hippocampal afferents to the rat prefrontal cortex: synaptic targets and relation to dopamine terminals. J Comp Neurol. 1996;369:1–15. doi: 10.1002/(SICI)1096-9861(19960520)369:1<1::AID-CNE1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Compte A, Brunel N, Goldman-Rakic PS, Wang XJ. Synaptic mechanisms and network dynamics underlying spatial working memory in a cortical network model. Cereb Cortex. 2000;10:910–923. doi: 10.1093/cercor/10.9.910. [DOI] [PubMed] [Google Scholar]
- Conde F, Maire-Lepoivre E, Audinat E, Crépel F. Afferent connections to the medial prefrontal cortex of the rat. II. Cortical and sub-cortical afferents. J Comp Neurol. 1995;352:567–593. doi: 10.1002/cne.903520407. [DOI] [PubMed] [Google Scholar]
- Crain SK, Shoemaker JM, Goins J, Ma L, Noh HR, Swerdlow NR. D1 blockade in prefrontal cortex disrupts startle gating in rats: Anatomical characterization. Abstr Soc Neurosci. 2003:858.18. [Google Scholar]
- Delatour B, Witter MP. Projections from the parahippocampal region to the prefrontal cortex in the rat: evidence of multiple pathways. Eur J Neurosci. 2002;15:1400–1407. doi: 10.1046/j.1460-9568.2002.01973.x. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, Budde S, Cools AR. Prepulse inhibition and latent inhibition: the role of dopamine in the medial prefrontal cortex. Neuroscience. 1996;75:535–542. doi: 10.1016/0306-4522(96)00307-7. [DOI] [PubMed] [Google Scholar]
- Finch DM, Gigg J, Tan AM, Kosoyan OP. Neurophysiology and neuropharmacology of projections from entorhinal cortex to striatum in rat. Brain Res. 1995;670:233–247. doi: 10.1016/0006-8993(94)01279-q. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Grace AA. Gating of hippocampal-evoked activity in prefrontal cortical neurons by inputs from the mediodorsal thalamus and ventral tegmental area. J Neurosci. 2003;23:3930–3943. doi: 10.1523/JNEUROSCI.23-09-03930.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DP, Aggleton JP, Saunders RC. Comparison of hippocampal, amygdala, and perirhinal projections to the nucleus accumbens: combined anterograde and retrograde tracing study in the macaque brain. J Comp Neurol. 2002;450:345–365. doi: 10.1002/cne.10336. [DOI] [PubMed] [Google Scholar]
- Gabbott P, Headlam A, Bubsy S. Morphological evidence that CA1 hippocampal afferents monosynaptically innervate PV-containing neurons and NADPH-diaphorase reactive cells in the medial prefrontal cortex (areas 25/32) of the rat. Brain Res. 2002;946:314–322. doi: 10.1016/s0006-8993(02)02487-3. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Selemon LD, Schwartz ML. Dual pathways connecting the dorsolateral prefrontal cortex with the hippocampal formation and parahippocampal cortex in the rhesus monkey. Neuroscience. 1984;12:719–743. doi: 10.1016/0306-4522(84)90166-0. [DOI] [PubMed] [Google Scholar]
- Goto K, Ueki A, Iso H, Morita Y. Involvement of nucleus accumbens dopaminergic transmission in acoustic startle: observations concerning prepulse inhibition in rats with entorhinal cortex lesions. Psychiatry Clin Neurosci. 2004;58:441–445. doi: 10.1111/j.1440-1819.2004.01281.x. [DOI] [PubMed] [Google Scholar]
- Goto K, Ueki A, Iso H, Morita Y. Reduced prepulse inhibition in rats with entorhinal cortex lesions. Behav Brain Res. 2002;134(1–2):201–207. doi: 10.1016/s0166-4328(02)00039-6. [DOI] [PubMed] [Google Scholar]
- Graham F. The more or less startling effects of weak prestimuli. Psychophysiology. 1975;12:38–248. doi: 10.1111/j.1469-8986.1975.tb01284.x. [DOI] [PubMed] [Google Scholar]
- Groenewegen HJ, Vermeulen-Van der Zee E, Te Kortschot A, Witter MP. Organization of the projections from the subiculum to the ventral striatum in the rat. A study using anterograde transport of Phaseolus vulgaris leucoagglutinin. Neuroscience. 1987;23:103–120. doi: 10.1016/0306-4522(87)90275-2. [DOI] [PubMed] [Google Scholar]
- Insausti R, Herrero MT, Witter MP. Entorhinal cortex of the rat: Cytoarchitectonic subdivisions and the origin and distribution of cortical efferents. Hippocampus. 1997;7:146–183. doi: 10.1002/(SICI)1098-1063(1997)7:2<146::AID-HIPO4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Irle E, Markowitsch HJ. Widespread cortical projections of the hippocampal formation in the cat. Neuroscience. 1982;7:2637–2647. doi: 10.1016/0306-4522(82)90088-4. [DOI] [PubMed] [Google Scholar]
- Japha K, Koch M. Picrotoxin in the medial prefrontal cortex impairs sensorimotor gating in rats: reversal by haloperidol. Psychopharmacology. 1999;144:347–354. doi: 10.1007/s002130051017. [DOI] [PubMed] [Google Scholar]
- Jay TM, Thierry AM, Wiklund L, Glowinski J. Excitatory amino acid pathway from the hippocampus to the prefrontal cortex. Contribution of AMPA receptors in hippocampo-prefrontal cortex transmission. Eur J Neurosci. 1992;4:1285–1295. doi: 10.1111/j.1460-9568.1992.tb00154.x. [DOI] [PubMed] [Google Scholar]
- Jay TM, Witter MP. Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgarisleucoagglutinin. J Comp Neurol. 1991;313:574–586. doi: 10.1002/cne.903130404. [DOI] [PubMed] [Google Scholar]
- Kelley AE, Domesick VB. The distribution of the projection from the hippocampal formation to the nucleus accumbens in the rat: an anterograde- and retrograde-horseradish peroxidase study. Neuroscience. 1982;7:2321–2335. doi: 10.1016/0306-4522(82)90198-1. [DOI] [PubMed] [Google Scholar]
- Klarner A, Koch M, Schnitzler HU. Induction of fos-protein in the forebrain and disruption of sensorimotor gating following N-methyl-D-aspartate infusion into the ventral hippocampus of the rat. Neuroscience. 1998;84:443–452. doi: 10.1016/s0306-4522(97)00475-2. [DOI] [PubMed] [Google Scholar]
- Koch M, Schnitzler HU. The acoustic startle response in rats-circuits mediating evocation, inhibition and potentiation. Behav Brain Res. 1997;89:35–49. doi: 10.1016/s0166-4328(97)02296-1. [DOI] [PubMed] [Google Scholar]
- Kodsi MH, Swerdlow NR. Regulation of prepulse inhibition by ventral pallidal projections. Brain Res Bull. 1997;43:219–228. doi: 10.1016/s0361-9230(96)00440-6. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Cruz D, Eggan S, Erickson S. Postnatal development of prefrontal inhibitory circuits and the pathophysiology of cognitive dysfunction in schizophrenia. Ann N Y Acad Sci. 2004;1021:64–76. doi: 10.1196/annals.1308.008. [DOI] [PubMed] [Google Scholar]
- Mogensen GJ, Nielsen M. A study of the contribution of hippocampal-accumbens-subpallidal projections to locomotor activity. Behav Neural Biol. 1984;42:38–51. doi: 10.1016/s0163-1047(84)90412-6. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates, 4th edition. San Diego, CA: Academic Press Inc.
- Razi K, Greene KP, Sakuma M, Ge S, Kushner M, DeLisi LE. Reduction of the parahippocampal gyrus and the hippocampus in patients with chronic schizophrenia. Br J Psychiatry. 1999;174:512–519. doi: 10.1192/bjp.174.6.512. [DOI] [PubMed] [Google Scholar]
- Rosene DL, Van Hoesene GW. Hippocampal efferents reach widespread areas of the cerebral cortex and amygdala in the rhesus monkey. Science. 1977;198:315–317. doi: 10.1126/science.410102. [DOI] [PubMed] [Google Scholar]
- Schmadel S, Schwabe K, Koch M. Effects of neonatal excitotoxic lesions of the entorhinal cortex on cognitive functions in the adult rat. Neuroscience. 2004;128:365–374. doi: 10.1016/j.neuroscience.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Schwabe K, Koch M. Role of the medial prefrontal cortex in N-methyl-D-aspartate receptor antagonist induced sensorimotor gating deficit in rats. Neurosci Lett. 2004;355:5–8. doi: 10.1016/j.neulet.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Seidman LJ, Faraone SV, Goldstein JM, Goodman JM, Kremen WS, Toomey R, Tourville J, Kennedy D, Makris N, Caviness VS, Tsuang MT. Thalamic and amygdala-hippocampal volume reductions in first-degree relatives of patients with schizophrenia: an MRI-based morphometric analysis. Biol Psychiatry. 1999;46:941–954. doi: 10.1016/s0006-3223(99)00075-x. [DOI] [PubMed] [Google Scholar]
- Shenton ME, Kikinis R, Jolesz FA, Pollak SD, LeMay M, Wible CG, Hokama H, Martin J, Metcalf D, Coleman M, et al. Abnormalities of the left temporal lobe and thought disorder in schizophrenia. N Engl J Med. 1992;327:604–612. doi: 10.1056/NEJM199208273270905. [DOI] [PubMed] [Google Scholar]
- Smith GN, Lang DJ, Kopala LC, Lapointe JS, Falkai P, Honer WG. Developmental abnormalities of the hippocampus in first-episode schizophrenia. Biol Psychiatry. 2003;53:555–61. doi: 10.1016/s0006-3223(02)01977-7. [DOI] [PubMed] [Google Scholar]
- Sorensen KE. Projections of the entorhinal area to the striatum, nucleus accumbens, and cerebral cortex in the guinea pig. J Comp Neurol. 1985;238:308–322. doi: 10.1002/cne.902380306. [DOI] [PubMed] [Google Scholar]
- Stefanis N, Frangou S, Yakeley J, Sharma T, O’Connell P, Morgan K, Sigmudsson T, Taylor M, Murray R. Hippocampal volume reduction in schizophrenia: effects of genetic risk and pregnancy and birth complications. Biol Psychiatry. 1999;46:697–702. doi: 10.1016/s0006-3223(99)00089-x. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA. Animal models of deficient sensorimotor gating: What we know, what we think we know, and what we hope to know soon. Behav Pharmacol. 2000a;111:185–204. doi: 10.1097/00008877-200006000-00002. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Caine SB, Braff DL, Geyer MA. Neural substrates of sensorimotor gating of the startle reflex: Preclinical findings and their implications. J Psychopharmacol. 1992;6:176–190. doi: 10.1177/026988119200600210. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL. Neural circuitry of prepulse inhibition of startle in the rat: Current knowledge and future challenge. Psychopharmacology. 2001a;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Hanlon FM, Henning L, Kim YK, Gaudet I, Halim ND. Regulation of sensorimotor gating in rats by hippocampal NMDA: Anatomical localization. Brain Res. 2001b;898:195–203. doi: 10.1016/s0006-8993(01)02143-6. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Light GA, Cadenhead KS, Sprock J, Hsieh MH, Haugland B, Holmes S, Braff DL. Startle gating deficits in a large cohort of schizophrenia patients: Relationship to clinical symptoms, neurocognitive and functional impairment. Biol Psychiatry. 2005;57:115S. [Google Scholar]
- Swerdlow NR, Lipska BK, Weinberger DR, Braff DL, Jaskiw GE, Geyer MA. Increased sensitivity to the gating-disruptive effects of apomorphine after lesions of the medial prefrontal cortex or ventral hippocampus in adult rats. Psychopharmacology. 1995;122:27–34. doi: 10.1007/BF02246438. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Pitcher L, Noh HR, Shoemaker JM. Startle gating in rats is disrupted by chemical inactivation but not D2 stimulation of the dorsomedial thalamus. Brain Res. 2002;953:246–254. doi: 10.1016/s0006-8993(02)03298-5. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Shoemaker JM, Auerbach PP, Pitcher L, Goins J, Platten A. Heritable differences in the dopaminergic regulation of sensorimotor gating: II. Temporal, pharmacologic and generational analyses of apomorphine effects on prepulse inhibition. Psychopharmacology. 2004a;174:452–462. doi: 10.1007/s00213-003-1480-4. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Shoemaker JM, Noh HR, Ma L, Gaudet I, Munson M, Crain S, Auerbach PP. The ventral hippocampal regulation of prepulse inhibition and its disruption by apomorphine in rats are not mediated via the fornix. Neuroscience. 2004b;123:675–685. doi: 10.1016/j.neuroscience.2003.08.028. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Taaid N, Halim N, Randolph E, Kim YK, Auerbach P. Hippocampal lesions enhance startle gating-disruptive effects of apomorphine in rats: A parametric assessment. Neuroscience. 2000b;96:523–536. doi: 10.1016/s0306-4522(99)00528-x. [DOI] [PubMed] [Google Scholar]
- Totterdell S, Meredith G. Topographical organization of projections from the entorhinal cortex to the striatum of the rat. Neuroscience. 1997;78:715–729. doi: 10.1016/s0306-4522(96)00592-1. [DOI] [PubMed] [Google Scholar]
- Vann SD, Brown MW, Erichsen JT, Aggleton JP. Using fos imaging in the rat to reveal the anatomical extent of the disruptive effects of fornix lesions. J Neurosci. 2000;20:8144–8152. doi: 10.1523/JNEUROSCI.20-21-08144.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velakoulis D, Pantelis C, McGorry PD, Dudgeon P, Brewer W, Cook M, Desmond P, Bridle N, Tierney P, Murrie V, Singh B, Copolov D. Hippocampal volume in first-episode psychoses and chronic schizophrenia: a high-resolution magnetic resonance imaging study. Arch Gen Psychiatry. 1999;56:133–141. doi: 10.1001/archpsyc.56.2.133. [DOI] [PubMed] [Google Scholar]
- Verwer RW, Meijer RJ, Van Uum HF, Witter MP. Collateral projections from the rat hippocampal formation to the lateral and medial prefrontal cortex. Hippocampus. 1997;7:397–402. doi: 10.1002/(SICI)1098-1063(1997)7:4<397::AID-HIPO5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Wan FJ, Caine SB, Swerdlow NR. The ventral subiculum modulation of prepulse inhibition is not mediated via D2 dopamine or nucleus accumbens non-NMDA glutamate activity. Eur J Pharmacol. 1996;314:9–18. doi: 10.1016/s0014-2999(96)00535-3. [DOI] [PubMed] [Google Scholar]
- Wan FJ, Swerdlow NR. Sensorimotor gating in rats is regulated by different dopamine-glutamate interactions in the nucleus accumbens core and shell subregions. Brain Res. 1996a;722:168 –176. doi: 10.1016/0006-8993(96)00209-0. [DOI] [PubMed] [Google Scholar]
- Wan FJ, Swerdlow NR. The basolateral amygdala regulates sensorimotor gating of acoustic startle in rats. Neuroscience. 1996b;76:715–724. doi: 10.1016/s0306-4522(96)00218-7. [DOI] [PubMed] [Google Scholar]
- Zavitsanou K, Cranney J, Richardson R. Dopamine antagonists in the orbital prefrontal cortex reduce prepulse inhibition of the acoustic startle reflex in the rat. Pharmacol Biochem Behav. 1999;63:55–61. doi: 10.1016/s0091-3057(98)00234-2. [DOI] [PubMed] [Google Scholar]
- Zhang J, Engel JA, Hjorth S, Svensson L. Changes in the acoustic startle response and prepulse inhibition of acoustic startle in rats after local injection of pertussis toxin into the ventral tegmental area. Psychopharmacology. 1995;119:71–78. doi: 10.1007/BF02246056. [DOI] [PubMed] [Google Scholar]
- Zhang W, Pouzet B, Jongen-Relo A, Weiner I, Feldon J. Disruption of prepulse inhibition following N- methyl-D-aspartate infusion into the ventral hippocampus is antagonized by clozapine but not by haloperidol: A possible model for the screening of atypical antipsychotics. Neuroreport. 2000;10:2533–2538. doi: 10.1097/00001756-199908200-00018. [DOI] [PubMed] [Google Scholar]