

Summary
Copolymers of N-isopropylacrylamide, 2-hydroxyethyl methacryl lactate and acrylic acid were prepared with varying mole ratios of monomers to develop copolymers with gelation properties above a certain concentration for a bioerodible, in-situ gelling material. The copolymers formed gels in situ under physiological condition. The gelation temperature of the copolymers decreased as the HEMA-lactate content of the copolymers increased due to the hydrophobicity of HEMA-lactate, and increased as the AAc content increased due to the hydrophilicity of AAc. The gels redissolve at 37 °C as their LCSTs increase above 37°C due to the hydrolysis of the HEMA-lactate pendant groups.
Keywords: bioerodible, gelation, injectable, PNIPAAm, stimuli-sensitive polymers
Introduction
Recently, injectable, in situ-forming devices have been intensively researched for tissue engineering and drug delivery. Biodegradable polymeric biomaterials with thermosensitive properties are particularly useful for these purposes because they are aqueous based and because their fabrication is so facile.[1] These biomaterials include poly(ethylene glycol)/poly(lactic acid-co-glycolic acid) (PEG/PLGA) based copolymers,[1a–1b] copolymers of methoxy poly(ethylene glycol) and poly(propylene fumarate),[1c] modified chitosan,[1d] and some poly(organophosphazenes).[1e]
Poly(N-isopropylacrylamide) (PNIPAAm) is a polymer with a LCST (lower critical solution temperature) of around 32 °C.[2] At temperatures below the LCST, the polymer is soluble in aqueous solution, preferring an expanded or hydrated coil conformation. Above this temperature, the polymer becomes insoluble, preferring a folded globule conformation. The phase transition occurs over a narrow temperature range and is reversible by lowering the temperature. The phase transition is due to a change in balance of hydrogen bonding and hydrophobic interactions. This polymer is easily injectable while below its LCST, as a solution in aqueous solution. However, this material is nonbiodegradable, although the temperature transition can be used to remove the material if the temperature can be lowered below the LCST.
To overcome this drawback, some groups have studied copolymers of NIPAAm with hydrolysable and proteolytically degradable side groups.[3] Shah et al.[3a] have reported on copolymers with NIPAAm and N-acryloxy succinimide, and Kim et al.[3b] have reported on injectable poly(N-isopropylacrylamide-co-acrylic acid) hydrogels with proteolytically degradable cross-links. Also, Neradovic et al. have studied NIPAAm-based copolymers including 2-hydroxyethyl methacryl lactate (HEMA-lactate), N-(2-hydroxypropyl)methacrylamide lactate (HPMAm-lactate) and PEG.[4] These NIPAAm-copolymers with hydrolysable moieties showed time-dependent LCST properties.[4a] Especially NIPAAm-based copolymer with above 35 mol-% HPMAm-lactate exhibited the cloud point above body temperature after hydrolysis and NIPAAm-based copolymers with HPMAM-lactate and PEG showed micelle properties.[4b] However, they have not yet reported in situ gelling properties of their copolymers.
The aim of this research is to make a NIPAAm-based, injectable, in situ-forming bioerodible copolymer by designing a time-dependent LCST into the polymer. Polymers with time-dependent LCST properties can be dissolved in aqueous buffers at temperatures below the LCST and will then gel upon injection at 37 °C (body temperature). These polymers will then redissolve when the LCST increases above 37 °C. In our previous report, by introducing acrylic acid into copolymers of NIPAAm and HEMA-lactate, copolymers with an LCST above 37 oC after hydrolysis were synthesized and characterized.[5] Now we study the detailed characterization and gelation properties of these polymers for in situ-forming, injectable, and bioerodible carriers.
Experimental Part
Materials
N-isopropylacrylamide (NIPAAm; Aldrich) was purified by recrystallization from n-hexane and dried under vacuum for four days. Acrylic acid (AAc; Aldrich) was distilled at 39°C/10mmHg. 2,2′-Azobisisobutyronitrile (AIBN; Aldrich 98 %) was purified by recrystallization from methanol. 2-Hydroxyethyl methacrylate (HEMA; Aldrich, 97 %), lactide ((3S-cis)-3,6-dimethyl-1,4-dioxane-2,5-dione, Aldrich 98 %) and stannous 2-ethylhexanoate (SnOct2, Aldrich) were used as received. Anhydrous 1,4-dioxane (Aldrich) and Tetrahydrofuran (Aldrich) were used as received. Other solvents used in this experiment were reagent grade and were used as received.
Synthesis of HEMA-lactate and poly(NIPAAm-co-HEMA-lactate-co-AAc)
HEMA-lactate was synthesized using the process in the previously reported literature. [5–6] The degree of polymerization (DP) of the lactate graft in HEMA-lactate was calculated from the ratio of the integrals of 1-ethylene and methine peaks at 5.6 and 5.1-5.3 ppm, respectively. The average degree of polymerization of lactate obtained was 2.3 and 2.5. Purity was above 93 %.
NIPAAm-based copolymers with varying feed ratios (NIPAAm/HEMA-lactate/AAc = 84:10:6, 79:15:6, 74:20:6, and 70:20:10) were synthesized by radical polymerization in dioxane or THF as in the literature.[5] In brief, the total monomer concentration was 0.1 g·mL−1 in 1, 4-dioxane (10 wt.-%). AIBN was used as an initiator (initiator/total amount of monomers = 7 × 10−3 mol/mol). Batches of approximately10 g were prepared. Nitrogen was bubbled through the solution at room temperature for 15 minutes prior to the addition of the initiator to reduce oxygen content in the polymerization reaction. The copolymerization was conducted at 65 °C for 20 h in a nitrogen atmosphere. Subsequently, the solvent was removed under reduced pressure. The copolymers were dissolved in acetone and precipitated in an excess of diethyl ether. The precipitated polymers were isolated by filtration and dried in a vacuum oven at 40 °C. 1H-NMR (CDCl3): δ = 5.2 (b, H10) 4.5 − 4.2 (H8, H9, H12), 4.0 (b, H3), 2.4 -1.2 (H1, H2, H6, H7, H11, H13, H14, H15), 1.1 (b, H4, H5).
Molecular weight and acrylic acid content
The molecular weights of the synthesized polymers were determined by gel permeation chromatography (Shimadzu HPLC) in conjunction with Static light scattering (Wyatt miniDawn® -Santa Barbara, CA). The copolymers were dissolved in THF at 3 mg·ml−1 and gel permeation chromatography (GPC) was performed with Waters ultrastyragel columns HR4® and HR6® in series. Spectrometric grade THF was used as the mobile phase flowing at 1 ml/·min-1.
The content of AAc was determined by titration. Each copolymer (0.1 g) was dissolved in 20 ml of deionized water by cooling at 4 oC and the solution was brought to room temperature and then titrated with 0.01N NaOH using phenolphthalein as an indicator. The amount of AAc in the copolymer was calculated from the amount of NaOH required for the neutralization. Samples were tested in triplicate.
Gelation properties
Differential Scanning Calorimetry (MCDSC, Calorimetry Sciences Corp, American Fork, Utah) was used to estimate the gelation temperature of all the copolymers. Each copolymer was dissolved in 0.1M Phosphate Buffered Saline (PBS) of pH 7.4 at the concentration of 21 wt.-% except copolymer 6 (25 wt.-%). The solution pH was adjusted to 7.4 with 0.1 N NaOH and 0.500 ± 0.0005 grams of the solution was placed in DSC ampoules and then tested in triplicate from 0 to 80 °C at 1 °C·min−1. 0.1 M PBS was used as the baseline. The temperature at the maxima of the DSC endotherms is reported as the sol-to-gel transition temperature for each copolymer.
Rheology
Quantification of the elastic or storage modulus for these materials was investigated using a TA Inst rheometer.1.26 ml of polymer solution was placed between parallel plate geometry with a 40 mm diameter and a gap of 1.0 mm at various temperatures. Frequency sweep studies were performed at 5, 22, and 37 °C, from 0.1 to 20 Hz, at an oscillation stress of 10 Pascal (Pa). Also, temperature sweep was performed from 5 to 45 °C with a heating rate of 1 °C·min−1. The data were collected under a controlled stress of 10 Pa and a frequency of 1 Hz. The sol to gel transition temperature was defined as the temperature at which storage modulus (G′) is equal to loss modulus (G″). Also the gelation temperature was defined as the point where the G′ and G″ are frequency independent.[7]
Hydroylsis
21 wt.-% solutions of poly(NIPAAm-co-HEMA-lactate-co-AAc) in 0.1 M PBS were prepared and incubated in a water bath at 37 °C. After the solution formed a shrunken gel, the supernatant liquid was replaced with the same amount of 0.1 M PBS of pH 7.4 on a daily basis to minimize pH change of the pH of the polymer solution during hydrolysis. Time-dependent hydrolytic behavior of the polymers was investigated by macrograph. After the polymer completed hydrolysis, some samples were taken, dialyzed in distilled water at 4 °C for 12 hours to remove salts and degradation products, and lyophilized for GPC. Complete hydrolysis was confirmed by the disappearance of the methine proton of the lactate.
Results and Discussion
Copolymers with different mole ratios (NIPAAm/HEMA-lactate/AAc) were prepared by free radical polymerization in Scheme 1. A list of the copolymers synthesized is found in Table 1. The compositions of the copolymers were calculated from the 1H-NMR spectra and by acid titration. The mole ratio of NIPAAm and HEMA-lactate was calculated from the integration ratio between the methyl proton (6H) of NIPAAm and the methine proton (1H) of HEMA-lactate appearing at 1.1 and 5.3 ppm, respectively, in Figure 1. The actual content of acrylic acid (AAc) was determined for each sample by acid titration. As shown in Table 1, preparation of the polymers in dioxane resulted in weight-average molecular weights in the range (1.1-7.8) × 105, while synthesis of copolymers in THF resulted in lower molecular weight less than 50,000 g·mol−1.
Scheme 1.

Synthesis of copolymers with NIPAAm, HEMA-lactate, and AAc.
Table 1.
Characteristics of poly(NIPAAm-HEMA-lactate-co-AAc) copolymers.
| N | Feed ratio (mol %) | Mol ratio (mol %) | Gel Temperature (oC)a) | MW (×10−5) | |||||
|---|---|---|---|---|---|---|---|---|---|
| NIPAAm | HEMA – lactate | Acrylic acid | NIPAAm | HEMA – lactate | Acrylic Acid | DSC | Rheology | ||
| 1 | 84 | 10 | 6 | 84.4 | 9.4 (dp=2.3) | 6.2 ± 0.1 | 33.6 ± 0.3 | 34 | 2.3 |
| 2 | 79 | 15 | 6 | 81.7 | 12.3 (dp=2.3) | 6.0 ± 0.1 | 26.8 ± 0.4 | 28 | 2.4 |
| 3 | 74 | 20 | 6 | 75.8 | 18.5 (dp=2.3) | 5.7 ± 0.1 | 22.4 ± 0.2 | 24 | 1.6 |
| 4 | 79 | 15 | 6 | 80.8 | 13.5 (dp=2.5) | 5.8 ± 0.1 | 26.9 ± 0.3 | 24 | 7.8 |
| 5 | 70 | 20 | 10 | 71 | 19.2 (dp=2.3) | 9.8 ± 0.1 | 27.1 ± 0.6 | 28 | 1.1 |
| 6 | 74 | 20 | 6 | 73.6 | 20.0(dp=2.3) | 6.4 ± 0.1 | 23.6 ± 1.0 | 22 | 0.4 |
Each copolymer was prepared in 0.1M Phosphate Buffered Saline (PBS) at the concentration of 21 wt.-% except copolymer 6 (25 wt.-%).
Figure 1.
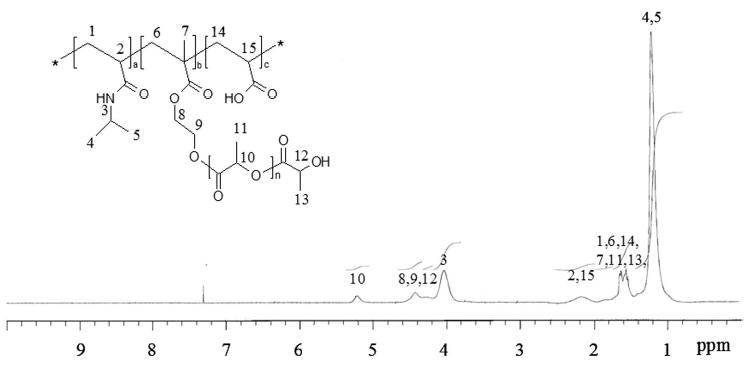
1H-NMR spectrum of copolymer 1
Gelation properties
Some thermosensitive polymers,[8] such as methylcellulose,[8a,8b] Pluronic or Poloxamer (polyethylene oxide(PEO)-polypropylene oxide(PPO)-polyethylene oxide(PEO)),[8c] and poly(ethylene glycol)/poly(lactic acid-co-glycolic acid) (PEG/PLGA) based copolymers,[1a–1b] and some polyphosphazene gels,[1e] undergo a sol-gel transition (thermally reversible gelation) upon heating. The sol to gel transition temperature of the thermal gelling materials can be determined from methods such as dynamic mechanical analysis, differential scanning calorimetry (DSC), the test tube inverting method, and the falling ball method.[8b,9] In this paper, DSC and dynamic mechanical analysis were employed to determine the gelation temperature. When the copolymer solution formed a gel in the heating process, an endothermic peak observed by DSC was regarded as the transition temperature. Further, the storage modulus (G′) and loss modulus (G″) were measured by dynamic mechanical analysis. The gelation temperature was defined as the temperature of crossover between G′ and G″ (G′=G″) of the copolymer. A gel (elastic solid) can be defined when G′ is higher than G″ as storage modulus (G′) is dominant at gel phase while loss modulus is dominant at the sol phase.
The gelation temperatures of the copolymers are presented in Table 1. The gelation temperatures obtained from DSC and dynamic mechanical analysis showed a similar trend and did not differ significantly. All the copolymers exhibited a gelation temperature below body temperature. Generally, the gelation temperature of thermosensitive polymers is largely affected by the hydrophobicity of comonomers.[le] The gelation temperature of the copolymer solutions (21 wt.-%) decreased as the content of the hydrophobic HEMA-lactate was raised. The trend between HEMA-lactate content and the transition temperature can be seen in data obtained from DSC. As the content of HEMA-lactate (dp=2.3) increased in the polymer in the order 1 (9.4 mol-%) < 2 (12.3 mol-%) < 3 (18.5 mol-%), the gelation temperature of copolymers decreased in the polymer in the order 1 (33.6 oC) > 2 (26.8 oC) > 3 (22.4 oC) for polymers in PBS (pH 7.4) at a concentration of 21 wt %. On the other hand, acrylic acid as a hydrophilic monomer raised the gelation temperature. Copolymer 3, with around 6 mol-% of AAc, formed a gel at 22.4 oC while copolymer 5, with around 10 mol-% AAc, gelled at 27.1 oC.
Usually polyNIPAAm in aqueous solution undergoes a sol to precipitate transition when there is not sufficient concentration for significant chain entanglement. At higher concentrations, polyNIPAAm undergoes a sol to shrunken gel transition when the temperature is raised above its LCST. Bae et al. determined that poly(N-isopropylacrylamide-co-acrylic acid)(Mw~1,000,000) which was synthesized in benzene underwent a sol to gel transition above a critical concentration in aqueous solution.[10] Interestingly, our copolymers with NIPAAm, HEMA-lactate, and AAc synthesized in 1,4-dioxane or THF exhibited the sol-to-gel transition at sufficient concentration. Even copolymer 6 with its lower molecular weight (380000 ≅ 0.4 × 10 5 g·mol−1) underwent the sol-to-gel transition at concentrations above 25 wt.-%.
The temperature dependence of the dynamic moduli of 21wt.-% solution of polymers is shown in Figure 2 (a). The crossover point corresponds to the gelation point. The gelation temperature of the copolymers decreased as the content of hydrophobic HEMA-lactate increased, similar to the result obtained from DSC. The gelation temperatures of copolymers 1 (9.4 mol-%), 2 (12.3 mol-% HEMA-lactate), and 4 (18.5 mol-% HEMA-lactate) were 34, 28, and 24 oC, respectively. Also, solutions of copolymers with higher content of HEMA-lactate had higher initial G′ and G″ values and also exhibited lower onset temperatures in their endothermic transition, as seen in Figure 2 (b), indicating that hydrophobic HEMA-lactate made polymer solutions that might be partially aggregated by hydrophobic interaction even at temperatures below the gelation temperature.
Figure 2.

(a) Temperature dependence of the dynamic moduli of 21 wt % solution of copolymers 1, 2, and 3 in 0.1 M PBS of pH 7.4 at frequency (1 Hz). (b) DSC thermograms of copolymers 1, 2, and 3 in 0.1 M PBS of pH 7.4 (heating: 1 oC/min).
The frequency dependence of the dynamic moduli for copolymer 4 (21 wt.-% solution) at 5, 22, and 37 oC is presented in Figure 3. At 5 oC, the loss modulus, G″, was larger than the storage modulus, G′, in the frequency range 0.1 to 20 Hz. At 22 oC, G″ was larger than G′ when the frequency was less than approximately 3 Hz; G′ was larger than G″ when frequency was greater than 3 Hz. G′ and G″ were dependent upon frequency at the above temperatures. At 37 oC (body temperature), G′ was much higher that G″ in the whole frequency range and the curves became nearly parallel, indicating that the polymer solution was an elastic solid at this temperature. This result is consistent with the finding that the dynamic moduli of the polymers above the gel temperature are almost independent of frequency for other thermo-gelling systems.[11]
Figure 3.
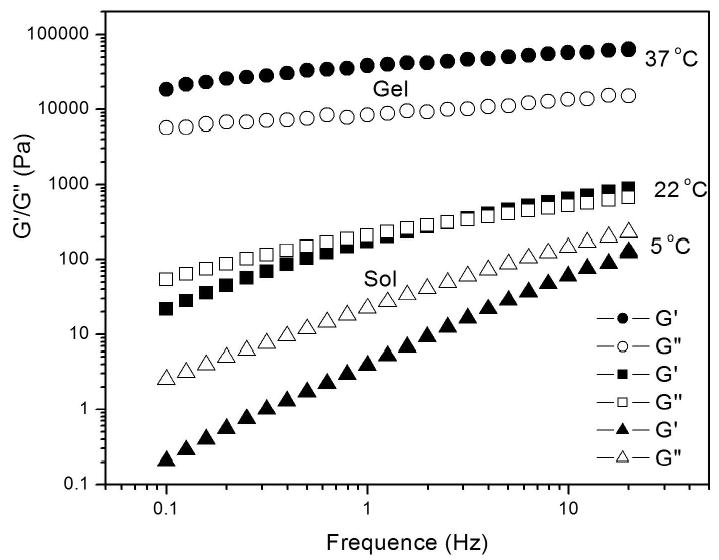
The frequency dependence of the dynamic moduli for copolymer 4 at 5, 22 and 37 oC.
Figure 4 shows the complex viscosity change for 15, 16 and 17 wt.-% aqueous solutions of copolymer 2 in 0.1 M PBS of pH 7.4 as a function of temperature. The gelation behavior of the polymers was dependent on their concentration in solution. As the concentration of the polymer solutions increased, so did the sharpness of the curves and the magnitude of their viscosity. The viscosity of each copolymer increased gradually up to around 19 oC, at which point the solution viscosity increased abruptly at temperatures above 22 oC for solutions with concentrations higher than 16-wt%. However, the viscosity of the 15 wt.-% solution of copolymer 2 decreased, implying that critical gelation concentration was 16 wt.-% for copolymer 2. This suggests that, below the critical gelation concentration, the polymer may undergo a sol to precipitation transition instead of a sol to gel transition.
Figure 4.
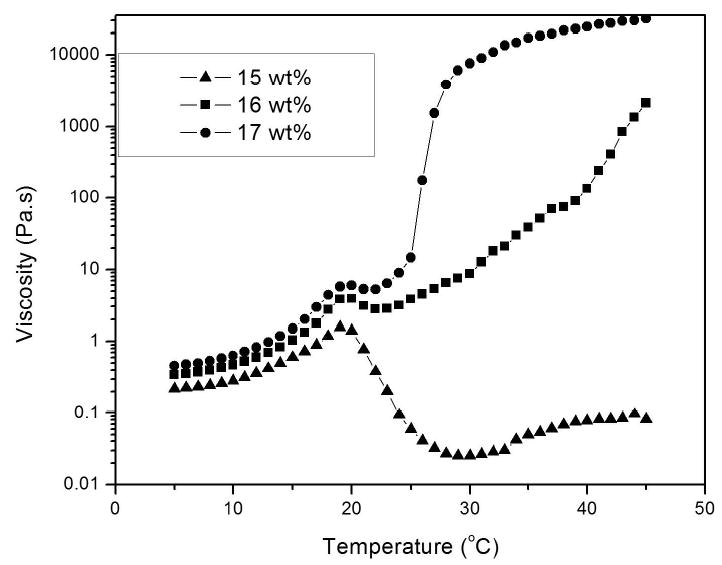
Complex viscosity change of 15, 16, and 17 wt % aqueous solutions of copolymer 2 in 0.1 M PBS of pH 7.4 as a function of temperature.
Hydrolysis
Figure 5 photographically demonstrates the sol-gel transition of copolymer 4. Copolymer 4 was dissolved at a concentration of 15 wt.-% in 0.1 M PBS solution of pH 7.4. As it was taken out of the cold chamber at 4 oC, the polymer solution was in a transparent sol state, as shown in Figure 5 (a). In contrast to this, after the temperature jumped from 4 to 37 oC, the polymer solution was in an opaque gel state (b) in-situ without syneresis. After 1-day incubation at 37 oC, the copolymer gel became a shrunken gel (c). The shrunken gel gradually transitioned into a transparent sol after 6 days. Previous work indicates that the LCST of the copolymer increased above 37 oC due to hydrolysis of HEMA-lactate during this same time period.[5]
Figure 5.

Demonstration photographs of the sol-gel transition of copolymer 4 at 15 wt % solution in 0.1 M PBS of pH 7.4 during the incubation of 6 days at 37 oC: (a) at 4 oC, (b) at 37 oC, (c) after 1 day at 37 oC, (d) after 2 days at 37 oC, (e) after 4 days at 37 oC, (f) after 6 days at 37 oC.
Figure 6 shows GPC chromatograms of copolymer 6 before and after complete hydrolysis. After complete hydrolysis, the GPC chromatogram showed a decrease in polydispersity index (PDI) from 2.2 to 1.4 upon hydrolysis, indicating that the polymer with the long side chain (HEMA-lactate) became much more linear after hydrolysis of HEMA-lactate. Before hydrolysis, the polydispersity is affected by both the composition and degree of polymerization, after hydrolysis, the affect of the composition on the polydispersity is reduced and the degree of polymerization becomes the main determinate of the polydispersity. Finally, the curve was obviously shifted from the molecular region (Mw= 38,000 g·mol −1) to the lower molecular region (26,000 g·mol −1) resulting from hydrolysis of HEMA-lactate. This suggests that these polymers can be cleared by the kidney when they become soluble because the molecular weight is below 40,000 g·mol −1.[12]
Figure 6.
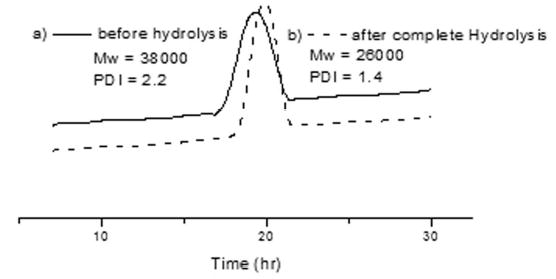
GPC Chromatograms of copolymer 6 (a) before hydrolysis (b) after complete hydrolysis.
Our previous report said that the present polymers with 6 mol % AAc exhibited a LCST beyond 37 oC after complete hydrolysis of HEMA-lactate regardless of the content of HEMA-lactate and that the time that it took for LCST values of the polymers to reach body temperature (37 oC) due to hydrolysis was dependent on the content of HEMA-lactate of the polymers: about 1 day for copolymer 1 (9.4 mol-% of HEMA-lactate), about 6 days for copolymer 2 (12.3 mol-% of HEMA-lactate), and about 10 days for copolymer 3 (18.5 mol-% of HEMA-lactate).[5] Polymers with time-dependent LCST properties, such as these, can be dissolved in aqueous buffer solutions at a temperature below their LCST. After temperature is raised to 37 oC, if the polymers are at a sufficient concentration, they can form gels in-situ and gradually redissolve as their LCST increased due to hydrolysis. This result might suggest that these copolymers have potential as an injectable drug carrier.
Conclusion
Copolymers with NIPAAm, HEMA-lactate and AAc were synthesized in dioxane and THF and their gelation temperatures were measured by DSC and rheometry. All copolymers had gelation properties above a critical concentration under physiological conditions. Even copolymers with a molecular weight less than 50,000 g·mol −1 formed gels at concentrations above 25 wt.-%. The gelation temperature of the copolymers depended on the hydrophobic-hydrophilic balance of comonomers. The formed gels redissolved as their LCSTs were raised above 37 oC owing to the hydrolysis of the HEMA-lactate. The copolymer with a molecular weight less than 50,000 g·mol −1 synthesized in THF may be useful as an injectable bioerodible material for applications such as drug delivery.
Acknowledgments
This work was funded by the National Institute of Health, grant number 5 R01 GM065917-02.
References
- 1.[1a] Jeong B, Bae YH, Lee DS, Kim SW.Adv Drug Del Rev 20025437. [DOI] [PubMed] [Google Scholar]; [1b] Zentner GM, Rathi R, Shih C, McRea JC, Seo MH, Oh HS, Rhee BG, Mestecky J, Moldoveanu Z, Morgan M, Weitman S.J Controlled Release 200172203. [DOI] [PubMed] [Google Scholar]; [1c] Behravesh E, Shung AK, Jo S, Mikos AG.Biomacromolecules 20023153. [DOI] [PubMed] [Google Scholar]; [1d] Chenite A, Chaput C, Wang D, Combes C, Bushchmann MD, Hoemann CD, Leroux JC, Atkinson BL, Binette F, Selmani A.Biomaterials 2000212155. [DOI] [PubMed] [Google Scholar]; [1e] Lee BH, Song SC.Macromolecules 2004374533 [Google Scholar]
- 2.Yoshida R, Kaneko Y, Sakai K, Okanno T, Bae YH, Kim SW. J Controlled Release. 1994;32:96. [Google Scholar]
- 3.[3a] Shah SS, Wertheim J, Wang CT, Pitt CG.J Controlled Release 19974595 [Google Scholar]; [3b] Kim S, Healy KE.Biomacromolecules 200341214. [DOI] [PubMed] [Google Scholar]
- 4.[4a] Neradovic D, Hinrichs WLJ, Kettenes-van den Bosch JJ, Hennink WE.Macromol Rapid Commun 199920577 [Google Scholar]; [4b] Neradovic D, van Nostrum CF, Hennink WE.Macromolecules 2001347589 [Google Scholar]
- 5.Lee BH, Vernon B. Polym Int. 2005;54:418. [Google Scholar]
- 6.van Dijk-Wolthuis WNE, Tsang SKY, Kettenesvan den Boshch JJ, Hennink WE. Polymer. 1997;38:6235. [Google Scholar]
- 7.Winter HH, Chambon F. J Rheol. 1986;30:367. [Google Scholar]
- 8.[8a] Takahashi M, Shimazaki M, Yamamoto J.J Poly Sci, Part B: Polym Phys 20013991 [Google Scholar]; [8b] Xu Y, Li L, Zheng P, Lam YC, Hu X.Langmuir 2004206134. [DOI] [PubMed] [Google Scholar]; [8c] Cohn D, Sosnik A, Levy A.Biomaterials 2003243707. [DOI] [PubMed] [Google Scholar]
- 9.Chung YW, Simmons K, Gutowska A, Jeong B. Biomacromolecules. 2002;3:511. doi: 10.1021/bm0156431. [DOI] [PubMed] [Google Scholar]
- 10.Han CK, Bae YH. Polymer. 1998;39:2809. [Google Scholar]
- 11.[11a] Nordby MH, Kjøniksen A, Nyström B, Roots J.Biomacromolecules 20034337. [DOI] [PubMed] [Google Scholar]; [11b] Carlfors J, Edsman K, Petersson R, Jörnving K.European Journal of Pharmaceutical Sciences 199861135. [DOI] [PubMed] [Google Scholar]
- 12.Seymour LW, Duncan R, Strohalm J, Kopexek J. J Biomed Mater Res. 1987;21:1341. doi: 10.1002/jbm.820211106. [DOI] [PubMed] [Google Scholar]