

Abstract
Structural allograft healing is limited because of a lack of vascularization and remodeling. To study this we developed a mouse model that recapitulates the clinical aspects of live autograft and processed allograft healing. Gene expression analyses showed that there is a substantial decrease in the genes encoding RANKL and VEGF during allograft healing. Loss-of-function studies showed that both factors are required for autograft healing. To determine whether addition of these signals could stimulate allograft vascularization and remodeling, we developed a new approach in which rAAV can be freeze-dried onto the cortical surface without losing infectivity. We show that combination rAAV-RANKL- and rAAV-VEGF-coated allografts show marked remodeling and vascularization, which leads to a new bone collar around the graft. In conclusion, we find that RANKL and VEGF are necessary and sufficient for efficient autograft remodeling and can be transferred using rAAV to revitalize structural allografts.
In contrast to soft tissue organ transplantation (i.e., heart, liver, kidney), which must be live from a histocompatible donor, structural musculoskeletal grafts (i.e., bone, ligament) are often derived from allogenic cadavers. Although this convenience makes structural allografts readily available, the utility of these transplants is limited by their lack of viability. This is most evident from experimental and clinical studies showing that fresh vascularized autogenous grafts are vastly superior to allograft in terms of healing and remodeling1,2. Structural bone grafts used to heal critical defects and bone fusions undergo a repair and remodeling process that closely resembles fracture healing3. In live autograft healing, cells from both the graft and the host contribute to mediate bony union4,5. In contrast, healing of a diaphyseal defect that has been allografted can only be accomplished by invasion of the graft by host tissue to obtain a cortex-to-cortex union6. Following union, autografts continue to remodel as a result of osteoclastic resorption of necrotic or disused cortical bone that is followed by osteoblastic formation of new woven bone, which is later remodeled into stronger lamellar bone. In this way, autografts are sustained through normal bone homeostasis. In contrast, once creeping callus from the host calcifies on the cortex of an allograft, healing ceases, leaving a large segment of necrotic bone that is unable to respond to normal mechanical loading. Thus, structural allografts have a limited life span because microfractures that occur in them over time cannot be remodeled and repaired, and negative outcomes include a 25–35% failure rate from infection, nonunion and fracture7,8.
Two central issues that must be addressed to improve structural allografting are elucidation of the factors that facilitate autograft healing and are absent in allografts and a method to introduce these factors onto allografts. Toward resolving these issues, we have developed a mouse femoral model that faithfully recapitulates the central features of clinical structural bone grafting. Gene expression profiling studies showed that allografts are deficient in several factors known to regulate vascular ingrowth of skeletal elements, osteogenesis, bone resorption and remodeling. The two most notable factors were vascular endothelial growth factor (VEGF) and receptor activator of nuclear factor κB ligand (RANKL), which are known to dominantly regulate angiogenesis9 and osteoclastic bone resorption10, respectively, during skeletal repair. Specifically, VEGF is expressed by the perichondrium and hypertrophic chondrocytes and recruits endothelial cells to promote blood flow to the avascular tissue11. In addition to essential nutrients, this blood supply brings in osteoclast precursors that differentiate in response to RANKL expressed by stromal cells12. Based on this information we used in vivo blockade and ex vivo gene transfer to show that RANKL and VEGF are necessary for complete autograft healing. These findings support the hypothesis that RANKL and VEGF are crucial factors for establishing remodeling of the cortical surface of the autografts and that introduction of these factors onto allografts could result in bone resorption, neovascularization and revitalization of the dead bone. Using a new approach to immobilize recombinant adeno-associated virus (rAAV) onto the cortical surface of the allografts we show that RANKL and VEGF signals are sufficient to revitalize processed cortical bone and could be a method to sustain clinical allografts long term.
RESULTS
Mouse femoral autograft and allograft healing
To elucidate the cellular and molecular mechanisms that govern structural bone graft healing, we developed an in vivo mouse model in which an ∼4-mm osteotomy of the middle of the femoral diaphysis is performed and placed back into the original host as an autograft or ‘processed’ with fixation in alcohol and freezing and then transplanted as an allograft into an allogenic host13. Figure 1 shows the radiographic and histologic healing of the two grafts. Consistent with current knowledge, the autografts heal through endochondral bone formation at the junctions with concomitant intramembranous bone formation derived from the periosteum of the cortex of the graft. This bone formation results in a new bone collar of cortical bone that partly or completely encircles the graft by 4 weeks. During this time, a new marrow space is created between the new bone collar and the autograft, and accelerated osteoclastic resorption of the graft occurs. In contrast, allografts heal by endochondral bone formation only. At 2 weeks, cartilage derived from the host is observed creeping onto the ends of allografts. Notably, the osteocartilaginous tissue seems to be separated from the periosteum by a fibrous tissue reaction that partly or completely encases the allograft as part of a foreign body reaction to it. By 4 weeks, healing is completed as a new cortical union at the graft-host junctions with a large middle segment of necrotic bone that is completely devoid of osteoclast activity.
Figure 1.
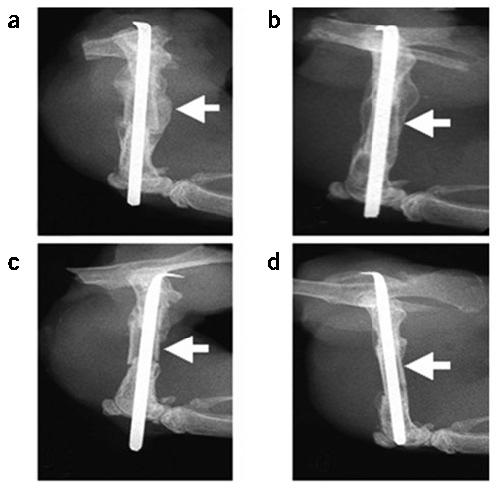
The mouse femoral allograft model. Mice received a femoral autograft or allograft, and were killed at 3 weeks (a,c), 4 weeks (b,d,f,h) or 2 weeks (e,g). Representative radiographs from an autografted (a,b) and an allografted (c,d) mouse are illustrated at 3 and 4 weeks after fracture. The arrows indicate the presence of callus on the cortical surface of the autograft at 3 weeks (a), which is remodeled by 4 weeks (b), and is completely absent in the allograft (c,d). Hematoxylin, eosin, orange G and acian blue–stained sections show the endochondral bone formation at the graft-host junctions (arrow heads) of both auto and allografts at 2 weeks (e,g), which is remodeled to form a bony union at 4 weeks (f,h). Of note is the periosteal intramembranous bone formation (*), which only occurs in autografts (e), producing a new cortical bone collar with bone marrow at four weeks (f). In contrast, allografts are encased by fibrous tissue (#), heal through creeping callus (g), and are dependent on dead cortical bone for structural integrity after remodeling (h).
Allografts are deficient in RANKL and VEGF
Over the last few years a wealth of information on the factors that regulate bone repair has been generated from microarray gene expression profiling studies on fracture callus tissue14. Based on this information, we performed a screen to identify dysregulated gene expression between autografts and allografts by RT-PCR. The two factors that showed the most substantial differential gene expression were Tnfsf11, which encodes RANKL, and Vegfa, which encodes VEGF (Fig. 2). The expression of both factors peaked 10 d after autografting, when Tnfsf11 and Vegfa levels were twofold and fivefold higher than those observed in allograft tissue, respectively. Whereas Tnfsf11 expression was deficient throughout the time course, Vegfa expression seemed to be delayed and peaked on day 14 when endochondral ossification is largely completed in stabilized fractures15. To follow up these findings, we performed microarray studies using RNA isolated from day 10 autografts and allografts, and found, according to the manufacturer's criteria, that these transcripts were present in the autografts and absent in the allografts.
Figure 2.

Altered Tnfsf11 and Vegfa gene expression during allograft healing. Total RNA was extracted from femoral autografts and allografts at the indicated time and processed for real time RT-PCR. The data are presented as the fold induction ± s.d., compared to the day 0 control, after standardization with the internal β-actin control. *P < 0.05 for autograft versus allograft at the same time point.
RANKL and VEGF in autograft healing
To assess the requirements of RANKL and VEGF signaling during autograft healing, we performed in vivo blockade experiments using systemic and local approaches. First, the autografted mice received control IgG, a soluble RANK decoy receptor (RANK:Fc) or neutralizing antibodies specific for VEGF (Fig. 3a-d). We also assessed the effects of blocking RANKL and VEGF locally by transducing the autografts ex vivo before implantation with AAV-β-gal, AAV- osteoprotegerin (OPG), AAV-sFlt1 (soluble Flt1, the receptor for VEGF) or a combination of AAV-OPG and AAV-sFlt1 (Fig. 3e-g). Radiographic and histologic analyses of the autograft healing showed that disruption of either RANKL or VEGF signaling, systemically or locally, significantly (P < 0.05) inhibited new bone formation on the cortical surface of the grafts. Notably, dual blockade did not induce additional inhibitory effects, indicating that these factors act in series to recruit and differentiate osteoclast progenitors to the cortical surface.
Figure 3.

Systemic and local loss of either RANKL or VEGF results in defective autograft healing. Mice received untreated autografts followed by injections of control IgG (a), RANK:Fc (b), or anti-VEGF (c) therapy, and were killed four weeks later. Representative hematoxylin and eosin–stained sections from these mice show a reduction in the amount of new bone formation around the autographs (arrow heads) and persistence of cartilage (blue in b,c). Representative radiographs from mice that received autografts transduced with rAAV-β-gal (e) or a combination of rAAV-OPG and rAAV-sFlt1 (f), 2 weeks after fracture are shown. Histomorphometry of the area of new bone formation on the autografts (d,g). *P < 0.05 compared to the IgG or rAAV-β-gal controls.
Transduction efficiency of freeze-dried rAAV
With the hypothesis that addition of crucial signals to the cortical surface of allografts will lead to autograft-like healing, we attempted to develop an approach to efficiently transfer these signals. Previously, a gene-activated matrix was developed for this purpose, in which naked plasmid DNA is immobilized onto osteoinductive materials16. Unfortunately, others and we have been unable to achieve effective transduction efficiencies in our models using gene-activated matrices. Based on the empirical advantages of rAAV vectors for orthopedic gene therapy17, and the clinical potential of this vector18, we evaluated the effects of freeze-drying and storage at −80 °C on rAAV transduction efficiency. Resuspension of rAAV in a sorbitol solution facilitates its application to various organic and inorganic implant materials (Fig. 4a,b). Application of the freeze-dried rAAV-β-gal to a monolayer of 293 human embryonic kidney cells leads to efficient transduction as indicated by X-gal staining of the culture plates, which showed a mosaic distribution of blue cells throughout the plates (Fig. 4c,d). No staining was detected in control cultures without virus (Fig 4e). This result suggests that the rAAV rehydrates and freely diffuses in the culture medium before infecting the cells. To assess the effects of freeze-drying and storage on the immobilized rAAV, 5 × 107 transducing units of rAAV-β-gal were directly placed on a monolayer of 293 cells, as a positive control; or freeze-dried onto pins and stored at −80 °C for various times before addition to the monolayer (Fig. 4f). Notably, we were able to recover ∼100% of the β-galactosidase activity in all of the samples. Thus, this coating process does not affect the infection capacity of the virus.
Figure 4.
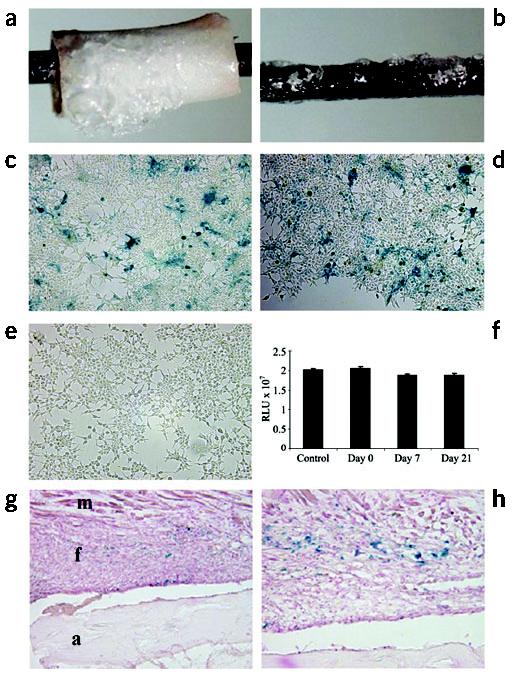
Transduction efficiency of rAAV-β-gal following freeze-drying onto allografts and implants in vitro and in vivo. 5 × 107 transducing units of rAAV-β-gal was lyophilized onto mouse femoral allografts (a) or stainless steel pins (b). The transduction efficiency was determined in vitro by incubating the coated pins on top of a monolayer of confluent 293 human embryonic kidney cells for 72 h. Photographs of the X-gal-stained cells distal (c) and proximal (d) to the coated pin, as well as an uncoated control pin (e) are shown. The transduction efficiency of the coated pins was also quantified after the indicated storage time at −80 °C. RLU, relative light units. (f). As a control, 5 × 107 transducing units of rAAV-β-gal in 50 μl PBS was directly placed on a monolayer of 293 cells. The β-galactosidase activity in the cultures was determined using the Galacto-Light system. No significant differences were observed. The efficiency of in vivo transduction 14 d after transplantation is shown at ×10 (g) and ×40 (h) magnification, where the blue staining indicates transduction of the fibroblasts (f) between the allograft (a) and the muscle (m).
To assess the transduction efficiency of freeze-dried rAAV-β-gal in vivo, we performed a dose-response experiment in which we coated femoral allografts with various doses of virus and transplanted them into mice. X-gal staining of histological sections showed that fibroblasts in the inflammatory tissue between the bone and muscle were readily transduced (Fig. 4g,h). The number of blue cells per section peaked at a dose of 5 × 107 particles/allograft. Thus, we used this as our effective dose in our gain-of-function studies.
Revitalization of rAAV-RANKL and rAAV-VEGF coated allografts
To evaluate the effect of exogenous RANKL and VEGF on processed cortical bone healing, allografts were coated with freeze-dried rAAV-β-gal, rAAV-RANKL, rAAV-VEGF or a combination of rAAV-RANKL and rAAV-VEGF. First, we confirmed the in vivo target gene expression following transplantation by determining serum levels of RANKL and VEGF over time (Fig. 5a). By day 4 a substantial increase in VEGF was detected, which peaked at day 8 before returning to baseline levels at 3 weeks. Analysis of RANKL did not detect levels above the detection limit (∼30 pg/ml) in any of our samples.
Figure 5.

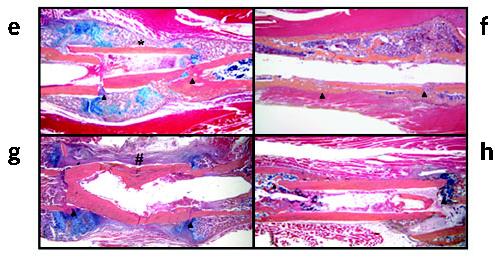
Revitalization of processed allografts via rAAV mediated-RANKL and VEGF gene transfer. Allografts containing 5 × 107 particles of rAAV-β-gal, rAAV-RANKL, rAAV-VEGF or a combination of rAAV-RANKL and rAAV-VEGF were transplanted into mice and evaluated 28 d after surgery. In vivo VEGF expression was analyzed in sera taken from the combined coated allografts at the indicated time after surgery (a). VEGF levels in uncoated allografts were consistently >50 pg/ml throughout the time course. Representative histology from the medial segment of the lateral cortex of a rAAV-β-gal (b) and rAAV-RANKL + rAAV-VEGF (c) coated allograft on day 28. Of note is the considerable amount of new bone on the rAAV-RANKL + rAAV-VEGF–coated allograft highlighted by a reversal line (arrows) and its similarity in cortical thickness to the rAAV-β-gal coated allograft. The new bone that formed on the allografts was quantified by histomorphometry (d) and the data are presented as the area of new bone formation on the graft ± s.d. (*P < 0.05 versus β-gal control).
Next, we histologically analyzed the treatment effects on allograft healing. Although there were no obvious effects of the first three treatments based on the appearances of the allografts, we observed a marked amount of live new bone on the periosteal surfaces, and focally on the endosteal surfaces of the rAAV-VEGF + rAAV-RANKL− coated allografts that was never observed in uncoated allografts in this model (Fig. 5b-d). The presence of the new bone on the outer surfaces of these allografts outside irregular reversal lines suggested that it had been laid down at sites of previous resorption of the allograft periosteal bone (Fig. 5b), showing that regions of the allografts have been partially resorbed and replaced by new bone, whereas others were still being resorbed at the time of killing of the mice as a result of the combined therapy. They include: (i) resorption of up to 50% of the thickness the allograft cortical bone, predominantly from the periosteal surface, and replacement of the resorbed dead bone with viable new bone (Fig. 5c and Fig. 6e,f), which in some cases extended the entire length of the allograft; (ii) reversal lines that clearly delineate the depth of dead allograft resorption and the sites of new bone formation (Fig. 6e,f), and indicate the location of a new bony union between the live host bone and the allograft surfaces; (iii) continuing active resorption of the dead cortical bone (Fig. 6b,d,g) with new bone formation (Fig. 6c,d,g) representative of tunneling remodeling, which does not occur physiologically in mouse cortical bone; (iv) neovascularization of the marrow cavity along the length of the allograft (Fig. 6e,f), in contrast to the necrotic marrow seen in samples from the other 3 groups (Fig. 6h); and (v) the complete absence of the fibrotic tissue reaction that typically surrounds allografts (Fig. 1g, Fig. 4g,h and Fig. 6h) as evidenced by the thin periosteal layer between the muscle and cortical bone (Fig. 6a,f).
Figure 6.

rAAV-mediated gene transfer of RANKL and VEGF induces cortical bone resorption, vascularization and remodeling in processed allografts in vivo. Representative TRAP-stained histologic sections from mice in the combination group (a–d). An example of a rAAV-VEGF + rAAV-RANKL–coated allograft in which remodeled bone extends the entire length of the graft (arrow heads) is shown (a). The novel histologic features of the combination group were characterized by osteoclastic resorption of the necrotic bone (black arrows in b,d,g), osteoblastic new bone formation in the resorption lacunae (white arrows in c,d) and osteoclastic remodeling of the new woven bone (yellow arrows in d). Hematoxylin and eosin–stained sections of allografts from the combination group revealed asymmetric reversal lines (dashed line in e and shown at higher magnification without the lines in g, and black arrow in c) between dead bone and newly formed live bone, new blood vessel formation inside the marrow cavities (* in e,f), and active tunneling resorption (arrows in f) in the necrotic bone. In contrast, none of the other groups showed these features and were all characterized by a fibrotic tissue (f) that covered the periosteal surface and necrotic tissue (n) that filled the marrow cavity (h).
Considering that none of these observations was made in any of the other groups of allografted mice we have studied (n >300), our findings provide evidence that the combination of exogenous RANKL and VEGF can induce vascularization and remodeling of processed structural allografts. This impression was supported by histomorphometry, which showed that the mean cortical thickness in the rAAV-VEGF + rAAV-RANKL–coated allografts was similar to that of the allografts that did not receive the RANKL-VEGF combination (Fig. 5b), suggesting that this new bone must have followed the removal of up to ∼100 μm of cortical bone. Consistent with the idea that RANKL and VEGF function in series during cortical graft healing, we did not observe any substantial effects of transferring either one of these factors alone. Although the resorption and new bone formation was observed in all of the mice given rAAV-VEGF + rAAV-RANKL –coated allografts, the amount and extent of resorption and new bone formation were variable, as evidenced by the observation that parts of some of the grafts were being resorbed for the first time at the time of killing, 4 weeks after surgery.
DISCUSSION
Although major progress has been made in many aspects of musculoskeletal repair procedures19,20, including the use of bone morphogenetic proteins as adjuvants for spinal fusion and fracture union21,22, processed structural allografts and nonremodeling bone substitutes remain the materials of choice for reconstructive orthopedic surgery. Although bone morphogenetic proteins represent a great advance for these indications, it has long been recognized that they are not useful for large critical defects because of their short half-life. As an alternative, many groups have been working on gene therapy approaches for skeletal healing23-27. Although gene therapy offers the potential of local, sustained gene expression, the development of a safe and effective delivery vector remains elusive28.
Over the last two decades, much work has been done to understand the critical differences between the efficacy of live autografts and processed allografts during bone healing5,29,30. Although these studies show that there is a host-acquired immune response to the allograft, it is clear that the most notable difference between the two grafts is the presence of live cells in the autograft, which directly contribute to angiogenesis and subsequent remodeling. We have addressed two central questions to advance our understanding of these differences: what prominent signals are present in autografts and absent in allografts that induce revascularization and remodeling and can introduction of these signals onto the cortical surface of structural allografts induce revascularization and remodeling? The first notable finding with the mouse femoral graft model was that bone morphogenetic protein expression is not substantially different between autografts and allografts. This result is somewhat contrary to the current thinking that introducing osteogenic signals to an osteoinductive and osteoconductive biomaterial is the best approach to improve bone healing. In contrast, our findings led us to explore an alternative hypothesis that stimulation of resorption of the graft through angiogenesis and osteoclast formation and activation leading to new bone formation on the cortical surface of allografts is a superior method to improve graft incorporation. In support of this hypothesis, we show that disruption of RANKL and VEGF signaling results in a decrease in new bone formation on the autograft cortical surface (Fig. 3).
To evaluate gain of RANKL and VEGF function in our model, we developed a technique in which rAAV can be immobilized to the cortical surface by freeze-drying (Fig. 4). There are many potential methods by which rAAV could be immobilized onto the allografts, including simple electrostatic interactions and more sophisticated bonding. Here we chose virus freeze-drying because of its ease and practicality. Although this method does not alter the infectivity of the virus and allows for effective transduction, the overall in vivo transduction efficiency is modest (1–5% of cells in immediate proximity to the allograft). It is likely that this low efficiency combined with transduction of cells that are rapidly turning over resulted in undetectable levels of RANKL and transient elevation of VEGF concentrations in the blood of the mice (Fig. 5a). The kinetics of the exogenous VEGF expression are also interesting from the standpoint that the rAAV-delivered VEGF compensates for the allograft VEGF deficiency at this crucial time point compared to autografts (Fig. 2). Despite this modest transduction, the effects of combination gene transfer were evident and significant (Figs. 5 and 6; P < 0.05). In so doing, we show that RANKL and VEGF are sufficient signals to markedly alter allograft healing to generate a live, vascularized, remodeling, bony union.
In retrospect, the identification of VEGF and RANKL as critical regulators of autograft healing in our microarray screen is not notable. In addition to its role in angiogenesis, a VEGF gradient produced by hypertrophic chondrocytes is needed for directional growth and invasion of cartilage by blood vessels in endochondral ossification during development11 and in fracture healing9. Because remodeling of bone requires osteoclastic resorption, and RANKL is the final effector molecule that differentiates mononuclear precursors into osteoclasts12, a crucial role of RANKL is obvious. Furthermore, it is believed that the induction of RANKL in stromal cells in response to hypocalcemia or microfracture is the most proximal event that triggers de novo bone remodeling12. Our microarray screen also identified many other putative players that could similarly affect allograft healing including transcription factors, signaling molecules, receptors and other cytokines, but further studies will be required to determine if they can increase the efficacy of our current approach.
Here we show a new method of freeze-drying rAAV onto a surgically implantable surface, which is a safe and effective approach that could potentially be used in other conditions in which local delivery of gene products may be indicated. The resorption and subsequent formation of new bone did not occur uniformly on the allograft surfaces with parts of some allografts only being resorbed for the first time at killing, 4 weeks after surgery. This variable response probably reflects variation in the levels and temporal expression of the target genes, and in local tissue damage and subsequent infection of cells around the grafts. To show the clinical utility of our coated allografts we have committed to the development of a large animal model of structural allografting in which functional in vivo radiology can formally prove complete vascular invasion. Biomechanical testing in this model will also be necessary to show the advantage of remodeling versus nonremodeling allografts.
METHODS
Mouse segmental femoral graft model. All animal studies were conducted in accordance with principles and procedures approved by the University of Rochester Committee for Animal Resources. We used 8-week-old C57BL/6 mice for femoral grafting as we have previously described13. We cleaned allografts from ICR mice with 70% ethanol, rinsed them three times in saline to remove residual ethanol, and then froze them at −80 °C for at least 24 h before use. This procedure is based on the methods used by the Musculoskeletal Transplant Foundation to prepare clinical allografts. Graft healing was followed radiographically using a Faxitron X-ray system as described previously15.
In vivo treatments. For loss-of-function studies with biologics, we administered antibodies against VEGF (R&D Systems, Inc.), RANK:Fc fusion protein (a gift from Immunex, Inc.), or goat IgG (Sigma) in phosphate-buffered saline (PBS) by intraperitoneal injection every 3 d until mice were killed, as described previously31. Ex vivo gene transfer to live autografts were performed by harvesting the specimen, incubating it in 20 μl of sterile PBS containing 5 × 107 transducing units of rAAV for 10 min at room temperature and rapidly placing it back in the original donor. We performed in vivo gene transfers to processed allografts by pipetting 5 × 107 particles of rAAV in 50 μl of a 1% sorbitol-PBS solution onto the cortical surface of the grafts. The allografts were then frozen at −80 °C, lyophilized and stored at −80 °C until they were transplanted. We used at least six mice in each treatment group.
Histological and histomorphometric analysis. Following killing of mice, the grafted femurs were processed and stained with hematoxylin, eosin, Orange G and alcian blue (H&E), or for tartrate-resistant acid phosphatase (TRAP) activity and counterstained with hematoxylin as we have described previously31,32. We performed X-gal (Sigma) staining on sections counterstained with eosin as we have described previously32. Histomorphometric analysis was carried out using Osteometrics software as we have described previously13.
Real-time quantitative RT-PCR assays and microarrays. We harvested autografts and allografts from killed mice, immediately froze them in liquid nitrogen, minced them using a 6750 Freezer/Mill (SPEX CertiPrep, Inc.), and extracted total RNA using TRIsol (Invitrogen Corp.). We made single-stranded cDNA using a reverse transcription kit (Invitrogen) and used it as template for real-time PCR with SYBR Green PCR Master Mix (Applied Biosystems) and gene-specific primers in a Rotor-Gene 2000 (Corbett Research) as previously described15. The mean cycle threshold (Ct) values from quadruplicate measurements were used to calculate the gene expression, with normalization to β-actin as an internal control. The primer sequences for Tnfsf11 are: forward, 5′-TCTCATAACCTGATGAAAGG-3′; reverse 5′-GCATCTTGATCCGGATCCAG-3′. The primer sequences for Vegfa are: forward, 5′-GATGTGAATGCAGACCAAAG-3′; reverse, 5′-CACATCTGCAAGTACGTTCG-3′. The Functional Genomics Core Facility performed the microarray experiments, under the direction of A. Brooks. The experiments were performed by pooling the RNA extracted from six independent samples per group (autografts or allografts) in duplicate. Total RNA from day 10 samples were biotinylated and amplified using the T7 linear amplification approach previously described33. Affymetrix m430_2.0 arrays, which represent approximately 45,000 mouse probe sets, were run following the manufacturer's protocol. Signal values were calculated using a probe level analysis normalization tool (Robust Multichip Analysis, RMA) before making pair-wise comparisons between allograft and autograft samples.
Preparation of rAAV vectors. The rAAV-β-gal34 and rAAV-OPG35 vectors have been described previously. Plasmids containing cDNA for Vegfa36, Tnfsf11 (ref. 37) and Flt1 (ref. 38) were used for subcloning into the pAAV-BGHA transfer vector using oligonucleotide primers containing restriction sites for NotI and EcoRI at the 5′ and 3′ end, respectively. After ligation and transformation, positive clones grown in E. coli were confirmed by restriction digests and DNA sequencing. The resulting plasmids were used to produce the rAAVs through a helper-virus-free method, which were titered by dot blot39. The function of each rAAV vector was verified by assessing protein production in vitro by enzyme-linked immunosorbent assay (ELISA; R&S systems) as described35. The functional activities of the rAAV-RANKL and rAAV-VEGF vectors were also confirmed by in vitro osteoclastogenesis35 and angiogenesis40 assays, respectively. In vivo expression of VEGF and RANKL was assessed by serum ELISA as we have described previously35. The transduction efficiency of rAAV-β-gal was determined in vitro by X-gal staining and by assaying for β-galactosidase activity using the Galacto-Light system (Tropix Inc.) as described previously34.
Statistical analysis. An observer blinded to the treatment performed the histomorphometry. Data were calculated as the mean ± s.d., and the groups were compared using two-tailed analysis of variance (ANOVA). Statistical significance was set at P < 0.05.
Accession numbers. The GEO accession numbers for the primary data files are GSM37204 and GSM37205.
ACKNOWLEDGMENTS
We thank: H. Burchardt (Musculoskeletal Transplant Foundation) for advice with this research. J. Huard (University of Pittsburgh) for providing us with the sFlt-1 cDNA, W. Min (Yale University) for providing us with the Vegfa cDNA, Amgen Inc. for providing us with the OPG and Tnfsf11 cDNA and the RANK:Fc. We also thank C. Hock and D. Reynolds for assistance with the serum ELISA studies, B, Fan, L. Gehan and B. Stroyer for assistance with the histology, H. Awad for assistance with manuscript preparation, and R. Guldberg and A. Lin for μCT analyses. This work was supported by research grants from the Orthopedic Research and Education Foundation, the Musculoskeletal Transplant Foundation, US National Institutes of Health grants AR51469, AR48149, AR48681, AR43510, ES011854 and HL066973, and unrestricted research grants from DePuy, J&J Inc.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests (see the Nature Medicine website for details).
References
- 1.Garbuz DS, Masri BA, Czitrom AA. Biology of allografting. Orthop. Clin. North. Am. 1998;29:199–204. doi: 10.1016/s0030-5898(05)70318-7. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg VM, Stevenson S. The biology of bone grafts. Semin. Arthroplasty. 1993;4:58–63. [PubMed] [Google Scholar]
- 3.Einhorn TA. The cell and molecular biology of fracture healing. Clin. Orthop. 1998:S7–S21. doi: 10.1097/00003086-199810001-00003. [DOI] [PubMed] [Google Scholar]
- 4.Burchardt H. Biology of bone transplantation. Orthop. Clin. North. Am. 1987;18:187–196. [PubMed] [Google Scholar]
- 5.Gould SE, Rhee JM, Tay B-B, Otsuka NY, Bradford DS. Cellular contribution of bone graft to fusion. J. Orthop. Res. 2000;18:920–927. doi: 10.1002/jor.1100180611. [DOI] [PubMed] [Google Scholar]
- 6.Enneking WF, Campanacci DA. Retrieved human allografts: a clinicopathological study. J. Bone Joint Surg. Am. 2001;83-A:971–986. [PubMed] [Google Scholar]
- 7.Lord CF, Gebhardt MC, Tomford WW, Mankin HJ. Infection in bone allografts. Incidence, nature, and treatment. J. Bone Joint Surg. Am. 1988;70:369–376. [PubMed] [Google Scholar]
- 8.Berrey BH, Jr., Lord CF, Gebhardt MC, Mankin HJ. Fractures of allografts. Frequency, treatment, and end-results. J. Bone Joint Surg. Am. 1990;72:825–833. [PubMed] [Google Scholar]
- 9.Colnot C, Thompson Z, Miclau T, Werb Z, Helms JA. Altered fracture repair in the absence of MMP9. Development. 2003;130:4123–4133. doi: 10.1242/dev.00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kon T, et al. Expression of osteoprotegerin, receptor activator of NF-kappaB ligand (osteoprotegerin ligand) and related proinflammatory cytokines during fracture healing. J. Bone Miner. Res. 2001;16:1004–1014. doi: 10.1359/jbmr.2001.16.6.1004. [DOI] [PubMed] [Google Scholar]
- 11.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat. Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 12.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 13.Tiyapatanaputi P, et al. A novel murine segmental femoral graft model. J Orthop Res. 2004;22:1254–1260. doi: 10.1016/j.orthres.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 14.Hadjiargyrou M, et al. Transcriptional profiling of bone regeneration. Insight into the molecular complexity of wound repair. J. Biol. Chem. 2002;277:30177–30182. doi: 10.1074/jbc.M203171200. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, et al. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J. Clin. Invest. 2002;109:1405–1415. doi: 10.1172/JCI15681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonadio J, Smiley E, Patil P, Goldstein S. Localized, direct plasmid gene delivery in vivo: prolonged therapy results in reproducible tissue regeneration. Nat. Med. 1999;5:753–759. doi: 10.1038/10473. [DOI] [PubMed] [Google Scholar]
- 17.Schwarz EM. The adeno-associated virus vector for orthopaedic gene therapy. Clin. Ortho. & Rel. Res. 2000;379S:S31–S39. doi: 10.1097/00003086-200010001-00005. [DOI] [PubMed] [Google Scholar]
- 18.Rabinowitz JE, Samulski RJ. Building a better vector: the manipulation of AAV virions. Virology. 2000;278:301–308. doi: 10.1006/viro.2000.0707. [DOI] [PubMed] [Google Scholar]
- 19.Wu D, Razzano P, Grande DA. Gene therapy and tissue engineering in repair of the musculoskeletal system. J. Cell. Biochem. 2003;88:467–481. doi: 10.1002/jcb.10332. [DOI] [PubMed] [Google Scholar]
- 20.Gamradt SC, Lieberman JR. Genetic modification of stem cells to enhance bone repair. Ann. Biomed. Eng. 2004;32:136–147. doi: 10.1023/b:abme.0000007798.78548.b8. [DOI] [PubMed] [Google Scholar]
- 21.Boden SD, Kang J, Sandhu H, Heller JG. Use of recombinant human bone morphogenetic protein-2 to achieve posterolateral lumbar spine fusion in humans: a prospective, randomized clinical pilot trial: 2002 Volvo Award in clinical studies. Spine. 2002;27:2662–2273. doi: 10.1097/00007632-200212010-00005. [DOI] [PubMed] [Google Scholar]
- 22.Friedlaender GE. OP-1 clinical studies. J. Bone Joint Surg. Am. 2001;83-A(Suppl 1):S160–S161. [PubMed] [Google Scholar]
- 23.Lieberman JR, Ghivizzani SC, Evans CH. Gene transfer approaches to the healing of bone and cartilage. Mol. Ther. 2002;6:141–147. doi: 10.1006/mthe.2000.0663. [DOI] [PubMed] [Google Scholar]
- 24.Baltzer AW, Lieberman JR. Regional gene therapy to enhance bone repair. Gene Ther. 2004;11:344–350. doi: 10.1038/sj.gt.3302195. [DOI] [PubMed] [Google Scholar]
- 25.Sandhu HS, Boden SD, An H, Kang J, Weinstein J. BMPs and gene therapy for spinal fusion: summary statement. Spine. 2003;28:S85. doi: 10.1097/01.BRS.0000076903.10966.86. [DOI] [PubMed] [Google Scholar]
- 26.Musgrave DS, et al. Ex vivo gene therapy to produce bone using different cell types. Clin. Orthop. 2000;378:290–305. doi: 10.1097/00003086-200009000-00040. [DOI] [PubMed] [Google Scholar]
- 27.Hidaka C, et al. Acceleration of cartilage repair by genetically modified chondrocytes over expressing bone morphogenetic protein-7. J. Orthop. Res. 2003;21:573–583. doi: 10.1016/S0736-0266(02)00264-4. [DOI] [PubMed] [Google Scholar]
- 28.Verma IM, Somia N. Gene therapy -- promises, problems and prospects. Nature. 1997;389:239–242. doi: 10.1038/38410. [DOI] [PubMed] [Google Scholar]
- 29.Bos GD, Goldberg VM, Powell AE, Heiple KG, Zika JM. The effect of histocompatibility matching on canine frozen bone allografts. J. Bone Joint Surg. Am. 1983;65:89–96. [PubMed] [Google Scholar]
- 30.Stevenson S, Li XQ, Davy DT, Klein L, Goldberg VM. Critical biological determinants of incorporation of non-vascularized cortical bone grafts. Quantification of a complex process and structure. J. Bone Joint Surg. Am. 1997;79:1–16. [PubMed] [Google Scholar]
- 31.Childs LM, et al. In vivo RANK signaling blockade using the receptor activator of NF- kappaB:Fc effectively prevents and ameliorates wear debris-induced osteolysis via osteoclast depletion without inhibiting osteogenesis. J. Bone Miner Res. 2002;17:192–199. doi: 10.1359/jbmr.2002.17.2.192. [DOI] [PubMed] [Google Scholar]
- 32.Childs LM, Goater JJ, O'Keefe RJ, Schwarz EM. Effect of anti-tumor necrosis factor-alpha gene therapy on wear debris- induced osteolysis. J. Bone Joint Surg. Am. 2001;83-A:1789–1797. doi: 10.2106/00004623-200112000-00004. [DOI] [PubMed] [Google Scholar]
- 33.Musatov S, et al. Inhibition of neuronal phenotype by PTEN in PC12 cells. Proc. Natl. Acad. Sci. USA. 2004;101:3627–3631. doi: 10.1073/pnas.0308289101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goater J, et al. Empirical advantages of adeno associated viral vectors in vivo gene therapy for arthritis. J. Rheumatol. 2000;27:983–989. [PubMed] [Google Scholar]
- 35.Ulrich-Vinther M, et al. Recombinant adeno-associated virus-mediated osteoprotegerin gene therapy inhibits wear debris-induced osteolysis. J. Bone Joint Surg. Am. 2002;84-A:1405–1412. doi: 10.2106/00004623-200208000-00016. [DOI] [PubMed] [Google Scholar]
- 36.Zhang R, et al. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J. Biol. Chem. 2003;278:51267–51276. doi: 10.1074/jbc.M310678200. [DOI] [PubMed] [Google Scholar]
- 37.Lacey DL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 38.Peng H, et al. Synergistic enhancement of bone formation and healing by stem cell-expressed VEGF and bone morphogenetic protein-4. J. Clin. Invest. 2002;110:751–759. doi: 10.1172/JCI15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deckers M, et al. Effect of angiogenic and antiangiogenic compounds on the outgrowth of capillary structures from fetal mouse bone explants. Lab. Invest. 2001;81:5–15. doi: 10.1038/labinvest.3780207. [DOI] [PubMed] [Google Scholar]