

Abstract
Ligand flexibility is an important problem in molecular docking and virtual screening. To address this challenge, we investigate a hierarchical pre-organization of multiple conformations of small molecules. Such organization of pre-calculated conformations removes the exploration of ligand conformational space from the docking calculation and allows for concise representation of what can be thousands of conformations. The hierarchy also recognizes and prunes incompatible conformations early in the calculation, eliminating redundant calculations of fit. We investigate the method by docking the MDL Drug Data Report (MDDR), an annotated database of 100,000 molecules, into apo and holo forms of seven unrelated targets. This annotated database allows us to track the ranking of tens to hundreds of annotated ligands in each of the docking systems. The binding sites and database are prepared in an automated fashion in an attempt to remove some human bias from the calculations. Many thousands of explicit and implicit ligand conformations may be docked in calculations not much longer than required for single conformer docking. As long as internal energies are not considered, recombination with the hierarchy is additive as the number of degrees of freedom is increased. Molecules with even millions of conformations can be docked in a few minutes on a single desktop computer.
Keywords: Molecular docking, combinatorial, flexibility, high-throughput
INTRODUCTION
Ligand-receptor interactions are central to most biological processes. Computational modeling of these interactions remains difficult because of the many ways molecules can fit together and the often small energy differences separating the most favored configurations from alternative structures and from aqueous solvation. Together these two challenges make up the "Docking Problem." A major component of the sampling problem in docking is that of conformational flexibility. Ignoring excluded volume,[1,2] the number of conformations accessible to docking molecules scales as the power of the number of their rotatable bonds. This becomes that much more problematic in screening large databases of diverse molecules for novel inhibitor discovery, where potential ligands must not only be fit correctly but must be distinguished from non-binders. Here we present a new method for addressing ligand flexibility. We evaluate the method by docking an annotated small-molecule database to seven different enzymes (Table 1). Both apo and holo receptor conformations are considered as docking targets.
Table 1.
Docking Systems and Annotated Ligands
| Crystallographic | Complexed | Uncomplexed | # of ligands | Average number of | ||||
|---|---|---|---|---|---|---|---|---|
| Docking target | Ligand | PDB | Res. (Å) | PDB | Res. (Å) | Rotors | Conformations | |
| Acetylcholinesterase | Galanthamine | 1DX6 | 2.3 | 1EA5 | 1.8 | 637 | 4.4 | 396.1 |
| Adenosine kinase | Adenosine | 1DGM | 1.8 | 1DH2 | 2.5 | 45 | 3.2 | 140.3 |
| Phospholipase C | --- | --- | --- | 1AH7 | 1.5 | 25 | 4.4 | 89.8 |
| Thymidylate synthase | Methotrexate | 1AXW | 1.7 | --- | --- | 185 | 7.8 | 1753.4 |
| Thrombin | thiazole inhibitor | 1A4W | 1.8 | --- | --- | 788 | 8.8 | 1047.3 |
| Neutral endopeptidase | Phosphoramidon | 1DMT | 2.1 | --- | --- | 356 | 8.4 | 283.9 |
| Dihydrofolate reductase | Methotrexate | 3DFR | 1.7 | 6DFR | 2.4 | 165 | 7.3 | 2696.7 |
Several methods have been introduced to dock flexible molecules. One class of docking programs including GOLD [3], DARWIN [4], implementations of AUTODOCK[5] and several others [6–8] use genetic algorithms to optimize ligand conformations in the context of the binding site. Other approaches to flexible ligand docking include optimizing the ligands in the context of the binding site, [9–11] enumeration of multiple low energy ligand minima followed by energy refinement [12,13] or placing fragments into the binding site for later connection [14–17]. A common technique for flexible docking is that of anchor-and-grow or incremental construction. Algorithms like FlexX, [18,19] DOCK4 [20,21] and DOCK5 place a fragment of the ligand in the binding site, and explore the conformational space accessible in the context of the binding site. This process is repeated for each orientation of the starting fragment, for each ligand, during each docking run, against each target. The program DREAM++[22] explores combinatorial chemistry in the context of the binding site using a similar schema.
These methods evade the conformational explosion in ligand docking by using the excluded volume of the site to prune the search tree at every step as the flexible fragments are grown off of the rigid fragment. In this they have been successful to the point where ligand flexibility is considered an essential feature of a docking program. Nevertheless, after ligands grow past a certain number of rotatable bonds the ability of the site to prune the search tree sufficiently is exceeded and even anchor-and-grow methods face a combinatorial explosion.
Here we introduce a method that attempts to reduce the exponential dependence of conformations on the number of rotatable bonds to an additive dependence. To do so, we assume that different side chains are completely independent of each other and we exploit the tremendous redundancy present in multi-conformer libraries by organizing ligand information hierarchically (see next section). We evaluate the performance of this hierarchical method by two criteria: 1. How computationally expensive is this multi-conformer or multi-analog approach relative to single molecule rigid docking? 2. How well are the structures of ligands predicted using this multi-conformer approach vs. rigid-ligand docking? We undertake docking screens against seven enzyme targets whose structures have been determined by x-ray crystallography, typically in both their apo (no ligand ray bound) and holo (ligand bound) conformations. For each of the seven enzymes there are annotated inhibitors in the compound database that is screened (below); the vast majority of the compounds in the database are thought not to bind to the enzymes. Because of our own interest in using docking as a screening tool, we will focus on calculation times, predicted structures, and ligand identification from docking calculations performed on the entire database, and not on single molecule calculations.
Overview of the Docking Method and its Evaluation
The annotated small-molecule database used to evaluate our docking method is the MDL Drug Data Report (MDDR, MDL Inc., San Leandro, CA). The MDDR contains drug-like small molecules annotated for biological activity and, like often, molecular target. It is commonly used to train and test cheminformatics methods for library design,[23,24] pharmacophore fingerprinting [25–28] and determining drug-likeness [29–31]. This annotated database provides a list of likeness ligands that we can track. The ability to select for true ligands is measured by the total number ligands identified as binding to the binding site, and the percent of the ligands ranked in the top percentages of the database.
We have previously described a flexible ligand docking method that uses pre-calculated ensembles of ligand conformations [32]. This method docked multiple pre-calculated conformations as a proxy for true flexible calculated docking, extending an idea first introduced by the FLOG program.[33] One innovation of the original ensemble method was to use a rigid anchor fragment to which all conformations of a given molecule were aligned. This alignment allowed a single translation-rotation matrix to be used for all conformations. Because only one rotation-matrix needed to be calculated for all N conformations for a given molecule, rather than N rotation-matricies, this ensemble method decreased time requirements by up to 100-fold.
The method presented here extends this ensemble method by representing conformational ensembles hierarchically, allowing for dramatic increases in sampling and aggressive pruning that largely eliminates the extra computational burden of evaluating multiple conformations. Like the ensemble method,[32] multiple low energy conformations for each molecule are calculated off-line; these conformations are in a common frame of reference so that they have a maximum number of atoms in identical positions (Fig. 1). This allows a single translation-rotation matrix to be applied to all conformations for any given orientation of the rigid, guide fragment of the ligand. Atoms outside the rigid fragment are rotated in discrete intervals to facilitate the recognition and removal of redundant atom coordinates. Atoms most proximal to the rigid fragment have fewer unique positions than more distal atoms. Each ensemble of pre-generated ligand conformations is processed into a hierarchical data structure such that atom connectivity is implicitly represented across all conformations of the ensemble (Fig. 1). These conformations are stored in a database and are not recalculated subsequently. The hierarchical data structure for representing small molecules is optimized for three features:
Fig (1).
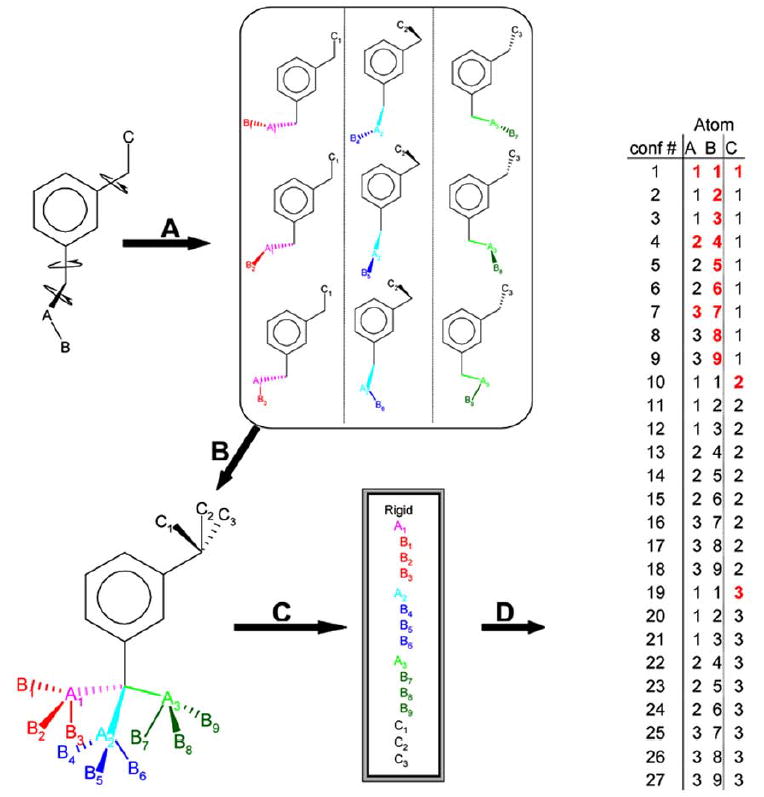
Generation of a conformational hierarchy and its representation in pseudo-database format A. A representative molecule, 3-ethyl propylbenzene, for which 27 low-energy conformations may be calculated using a torsion-library (9 are shown). B. These 27 conformations may be overlaid in a common reference frame defined by the largest rigid fragment (the m-xylene component). This largest rigid fragment is the anchor defining docked orientations for the entire ensemble. C. The coordinates are organized hierarchically to allow for rapid pruning of branches that clash with the protein. D. The coordinates of all 27 possible low-energy conformations for the three movable atoms are evaluated using only the subset of unique coordinates (shown in red).
Elimination of redundant calculations. For molecules that have branches, when one branch changes conformation, the other branches and rigid fragment need not be re-evaluated. Also, atoms closest to the rigid fragment have evaluated. fewer unique positions relative to the rigid fragment than do the more terminal atoms. For conformations where only the more terminal atoms change, all less terminal atoms need not be re-evaluated.
Rapid pruning of clashing branches. Atoms within each branch are ordered so that conformations that clash with the receptor can be pruned off as early as possible. Since, terminal atoms have more unique positions than atoms closer to the rigid fragment, avoiding evaluation of many terminal atom positions can greatly speed up calculations.
Recognition that branches can be moved independently. The size and number of calculations required for an ensemble can be greatly reduced if branches are evaluated combinatorially. Assuming complete additivity, an exponential number of conformations can be evaluated while computation time increases linearly. A feature of this method is that it allows conformations that may not be explicitly represented in the ensemble to be docked. This becomes most useful for molecules capable of adopting very large numbers of conformations. A limitation is the assumption of additivity, which will not always hold and which, in pathological cases, can lead to unreasonable conformations (see Discussion).
In the first step of the hierarchy method (Fig. 1A) rotatable bonds are identified, and rotated in fixed increments creating an ensemble of conformations. In the second step (Fig. 1B), the ensemble of conformations is written out in a common frame of reference. The first two steps can be performed by a variety of algorithms; we used the program Omega (OpenEye Scientific Software, Santa Fe NM). In the third step (Fig. 1C) the ensemble of conformations is digested into a non-redundant hierarchical representation. After conversion of the conformational ensembles, the hierarchies are stored on disk and not recreated for each docking calculation. During the docking itself, each ligand hierarchy is expanded as poses are sampled in the site. In any pose, only the atom positions that have changed from previous steps in the conformational expansion are evaluated (red atoms, Fig. 1D). For all other atoms, scores are simply retrieved from previous calculations. Docking targets are prepared using a semiautomatic script in an attempt to remove some human bias and to test the feasibility of extending docking from its current use in high-throughput ligand screening, to high-throughput target screening.
RESULTS
Database and Docking Statistics
Of the 113,842 molecules listed in the MDDR 2000.2, about 98,500 passed all of the filtering criteria and were included in the docking database. Due to multiple charge-state representations of particular ligands and “rigid” fragment ring pucker, the total number of unique entities in the database was about 135,000. For instance, molecules containing a cyclohexane ring as part of the rigid fragment were represented as two independent entities: the “boat” and “chair” ring conformations. Molecules with titratable groups around physiological pH, such as 4-aminopyridines, were represented in both their charged and neutral forms. The computer program, Omega, was used to produce about 34.8 million conformations, slightly fewer than 260 conformations per molecule, represented by 1.6 billion atom coordinates. Upon reorganization into a hierarchy, the number of conformations accessible through side chain recombination increased about 50-fold to 1.5 billion (average 11,000 conformations per molecule). Meanwhile, the number of atoms explicitly represented decreased 5-fold to 351 million through the elimination of redundant coordinates. These conformations all differ in the relative positions of their heavy atoms; upon inclusion of terminal hydrogen conformations, e.g., hydroxyls, the number of conformations in the database increases to 1.1 trillion. Several extremely flexible molecules, such as MDDR00246993 with over eight million conformations and MDDR00224885 with 197,760 conformations, and 12,882 rigid molecules, skew the number of conformers in the database. The median number of conformations for flexible molecules in the hierarchy is 64 and 98 including hydroxyl rotations. Approximately one quarter of the database is comprised of molecules with more than 1,000 conformations. The hierarchy database encodes the explicit coordinates and atomic information for 1.1 trillion conformations in 7.6GB. The uncompressed database in ASCII format, including atomic charges, van der Waals type, partial atomic desolvation, and three-dimensional coordinates, effectively encodes 145 conformations per byte. Generating this database required approximately three lab days using 10 Pentium III CPUs.
Overall, multi-conformer docking identified up to 65% more annotated ligands than rigid docking (Table 2). The largest hit rate improvements were observed in the three targets whose ligands had a large number of conformations. In these targets, where the ligands could adopt thousands of conformations, 25 to 65% of these ligands ranked in the top 5% of the dock results, compared to 5 to 9% in the top 5% of the rigid docking. Holo receptor conformations had better enrichment factors among the top ranking molecules than did their apo counterparts. Hierarchical docking took between two and five times longer than rigid docking, but fit up to four orders of magnitude more conformations. The longest rigid docking calculations took a little less than one CPU day, whereas the longest hierarchy calculations took a little over three CPU days. All timings are given in terms of a single 800 MHz Pentium III processor running Redhat Linux 7.0.
Table 2.
Docking Statistics for Hierarchy and Rigid Docking
| Hierarchy docking | Rigid docking | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| system | Moleculesa | Confsb | Orientationsc | Time (h)d | Moleculesa | Confsb | Orientationsc | Time (h)d | |
| apo | AChE | 74,922 | 1.7 × 109 | 3,605.20 | 10.57 | 35,053 | 35,053 | 4,468.10 | 5.92 |
| holo | AChE | 77,255 | 2.6 × 109 | 3,578.20 | 11.9 | 36,302 | 36,302 | 4,384.00 | 6.27 |
| apo | Aden. Kin. | 90,253 | 3.8 × 109 | 525.4 | 5.29 | 47,273 | 47,273 | 679.5 | 2.59 |
| holo | Aden. Kin. | 123,224 | 8.0 × 109 | 1,771.70 | 50.75 | 99,428 | 99,428 | 1,905.70 | 10.49 |
| apo | Phos. C | 122,876 | 8.8 × 109 | 1,237.10 | 30.31 | 105,076 | 105,076 | 1,419.00 | 14.28 |
| holo | TS | 53,917 | 6.8 × 108 | 2,440.90 | 11.33 | 22,835 | 22,835 | 2,783.00 | 4.29 |
| holo | thrombin* | 235,661 | 1.3 × 1010 | 7,648.40 | 122.49 | 194,575 | 194,575 | 7,808.10 | 30.46 |
| holo | Neut. Endo. | 130,445 | 7.0 × 109 | 5,040.30 | 77.57 | 126,117 | 126,117 | 5,006.80 | 18.8 |
| apo | DHFR | 101,990 | 4.6 × 109 | 3,765.30 | 36.99 | 72,166 | 72,166 | 4,079.00 | 7.8 |
| holo | DHFR | 49,540 | 1.2 × 109 | 3,985.80 | 10.69 | 19,405 | 19,405 | 4,472.60 | 5.1 |
Number of molecules successfully built into the binding site.
Total conformations docked.
Average orientations per molecule.
Total CPU time for the docking run, scaled to reflect docking time on a single 800 MHz Pentium III.
Results are combined from two independent calculations.
Dihydrofolate Reductase (DHFR)
About 50,000 of the 135,000 MDDR molecules could be fit into the holo conformation of DHFR as flexible molecules. These 50,000 molecules contained 78% of the 165 annotated ligands, with one-third of these scoring in the top 1% of the database (Table 3, Fig. 2A). For rigid docking only 19,000 molecules containing 27% of the annotated ligands fit the binding site. Here 2% of the ligands ranked in the top 1% of the docking results. The rigid docking required five hours, or about half the time required for the hierarchical docking (Table 2).
Table 3.
Identifying Ligands in Hierarchy and Rigid Searches
| 0.5%, 492 molecules # of hits (ef)* | 1.0%, 984 molecules # of hits (ef)* | 5.0%, 4,922 molecules # of hits (ef)* | 100%, 98,438 molecules # of hits (% of total) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Docking target | Number of ligands | Flexible | Rigid | Flexible | Rigid | Flexible | Rigid | Flexible | Rigid | ||||||||
| apo AChE | 637 | 8 | (3) | 40 | (13) | 22 | (3) | 51 | (8) | 105 | (3) | 114 | (4) | 551 | (86%) | 316 | (50%) |
| holo AChE | 637 | 40 | (13) | 72 | (23) | 57 | (9) | 78 | (12) | 130 | (4) | 121 | (4) | 551 | (86%) | 328 | (51%) |
| apo Aden. Kin. | 45 | 1 | (4) | 2 | (9) | 1 | (2) | 2 | (4) | 5 | (2) | 5 | (2) | 42 | (93%) | 30 | (67%) |
| holo Aden. Kin. | 45 | 1 | (4) | 1 | (4) | 2 | (4) | 1 | (2) | 10 | (4) | 10 | (4) | 45 | (100%) | 43 | (96%) |
| apo Phos. C | 25 | 5 | (40) | 0 | 0 | 5 | (20) | 0 | 0 | 9 | (7) | 2 | (2) | 24 | (96%) | 19 | (76%) |
| holo TS | 185 | 48 | (52) | 11 | (12) | 65 | (35) | 13 | (7) | 122 | (13) | 16 | (2) | 147 | (79%) | 25 | (14%) |
| holo thrombin | 788 | 35 | (9) | 2 | (1) | 66 | (8) | 3 | 0 | 175 | (4) | 30 | (1) | 763 | (97%) | 711 | (90%) |
| holo Neut. Endo. | 356 | 3 | (2) | 0 | 0 | 5 | (1) | 0 | 0 | 20 | (1) | 6 | 0 | 348 | (98%) | 343 | (96%) |
| apo DHFR | 165 | 9 | (11) | 2 | (2) | 18 | (11) | 3 | (2) | 65 | (8) | 14 | (2) | 147 | (89%) | 96 | (58%) |
| holo DHFR | 165 | 44 | (53) | 4 | (5) | 56 | (34) | 4 | (2) | 87 | (10) | 11 | (1) | 129 | (78%) | 44 | (27%) |
Number of active ligands identified within this fraction of the database (enrichment factor over random distribution for this fraction).
Fig (2).
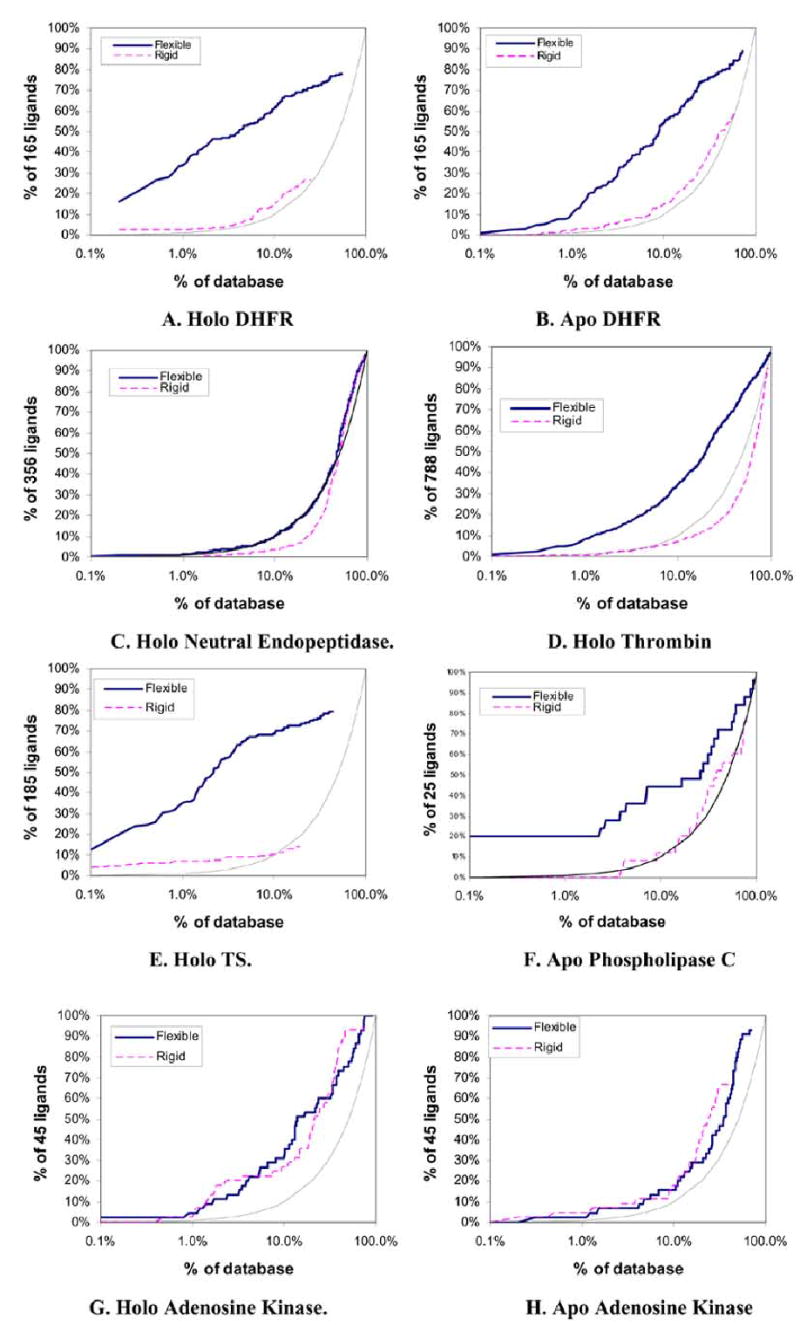
Semi-log enrichment curves evaluating the ability of flexible (solid dark) and rigid (dashed light) docking screens to highly rank annotated ligands against ten target structures. The performance of random selection is shown in as a solid gray line. The structures screened were A. Holo DHFR, B. Apo DHFR, C. Neutral endopeptdidase, D. Holo thrombin, E. Holo TS, F. Apo Phospholipase C, G. Holo Adenosine Kinase, H. Apo Adenosine Kinase, I. Holo acetylcholinesterase, J. Apo acetylcholinesterase.
The apo receptor is more open than the holo receptor allowing more molecules to be docked, increasing the total number of ligands identified, and increasing run times. About 100,000 flexible molecules were fit into the apo binding site. A total of 89% of the annotated ligands were identified in the flexible docking run, with 11% of these ranking in the top 1% of the docking results (Table 3, Fig. 2B). 58% of the annotated ligands were identified among the 72,000 molecules from rigid docking; 2% in the top 1% of the hit list. The flexible and rigid runs required 37 and eight hours of CPU time respectively (Table 2).
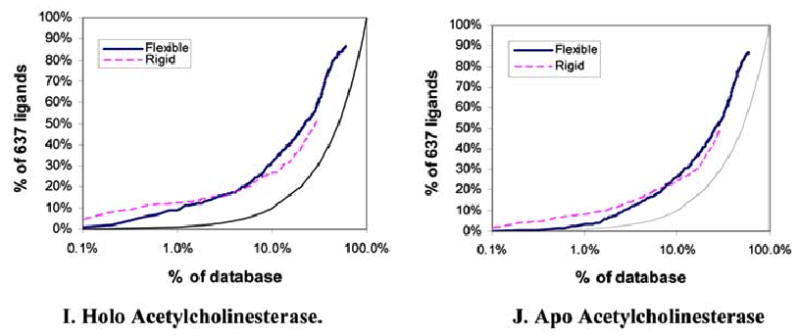
DHFR is well-suited to hierarchal docking because it has a constrained deep pocket that binds a relatively large, functionally important rigid group common to most DHFR ligands. The constraints of the deep binding site have the effect of pruning large portions of the hierarchy early in the calculation. Thus, although an average of 10.8 million complexes (2,700 conformations × 4,000 orientations) were evaluated for each of the annotated ligands, most of these were excluded early in the hierarchical expansion in the site. For instance, when an atom early in the expansion of methotrexate clashes with the enzyme, about 900 conformations are eliminated without the need to search further down the conformational tree. DHFR ligands are also well-suited to the hierarchy method. Most contained recognition rings such as pyrimidines, pteridines, quinazolines, or triazines, which also constituted the largest rigid fragment in the molecule, which is the first part of the molecule to be placed in the site, before the conformational expansion. Thus, most DHFR ligands, were ranked well in geometries similar to those observed crystallographically (Fig. 3A). However when the largest rigid fragment was not an important recognition element for the enzyme, few good docking solutions were found. This is a weakness in our method (see Discussion).
Fig (3).
The docked (carbons in magenta) vs. the crystallographic (carbons in cyan) poses of characteristic ligands in their target enzymes (molecular surface transparent gray). Oxygens are red and nitrogens blue. A. Methotrexate in DHFR. B. 3-{3-(4-(4-Carbamimidoyl-phenyl)- butyl)-ureido}-3-phenyl-propionic acid in thrombin. C. Methotrexate in thymidylate synthase; the carbons of the co-factor dUMP are colored gold. D. Adenosine in adenosine kinase. E. Galanthamine in the binding site of acetylcholineesterase.
Neutral Endopeptidase
More than 93% of the MDDR molecules fit into the large (20Å in diameter) binding site of the holo form of neutral endopeptidase with or without flexibility. On average, molecules were evaluated in 5,000 orientations in this binding site. The flexible docking required a little over three CPU days compared to slightly less than one day for the rigid calculation (Table 2). About 95% of the 356 annotated ligands were fit in both rigid and flexible searches, with about 1% scoring in the top 1% of the database, little better than random selection (Table 3, Fig. 2C). Unlike the DHFR ligands, neutral endopeptidase ligands are ill-suited for the hierarchical method. These ligands are peptide or peptide-like and the largest rigid fragment is rarely a key recognition like element.
Thrombin
When docked into a holo conformation of thrombin, over 90% of the MDDR molecules fit into the protease site. Flexible docking fit 97% of the 788 annotated ligands, ranking 8% of them in the top 1% of the hit list. Rigid docking fit 90% of the annotated ligands, but only three in the top 1% of the hit list (Table 3, Fig. 2D). The flexible docking calculation took about four times longer than the rigid calculation (Table 2).
The 788-member subset of MDDR thrombin ligands contained those likely to bind to the protease site of the enzyme; peptides likely to bind to the hiruden site were excluded from this list. Molecules were docked to the P1 and P3 sites in two separate docking calculations. Many of the top scoring ligands placed an amidinium or guanidinium group into P1 interacting with Asp189. The annotated ligands contained an average 8.8 rotatable bonds leading to about 1,000 conformations per ligand. On average, each MDDR molecule was docked in a little over 5,200 different orientations. Whereas the algorithm was unable to reproduce the crystallographic pose for the ligand NAPAP, many other ligands fit in reasonable geometries (Fig. 3B).
Thymidylate Synthase
The holo, ternary conformation of thymydylate synthase was used for docking, with the pyrimidine nucleotide included in the target structure. Flexible docking fit 54,000 molecules into the binding site including 79% of the 185 annotated ligands (Table 3, Fig. 2E). Over one-third of these scored in the top 1% of the docking hits when fit as flexible molecules. Rigid docking fit 23,000 molecules into the binding site and identified 14% of the annotated ligands. The top 1% of the docked hit list contained less than one-tenth of the ligands. Flexible docking took 2.5-fold longer than rigid docking (Table 2).
The 185-member subset of TS ligands excluded nucleotides and their analogs, since we were docking to the folate-binding site. The inhibitors in this subset contained an average of 1,750 conformations (an average of 7.8 rotatable bonds). Each molecule in the database was docked in about 2,500 different orientations. The algorithm was able to closely reproduce the crystallographic binding mode for methotrexate (Fig. 3C).
Phospholipase C
Twenty-four of 25 annotated ligands of phospholipase C, along with 123,000 molecules, were successfully fit to the enzyme using flexible docking. About 1,300 orientations were scored for each conformation of each molecule in the database. The 25 ligands had an average 4.4 rotatable bonds and about 90 conformations were sampled for each. Of the ligands, 20% ranked in the top 1% of the database (Table 3, Fig. 2F). The rigid search docked 105,000 molecules into the binding site, fitting 76% of the annotated ligands, but none among the top 1% of the database. The rigid docking took about half as long as the flexible docking.
Adenosine Kinase
Although adenosine kinase undergoes a 30º domain movement on ligand binding, the docking results against the apo and holo conformations of the enzyme were similar. Flexible docking to the holo structure successfully fit all 45 of the annotated ligands, ranking two in the top 1% of the hit list. Rigid docking to the same structure fit 43 of the annotated ligands, ranking one in the top 1% of the database. Both the rigid and flexible searches identified the same 10 ligands within the top 5% of the database (Table 3, Fig. 2G). Flexible docking to this site took five-times longer than the rigid docking (Table 2).
For the apo conformation of adenosine kinase, flexible docking fit 90,000 molecules including all 45 annotated ligands. One ligand was ranked in the top 1% of the docking hits (Table 2, Fig. 2H). Rigid docking fit 47,000 molecules into the site, including 30 of the annotated ligands; two ranked in the top 1% of the docking hits. Again, at 5% of the database, the rigid and flexible dockings were equally successful, each identifying five annotated ligands. The rigid docking took half as long as the flexible docking calculation (Table 2).
Over half of the 45 adenosine kinase ligands were pyrimidine analogs. In most cases the pyrimidine ring was selected as the rigid fragment of the ligand. The ligands contained an average 3.2 rotatable bonds and 140 conformations. Because the apo structure of adenosine kinase is more constrained than the holo structure, the number of orientations evaluated for the former was about one third of that for the latter, and the time required for the apo and holo calculations reflect this difference. The algorithm was able to closely reproduce the crystallographic binding mode for adenosine (Fig. 3D).
Acetylcholinesterase
About 77,000 MDDR molecules could be fit into the holo form of the enzyme. These included 86% of the annotated ligands; of these 9% ranked in the top 1% of the docked hit list. The rigid docking calculation fit about half as many ligands into the binding site as the flexible and found 50% of these. Here about 12% of the annotated ligands ranked in the top 1% of the results (Table 3, Fig. 2I).
The apo acetylcholinesterase structure accommodated 75,000 flexibly docked molecules. These included 87% of the annotated ligands; 3% of these ranked in the top 1% of the dock hit list (Table 3, Fig. 2J). In rigid docking to the apo conformation, only about half as many molecules could be fit into the site. These included 50% of the annotated ligands, 8% of which were found in the top 1% of top scoring molecules.
Overall, 637 annotated acetylcholinesterase ligands from the MDDR were docked. These ligands averaged about 400 conformations and 4.4 rotatable bonds. Similar docking statistics resulted for both the apo and holo conformations of the enzyme. Flexible docking evaluated about 3,600 orientations per molecule and rigid evaluated about 4,400. The algorithm was able to closely reproduce the crystallographic binding mode for galanthamine (Fig. 3E).
DISCUSSION
To overcome the exponential dependence of conformations on degrees of freedom, the hierarchical docking method assumes complete side chain independence, aggressively prunes the search using steric constraints from the enzyme, and eliminates redundancy inherent in multi-conformer databases, leading to a concise representation of conformer the molecules for both storage and evaluation. Returning to the questions we posed in the introduction, the method is fast: Database screens considering tens to millions of conformations for each molecule take only two to five-fold longer than single conformer database screens. In database screening, 40 to 200 poses are evaluated each microsecond. On an atomic level, the structures of the docked ligand complexes corresponded closely with the experimental structures, taking these results directly from the database screening calculations. Finally, for most of the seven enzymes targeted, annotated ligands were enriched among top scoring molecules in the docking screens of the MDDR database; the more flexible the ligands, the more the hierarchical docking outperformed rigid-ligand docking. Below we consider some of these points in detail, including methodological caveats and areas where the method fails.
More Flexible Versus More Rigid Ligands
In every target, flexible, hierarchical docking fit more of the known ligands, and more of the database overall, into the site than did rigid docking. A key question is how the ligands ranked compared to the vast number of other, presumably non-binding molecules in the MDDR. In five of the seven enzymes, thymidylate synthase, thrombin, neutral endopeptidase, DHFR, and phospholipase C, hierarchical docking returned much better ligand rankings for known ligands than did rigid docking; for two targets, acetylcholinesterase and adenosine kinase, hierarchical docking returned worse or equivalent rankings (Table 3). The relative performance of the hierarchical docking correlates with the flexibility of the ligands being fit. For thymidylate synthase, thrombin, neutral endopeptidase, and DHFR, the annotated ligands averaged between 7.3 and 8.8 rotatable bonds, so that without sampling multiple conformations, complementary fits were unlikely to be found. Conversely, the ligands for acetylcholinesterase and adenosine kinase are more rigid, averaging between 3.2 and 4.4 rotatable bonds per molecule, and sampling flexibility appears to have been much less important. For these latter systems, adding flexibility did not reduce the docking scores of the annotated ligands, but it did diminish their rankings relative to the vast number of decoy molecules that make up the MDDR. This suggests two possible failures. First, we are not sufficiently accounting for internal energy of the docking molecules (not penalizing molecules that have high internal energies). As a result, molecules that, through failure of our conformation-generation methods, assume high-energy structures may complement the binding site artificially well; these molecules are decoys for our algorithm. Second, as almost a signal to noise issue, we are challenging the discriminating power of our scoring function by expanding our database to 1.1 trillion combinatorially sampled conformations in hierarchical docking. On the other hand, when we dock ligands as rigid molecules we also dock the other non-binders in the MDDR rigidly, vastly reducing the chance that a non-binder will fit.
A target that performed poorly with hierarchical docking and was a catastrophe for rigid docking was neutral endopeptidase, where the average ligand had 8.4 rotatable bonds. Even for hierarchical docking we saw little enrichment over random. The ligands for this enzyme are peptidic with few of the key recognition functional groups in the rigid fragment used for initial anchoring of the ligand in the site; rather, most are in the flexible side chains. For these sorts of ligands, our “trick” of dividing the ligand into an anchor and flexible fragments fails us. Recent work suggests that this can be partly overcome by defining multiple rigid fragments for docking, [34] nevertheless, this remains a weakness of this approach.
Database
The elimination of redundant information means that frequently a new conformation for a molecule can be identified by the addition of a set of coordinates for a single atom. Four conformations for phenol can be encoded by representing the six carbon atoms, the oxygen, and the ring hydrogens a single time and then in a separate hierarchy level representing the four hydroxyl hydrogen positions. Benzene-1,3-diol can be used to demonstrate recombination. The eight heavy atoms and four ring hydrogens are represented a single time and both hydroxyl hydrogens are represented in four different conformations, positions A, B, C, and D. For this example, the input may contain only four conformations with hydrogens in positions AA, BB, CC, DD. The hierarchy would recombine these to create all 16 possibilities.
Calculation Run Time
The CPU time for a docking calculation correlates with the size of the binding site. Large sites (neutral endopeptidase) or solvent exposed (thrombin) require the longest time. For these sites very little of the conformational ensemble can be pruned and much time is spent expanding the entire conformational ensemble distal to the receptor.
Methodological Caveats
A potential problem for the method is the possibility of side chains from different conformations being recombined such that they clash (internal energies are not evaluated). Initial tests suggest that this occurs rarely, but does occasionally occur. Additionally, the organization of the hierarchy for specific molecules can be problematic. If the rigid fragment of the ligand does not bind in the region targeted for docking (in the DOCK series of programs, defined by matching “spheres”) [35], good poses will be missed. This was the problem encountered in neutral endopeptidase; as alluded to, the problem can be partly addressed by generating multiple ensembles based on multiple different rigid fragments, [34] or by altering the location of the docking spheres and possibly performing multiple docking calculations. Finally, this method had not been investigated heavily in the context of allowing for receptor flexibility, as other methods have done [11,13,36–40] (though see ref. [41]).
CONCLUSION
The hierarchical organization of molecular information described here allows for concise representation of multiple ligand conformations and allows us to aggressively prune the docking search tree. Especially in targets with flexible ligands, this improves our ability to identify ligands in docking screens of a diverse molecular database. For flexible molecules that can adopt many conformations, the method increases selectivity for ligands, improves sampling near the native binding modes, and does so at little computational cost. In our own lab, we now use this hierarchical method regularly, and it has lead to the discovery of new ligands in several targets [41–44].
METHODS
Generation of the Database
Conformations were generated using Filter and Omega version 0.99 from OpenEye Software. Molecules exported from the MDDR were filtered to remove reactive functionalities, aliphatic chains longer than hexane, and molecules containing silicon. We modified the default torsion and atom type libraries to generate conformations that contained the bond torsions and formal charges we thought most appropriate. For some molecules, whose pKa is likely to be near 7.2, multiple charge states were represented as independent molecules. We allowed for up to 2,500 conformations of any given molecule to be written out, many more might be implied via side chain recombination at the time of docking. We required that conformations have an RMS deviation of at least 0.8Å different from any other conformations and that the internal energy be less than 8 kcal/mol from the global minimum calculated by Omega. Molecules with more than 15 rotatable bonds were treated as rigid. Selected non-aromatic ring systems were puckered. If the puckered ring occurred in the largest “rigid” fragment the 2,500 conformations were divided among the different puckers, with each pucker representing a new ensemble. The rigid fragment of each molecule was identified as the largest group of internally rigid atoms. As conformations were generated, this rigid part of the molecule was kept in the same reference frame (Fig. 1B).
The hierarchy for each molecule was generated empirically based on the atom position in each ensemble (Fig. 1C). Looking at all conformations of a given molecule, the atoms in the same position in all conformations were identified as the first level of the hierarchy or rigid fragment. Next a recursive algorithm identified all groups that branch off from the current group. All atoms (based on number of unique positions in the ensemble) in the group were written out and again all branches were identified. After the hierarchy was created, conformations for rotatable hydrogens were sampled (OH in 120º increments and sp2NH in 180º increments). During docking this compact ensemble representation is expanded to efficiently dock all possible conformations (Fig. 1D).
Docking Scheme
The hierarchy-docking method is implemented in DOCK.3.5.54. The ligand hierarchy is fit one group at a time using the orient-and-evaluate scheme longstanding in DOCK [35,45]. Docking begins by generating a translation-rotation matrix for a specific set of matching atoms and the rigid fragment of the ligand is oriented in the binding site. If the rigid fragment does not clash into the receptor, flexible groups are added to the ligand according the same rotation-translation matrix. If the rigid fragment clashes with the translation receptor, a new orientation is tried. Looping over each flexible branch of the molecule, the branch is explored only until one complete conformation can be built. If a given branch cannot be built due to clashes with the receptor, alternate branches are considered. If no conformation of a branch fits, a new orientation is tried. Assuming at least one conformation of the molecule can be built, all remaining conformations are examined. After all conformations of all branches have been evaluated, the scores from the best scoring branches are added and the best conformation for this orientation is saved. This process is repeated for each orientation of each molecule in the binding site.
Docking
The enzyme structures were prepared systematically by a semi-automatic script. Initial steric fit was evaluated on a grid generated by the DOCK-associated program DISTMAP. The electrostatic component of the interaction energy was calculated on a grid generated with Delphi.[46] Prior to calculation of the DelPhi grids, the binding sites were filled with pseudo-atoms (spheres) to better model the dielectric in the occupied binding site. Chemgrid [47] was used to calculate a Van der Waals potential grid for each binding site [48].
Acknowledgments
This work was supported by GM59957 from the NIH (to BKS). We thank Matt Stahl and Geoff Skillman from OpenEye Scientific Software (Santa Fe NM) for their assistance with Omega and Filter, and Alan Graves for reading this manuscript. We thank MDL Inc. (San Leandro, CA) for providing the MDDR database.
References
- 1.Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 2.Kussell E, Shimada J, Shakhnovich EI. Excluded volume in protein side-chain packing. J Mol Biol. 2001;311:183–193. doi: 10.1006/jmbi.2001.4846. [DOI] [PubMed] [Google Scholar]
- 3.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 4.Taylor JS, Burnett RM. DARWIN: a program for docking flexible molecules. Proteins. 2000;41:173–191. [PubMed] [Google Scholar]
- 5.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19 [Google Scholar]
- 6.Hou T, Wang J, Chen L, Xu X. Automated docking of peptides and proteins by using a genetic algorithm combined with a tabu search. Protein Eng. 1999;12:639–648. doi: 10.1093/protein/12.8.639. [DOI] [PubMed] [Google Scholar]
- 7.Oshiro CM, Kuntz ID, Dixon JS. Flexible ligand docking using a genetic algorithm. J Comput Aided Mol Des. 1995;9:113–130. doi: 10.1007/BF00124402. [DOI] [PubMed] [Google Scholar]
- 8.Yang JM, Kao CY. A family competition evolutionary algorithm for automated docking of flexible ligands to proteins. IEEE Trans Inf Technol Biomed. 2000;4:225–237. doi: 10.1109/4233.870033. [DOI] [PubMed] [Google Scholar]
- 9.Totrov M, Abagyan R. Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins. 1997;Suppl:215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 10.Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: applications of AutoDock. J Mol Recognit. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez-Recio J, Totrov M, Abagyan R. ICM-DISCO docking by global energy optimization with fully flexible side-chains. Proteins. 2003;52:113–117. doi: 10.1002/prot.10383. [DOI] [PubMed] [Google Scholar]
- 12.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 13.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 14.Bohm HJ. The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J Comput-Aid Mol Des. 1992;6:61–78. doi: 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]
- 15.Caflisch A, Niederer P, Anliker M. Monte Carlo Docking of Oligopeptides to Proteins. Proteins. 1992;13:223–230. doi: 10.1002/prot.340130305. [DOI] [PubMed] [Google Scholar]
- 16.Aronov AM, Suresh S, Buckner FS, Van Voorhis WC, Verlinde CL, Opperdoes FR, Hol WG, Gelb MH. Structure-based design of submicromolar, biologically active inhibitors of trypanosomatid glyceraldehyde-3-phosphate dehydrogenase. Proc Natl Acad Sci USA. 1999;96:4273–4278. doi: 10.1073/pnas.96.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majeux N, Scarsi M, Apostolakis J, Ehrhardt C, Caflisch A. Exhaustive docking of molecular fragments with electrostatic solvation. Proteins. 1999;37:88–105. [PubMed] [Google Scholar]
- 18.Rarey M, Kramer B, Lengauer T. A Fast Flexible Docking Method using an Incremental Construction Algorithm. J Mol Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 19.Schellhammer I, Rarey M. FlexX-Scan: fast, structure-based virtual screening. Proteins. 2004;57:504–517. doi: 10.1002/prot.20217. [DOI] [PubMed] [Google Scholar]
- 20.Ewing TJA, Kuntz ID. Critical Evaluation of Search Algorithms for Automated Molecular Docking and Database Screening. J Comp Chem. 1997;18:1175–1189. [Google Scholar]
- 21.Ewing TJ, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 22.Makino S, Ewing TJ, Kuntz ID. DREAM++: flexible docking program for virtual combinatorial libraries. J Comput Aided Mol Des. 1999;13:513–532. doi: 10.1023/a:1008066310669. [DOI] [PubMed] [Google Scholar]
- 23.Walters WP, Ajay, Murcko MA. Recognizing molecules with drug-like properties. Curr Opin Chem Biol. 1999;3:384–387. doi: 10.1016/s1367-5931(99)80058-1. [DOI] [PubMed] [Google Scholar]
- 24.Oprea T. I. Current trends in lead discovery: are we looking for the appropriate properties? Mol Divers. 2002;5:199–208. doi: 10.1023/a:1021368007777. [DOI] [PubMed] [Google Scholar]
- 25.Miller MD, Sheridan RP, Kearsley SK. SQ: a program for rapidly producing pharmacophorically relevent molecular superpositions. J Med Chem. 1999;42:1505–1514. doi: 10.1021/jm9806143. [DOI] [PubMed] [Google Scholar]
- 26.McGregor MJ, Muskal SM. Pharmacophore fingerprinting. 2. Application to primary library design. J Chem Inf Comput Sci. 2000;40:117–125. doi: 10.1021/ci990313h. [DOI] [PubMed] [Google Scholar]
- 27.Jenkins JL, Glick M, Davies JW. A 3D similarity method for scaffold hopping from known drugs or natural ligands to new chemotypes. J Med Chem. 2004;47:6144–6159. doi: 10.1021/jm049654z. [DOI] [PubMed] [Google Scholar]
- 28.Sheridan RP, Shpungin J. Calculating similarities between biological activities in the MDL Drug Data Report database. J Chem Inf Comput Sci. 2004;44:727–740. doi: 10.1021/ci034245h. [DOI] [PubMed] [Google Scholar]
- 29.Ajay A, Walters WP, Murcko M. A. Can We learn To distinguish between "Drug-like" and "Nondrug-like" molecules? J Med Chem. 1998;41:3314–3324. doi: 10.1021/jm970666c. [DOI] [PubMed] [Google Scholar]
- 30.Frimurer TM, Bywater R, Naerum L, Lauritsen LN, Brunak S. Improving the odds in discriminating "drug-like" from "non drug-like" compounds. J Chem Inf Comput Sci. 2000;40:1315–1324. doi: 10.1021/ci0003810. [DOI] [PubMed] [Google Scholar]
- 31.Muegge I, Heald SL, Brittelli D. Simple selection criteria for drug-like chemical matter. J Med Chem. 2001;44:1841–1846. doi: 10.1021/jm015507e. [DOI] [PubMed] [Google Scholar]
- 32.Lorber DM, Shoichet BK. Flexible Ligand Docking Using Conformational Ensembles. Protein Sci. 1998;7:938–950. doi: 10.1002/pro.5560070411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller MD, Kearsley SK, Underwood DJ, Sheridan RP. FLOG: a system to select 'quasi-flexible' ligands complementary to a receptor of known three-dimensional structure. J Comput Aided Mol Des. 1994;8:153–174. doi: 10.1007/BF00119865. [DOI] [PubMed] [Google Scholar]
- 34.Brenk, R.; Irwin, J. J.; Shoichet, B. K. Here be dragons: docking and screening in an uncharted region of chemical space. J. Biol. Screening, 2005 [Accepted for publication]. [DOI] [PMC free article] [PubMed]
- 35.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A Geometric Approach to Macromolecule-Ligand Interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 36.Claussen H, Buning C, Rarey M, Lengauer T. FlexE: efficient molecular docking considering protein structure variations. J Mol Biol. 2001;308:377–395. doi: 10.1006/jmbi.2001.4551. [DOI] [PubMed] [Google Scholar]
- 37.Zavodszky MI, Lei M, Thorpe MF, Day AR, Kuhn LA. Modeling correlated main-chain motions in proteins for flexible molecular recognition. Proteins. 2004;57:243–261. doi: 10.1002/prot.20179. [DOI] [PubMed] [Google Scholar]
- 38.Cavasotto CN, Abagyan RA. Protein flexibility in ligand docking and virtual screening to protein kinases. J Mol Biol. 2004;337:209–225. doi: 10.1016/j.jmb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Taylor RD, Jewsbury PJ, Essex JW. FDS: flexible ligand and receptor docking with a continuum solvent model and soft-core energy function. J Comput Chem. 2003;24:1637–1656. doi: 10.1002/jcc.10295. [DOI] [PubMed] [Google Scholar]
- 40.Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 41.Wei BQ, Weaver LH, Ferrari AM, Matthews BW, Shoichet BK. Testing a flexible-receptor docking algorithm in a model binding site. J Mol Biol. 2004;337:1161–1182. doi: 10.1016/j.jmb.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 42.Powers RA, Morandi F, Shoichet BK. Structure-based discovery of a novel, noncovalent inhibitor of AmpC beta-lactamase. Structure (Camb) 2002;10:1013–1023. doi: 10.1016/s0969-2126(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 43.Soelaiman S, Wei BQ, Bergson P, Lee YS, Shen Y, Mrksich M, Shoichet BK, Tang WJ. Structure-based inhibitor discovery against adenylyl cyclase toxins from pathogenic bacteria that cause anthrax and whooping cough. J Biol Chem. 2003;278:25990–25997. doi: 10.1074/jbc.M301232200. [DOI] [PubMed] [Google Scholar]
- 44.Ferrari AM, Wei BQ, Costantino L, Shoichet BK. Soft docking and multiple receptor conformations in virtual screening. J Med Chem. 2004;47:5076–5084. doi: 10.1021/jm049756p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DesJarlais R, Sheridan RP, Seibel GL, Dixon JS, Kuntz ID, Venkataraghavan R. Using Shape Complementarity as an Initial Screen in Designing Ligands for a Receptor Binding Site of Known Three-Dimensional Structure. J Med Chem. 1988;31:722–729. doi: 10.1021/jm00399a006. [DOI] [PubMed] [Google Scholar]
- 46.Gilson MK, Honig BH. Calculation of electrostatic potentials in an enzyme active site. Nature. 1987;330:84–86. doi: 10.1038/330084a0. [DOI] [PubMed] [Google Scholar]
- 47.Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, Profeta S, Weiner P. A New Force Field for Molecular Mechanical Simulation of Nucleic Acids and Proteins. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 48.Meng EC, Shoichet B, Kuntz ID. Automated Docking with Grid-Based Energy Evaluation. J Comp Chem. 1992;13:505–524. [Google Scholar]