

Abstract
Biological and epidemiologic evidence suggest that androgen or its receptor may play a role in ovarian cancer pathogenesis. The most notable genetic factor influencing androgen receptor (AR) activity is the functional cytosine, adenine, guanine (CAG) repeat in which length is inversely proportional to its transactivational activity. Additional genetic variation due to single nucleotide polymorphisms in the AR gene may be captured through haplotypes. We genotyped the CAG microsatellite and six haplotype-tagging single nucleotide polymorphisms (rs962458, rs6152, rs1204038, rs2361634, rs1337080, rs1337082) of the androgen receptor gene in 987 ovarian cancer cases and 1,034 controls from a study conducted in New Hampshire and eastern Massachusetts between May 1992 and July 2003. We estimated haplotype frequencies and calculated odds ratios with 95% confidence intervals to evaluate the association between the haplotypes and the AR CAG microsatellite with ovarian cancer risk. We observed that carriage of two alleles with ≥22 CAG repeats was associated with an increased risk of ovarian cancer compared with carriage of two alleles with <22 CAG repeats (covariate-adjusted odds ratios, 1.31; 95% confidence intervals, 1.01-1.69). Five common haplotypes in the AR gene were identified, but no association between these and ovarian cancer risk was observed. Our results suggest that possession of two long AR alleles (≥22 CAG repeats) may be associated with increased risk of ovarian cancer compared with women with two short AR alleles (<22 CAG repeats).
Introduction
Epidemiologic and biological data suggest a role for androgens and perhaps their receptor in ovarian cancer development. Oral contraceptives, which suppress androgens, are inversely associated with ovarian cancer risk (1), whereas polycystic ovarian syndrome and central obesity, which are characterized by increased androgen levels, are associated with increased ovarian cancer risk (2, 3). Furthermore, in a nested case-control study, prediagnostic androgen levels were higher in ovarian cancer cases than in controls (4). In phase 2 clinical trials of antiandrogens as a treatment for ovarian cancer, investigators observed that antiandrogens reduced the tumor burden or stabilized the progression of ovarian cancer in some women who had previously failed at least one chemotherapy (5, 6). Although data from animal models and cell lines have been less consistent (7–9), showing both stimulation and inhibition of ovarian cell growth with androgen administration, the epidemiologic and clinical data seem to suggest a role for androgens in ovarian cancer.
Androgen receptors are present in both normal human ovarian epithelial cells and human ovarian cancer cell lines (10). Some studies reported between 65% and 90% of ovarian tumors possess androgen receptors (11, 12), whereas others reported low or decreased AR expression in ovarian tumors in comparison to normal ovarian cell lines (10, 13).
In exon 1 of the AR gene, a cytosine, adenine, guanine (CAG) trinucleotide repeat codes for a polyglutamine tract which normally ranges from 6 to 39 repeats (14). CAG repeat number varies by ethnicity with the longest AR alleles in Mexican-Americans (mean, 25; range, 16-32) and the shortest in African-Americans (mean, 20; range, 10-29; ref. 14). In vitro studies have shown that CAG repeat number is inversely proportional to AR transactivational activity, which initiates transcription of target genes in coordination with transcriptional cofactors (14, 15). Decreased transactivational activity is evident with each additional CAG repeat (14), and possession of >40 CAG repeats results in Kennedy’s disease, a motorneuron disease linked to androgen insensitivity (16). Therefore, we would expect longer AR alleles to be associated with reduced expression of AR target genes compared with short AR alleles. Epidemiologic studies have shown an association between CAG allele length and a variety of androgen-related conditions, including benign prostatic hyperplasia with shorter length (17), impaired spermatogenesis with longer length (18), hirsutism with shorter length (19), and polycystic ovarian syndrome accompanied by high testosterone levels with shorter length (20).
AR allele length has also been studied in connection with hormonally related cancers in both men and women. Multiple studies have established an association between short AR allele length and increased prostate cancer risk (reviewed in ref. 21) and data on associations with other cancers is emerging, including breast cancer (22, 23) and endometrial cancer (24, 25). The association between the AR CAG repeat polymorphism and ovarian cancer has been previously evaluated in four studies restricted to high-risk populations (26–29) and two case-control studies (30, 31), but results were inconsistent due to population differences and sample size.
Although the CAG repeat polymorphism in exon 1 has received most of the attention regarding genetic variation in the AR gene, analysis of single nucleotide polymorphisms (SNPs) may also prove to be informative. Several single nucleotide changes have been previously identified in the AR gene (32, 33), however, these genetic changes are rare and have not been evaluated in relation to disease risk on a population level. Several studies have shown that the use of haplotypes, the linear combination of linked SNPs on a chromosome, is a powerful way to identify genetic variation (34, 35). In this study, we evaluate the association between ovarian cancer risk and genetic variation in the AR gene, using both haplotypes and the CAG repeat polymorphism in 987 ovarian cancer cases and 1,034 controls from New Hampshire and eastern Massachusetts.
Materials and Methods
This population-based study was approved by the Human Subjects Review Committees at both Brigham and Women’s Hospital and Dartmouth Medical School, and each participant provided a signed informed consent. Data and specimens from this New England–based case-control study of ovarian cancer come from two enrollment phases corresponding to two funding periods. Phase 1 began in May 1992 and ended in March 1997, whereas phase 2 began in July 1998 and ended in July 2003. During the combined study phases, we identified 2,347 women (1,080 from phase 1 and 1,267 from phase 2) residing in eastern Massachusetts or New Hampshire with a diagnosis of incident ovarian cancer from hospital tumor boards and statewide cancer registries. Of these, 502 (203 phase 1 and 299 phase 2) could not be contacted because they had died (n = 210), moved or had no telephone (n = 160), did not speak English (n = 37), had a non–ovarian primary tumor after review (n = 93), or lived outside the study area (n = 2). Physicians declined permission to contact 232 (126 phase 1 and 106 phase 2) of the remaining cases, and 307 cases (136 phase 1 and 171 phase 2) declined or were too ill to participate. Of these, 1,231 cases had epithelial ovarian tumors, including tumors of borderline malignancy (563 phase 1 and 668 phase 2). Of these, 1,075 cases (463 phase 1 and 612 phase 2) gave a blood specimen at the time of enrollment, and at the time of this study, 987 case specimens were available for genotyping.
Controls in phase 1 were selected using random digit dialing supplemented with residents lists for older controls and has been described previously (36). Briefly, for the random digit dialing, in ~10% of households the answerer declined to provide a household census and 80% of households an age- and sex-matched control for a case could not be made or was ineligible because of a previous oophorectomy. Of the remaining 10% of screened households containing a potentially eligible control, 72% agreed to participate. Because random digit dialing proved inefficient for identifying controls >60 years old in Massachusetts, we identified older controls in Massachusetts by randomly selecting women from the residents’ lists (townbooks) matched to cases by community and age within 4 years. Of 328 sampled townbook controls, 21% could not be reached, 18% were ineligible, and 30% declined to participate. A total of 523 (421 random digit dialing and 102 townbook) controls were enrolled from phase 1 of the study.
Controls for phase 2 were identified through townbooks in Massachusetts and drivers’ license lists in New Hampshire. Age matching was accomplished by sampling controls based upon the age distribution of cases in the previous phase of the study with adjustment as current cases were enrolled. Of the 1,843 potential controls identified in phase 2, 576 were ineligible because they had died, moved, had no telephone, did not speak English, had no ovaries, or were seriously ill, and 546 potential controls declined participation either by phone or by “opt out” postcard. Of the 1,244 controls (523 phase 1 and 721 phase 2) that enrolled in the study, 1,098 donated a blood specimen. For the purposes of this study, 1,034 control specimens were available for genotyping.
Questionnaire data.
Risk factors for ovarian cancer and other potential confounders were collected by questionnaires administered in-person. To avoid the possible influence of preclinical disease on exposure status, cases were asked about exposures that occurred at least 1 year before diagnosis, and controls were asked about exposures that occurred >1 year before the interview date.
Genotyping methods.
Heparinized blood was collected at the time of enrollment, and separated into plasma, red cell, and buffy coat (white cell) components. DNA was extracted from buffy coat using a QIAamp 96 DNA blood kit (Qiagen, Valencia, CA).
Genotyping was done at the Dana-Farber Cancer Institute/Harvard Cancer Center High-Throughput Genotyping Core, a unit of the Harvard Partners Genotyping Facility. For the AR CAG repeat polymorphism, genomic DNA was PCR-amplified using fluorescently labeled primers. The length of these fragments varied by the number of CAG repeats. Fragments were run on denaturing polyacrylamide gels on the Applied Biosystems Prism 3100 and analyzed by Applied Biosystems Prism Genescan automated fluorescence detection (Applied Biosystems, Foster City, CA). Fragment lengths were determined from a series of sequenced PCR samples of varying size.
Haplotype tagging SNPs (htSNP) were selected by the National Cancer Institute Cohort Consortium and are available on the world wide web.6 Their method of htSNP selection has been described elsewhere (37). Briefly, SNPs identified through previous report and resequencing were genotyped, and haplotypes were estimated in a multiethnic sample, including African-Americans, Japanese, Latinos, and Caucasians.
Haplotype block structure was determined for each ethnic group based on criteria outlined by Gabriel et al. (38) using 90% confidence bounds of D′ to define sites of historical recombination between SNPs. Six htSNPs were selected for Caucasians in three haplotype blocks based on the squared correlations (Rh2) between the true haplotypes and estimated haplotype probabilities for each subject (37). The first block contains two SNPs (rs962458 and rs6152), the second block contains three SNPs (rs1204038, rs2361634, and rs1337080), and the third block contains only one SNP (rs1337082).
We then genotyped these six SNPs in the 987 cases and 1,034 controls from our case-control study. Genotyping assays for all six SNPs were done by the 5′ nuclease assay (TaqMan) on the Applied Biosystems Prism 7900HT Sequence Detection System (Applied Biosystems). TaqMan primers, probes, and conditions for genotyping assays are available upon request. Genotyping was done by laboratory personnel blinded to case-control status, and blinded quality control samples were included to validate genotyping procedures. Over 95% of the samples were successfully genotyped for each of the polymorphisms. Genotyping failures were considered missing data.
Statistical analysis.
The distribution of CAG repeats for the short allele, the long allele and the average of the short and long alleles were compared between cases and controls using the Wilcoxon rank sum test. Binary categories of CAG repeat length (≥22 versus <22, ≥27 versus <27, ≥29 versus <29, ≥19 versus <19) were created in order to replicate analyses done previously in other study populations (30, 31). In addition, CAG length was evaluated in categories separately for the longer allele (≤21, 22-23, 24-25, ≥26) and the shorter allele (≥22, 21, 20, ≤19) based on work done by Santarosa and colleagues (30). Because transcriptional activity of the androgen receptor decreases linearly with CAG number (14), CAG length was also evaluated as a continuous variable in relation to ovarian cancer risk. Odds ratios (OR) and 95% confidence intervals (CI) were calculated using unconditional logistic regression with adjustment for age (continuous) and study center (Massachusetts or New Hampshire) to evaluate the association between CAG repeat number and ovarian cancer risk. In order to evaluate the association between variation in the AR gene and ovarian cancer risk independent of ovarian cancer risk factors, multivariate models were run with adjustment for parity (0, 1, 2+ livebirths), oral contraceptive use (<3 months or never, ≥3 months), and family history of a mother or sister with breast or ovarian cancer (yes, no).
Furthermore, we assessed possible effect modification by a priori variables that may influence ovarian cancer risk or androgen levels including: family history of breast or ovarian cancer in mother or sister (yes, no), parity (nulliparous, parous), oral contraceptive use (<3 months or never, ≥3 months), menopausal status (premenopausal, postmenopausal), fertility status (no reported infertility, infertile), body mass index (≤24.4, >24.4 kg/m2), and polycystic ovarian syndrome (yes, no). Jewish or non-Jewish religion (as a marker for likelihood of being a BRCA1/2 carrier) was also examined. In addition, whether the association between CAG length and ovarian cancer risk differed by age of onset (≤40, >40 years) was evaluated. To test for statistical significance of interactions between these a priori exposures and AR genotypes, likelihood ratio tests were used to compare nested models that included terms for all combinations of the AR genotype and potential effect modifiers to the models with only the main effects. The SAS version 8 statistical package (SAS Institute, Cary, NC) was used for all statistical analyses of the CAG microsatellite.
For the haplotype analysis, we first determined whether the SNP genotypes were in Hardy-Weinberg equilibrium using χ2 tests. Haplotypes were estimated and evaluated within predetermined haplotype blocks, reported by the National Cancer Institute Cohort Consortium.7 Haplotype frequencies were estimated in cases and controls together using the expectation-maximization (E-M) algorithm (39). Posterior probabilities of the haplotypes given the observed genotypes were calculated for each individual, as previously described by Zaykin and colleagues (40). To ensure adequate statistical power, only haplotypes with >5% frequency were used to evaluate the association between AR haplotypes and ovarian cancer. To evaluate linkage disequilibrium across the entire gene, the linkage disequilibrium between all pairwise combinations of AR htSNPs was calculated, using Lewontin’s D′ statistic (41).
To evaluate risk associated with carriage of each haplotype compared with carriage of the most common haplotype, ORs and 95% CIs for the haplotype analyses were calculated using unconditional logistic regression with adjustment for age (continuous) and study center (Massachusetts or New Hampshire). We further adjusted for the following ovarian cancer risk factors: parity (0, 1, 2+ livebirths), oral contraceptive use (<3 months or never, ≥3 months), and family history defined as having a mother or sister with breast or ovarian cancer (yes, no). Likelihood ratio tests were used to evaluate the global association between all common (>5% frequency) haplotypes together and disease status, adjusting for the above covariates. The SAS Genetics statistical package (SAS/Genetics, SAS Institute) was used for all haplotype analyses.
Allele frequencies for the AR polymorphisms are known to vary dramatically by ethnicity (42), which could lead to a spurious association because ovarian cancer risk also varies by ethnicity. We were underpowered to evaluate the association between AR polymorphisms and ovarian cancer risk among non-Caucasians (n = 82); consequently, all our analyses were restricted to Caucasians.
Results
The population characteristics of this case-control study population have been described previously (43). The AR CAG repeat polymorphism was successfully genotyped in 905 cases and 976 controls. CAG repeat length ranged from 11 to 34 repeats in cases and 8 to 3 6 repeats in controls. When only the longer allele for each participant was considered, we observed a bimodal distribution of allele lengths (Fig. 1); this bimodal distribution was not observed for shorter allele lengths (Fig. 2). For the shorter allele, we observed significantly fewer CAG repeats in controls (median, 20; range, 8-29) compared with cases (median, 21; range, 11-29; P = 0.002). There was no significant difference in the longer allele length between cases (median, 23; range, 18-34) and controls (median, 24; range, 13-36) or in the average of the two allele lengths between cases (median, 22; range, 15-30) and controls (median, 22; range, 13-29).
Figure 1.
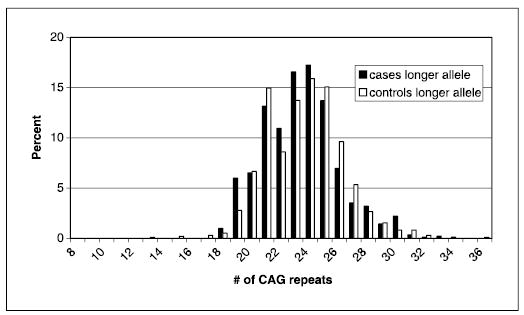
Distribution of AR CAG repeat number for the longer allele among ovarian cancer cases and controls (restricted to Caucasians).
Figure 2.

Distribution of AR CAG repeat number for the shorter allele among ovarian cancer cases and controls (restricted to Caucasians).
Compared with women who possessed two alleles with <22 CAG repeats, those who carried one allele with ≥22 CAG repeats had no increased risk of ovarian cancer (covariate-adjusted OR, 1.07; 95% CI, 0.85-1.35), but women who carried two alleles with ≥22 CAG repeats had a significantly increased risk of ovarian cancer (covariate-adjusted OR, 1.31; 95% CI, 1.01-1.69). Other CAG cutpoints (≥27 versus <27, ≥29 versus <29, ≥19 versus <19) were evaluated based on previous literature (30, 31), but these did not yield any significant associations (data not shown). We also considered the longer and shorter allele possessed by participant separately with respect to ovarian cancer risk. Compared with women whose shorter allele contained ≤19 CAG repeats, women whose shorter allele contained 20 or 21 repeats had a nonsignificant increase in risk of ovarian cancer, whereas those women with ≥22 CAG repeats on the shorter allele had a significant increase in risk (covariate-adjusted OR, 1.35; 95% CI, 1.07-1.71). We observed no association between length of the longer alleles and ovarian cancer risk (Table 1).
Table 1.
Association between AR CAG polymorphism and ovarian cancer risk
| Cases, n (%) | Controls, n (%) | Adjusted OR* (95% CI) | |
|---|---|---|---|
| ≥22 CAG repeats | |||
| 0 alleles | 212 (23) | 249 (26) | 1.00 |
| 1 allele | 432 (48) | 488 (50) | 1.07 (0.85-1.35) |
| 2 alleles | 261 (29) | 239 (24) | 1.31 (1.01-1.69) |
| 1 or 2 alleles | 693 (77) | 727 (75) | 1.15 (0.92-1.43) |
| CAG repeats in shorter allele | |||
| ≤19 | 282 (31) | 364 (37) | 1.00 |
| 20 | 170 (18) | 174 (18) | 1.22 (0.94-1.59) |
| 21 | 192 (21) | 199 (20) | 1.23 (0.95-1.58) |
| ≥22 | 261 (29) | 239 (24) | 1.35 (1.07-1.71) |
| CAG repeats in longer allele | |||
| ≤21 | 212 (23) | 249 (26) | 1.00 |
| 22-23 | 249 (28) | 218 (22) | 1.32 (1.01-1.72) |
| 24-25 | 280 (31) | 302 (31) | 1.12 (0.87-1.44) |
| ≥26 | 164 (18) | 207 (21) | 1.00 (0.75-1.32) |
| 1 CAG increase in shorter allele length | 1.08 (1.04-1.12) | ||
| 1 CAG increase in longer allele length | 1.00 (0.97-1.04) | ||
| 1 CAG increase in average allele length | 1.05 (1.01-1.10) | ||
NOTE: Restricted to Caucasians.
Adjusted for age (continuous), center (Massachusetts or New Hampshire), oral contraceptive use (<3 months or never, ≥3 months), parity (0, 1, 2+), family history of breast or ovarian cancer in mother or sister.
Because transactivational activity of the AR decreases linearly with each additional CAG repeat (14), we evaluated the association between a single trinucleotide increase in allele length and ovarian cancer risk. A single CAG increase length in the long allele did not significantly influence ovarian cancer risk. In contrast, a single trinucleotide increase in the short allele was associated with a significant increase in ovarian cancer risk (covariate-adjusted OR, 1.08; 95% CI, 1.04-1.12). We observed similar results with each one unit increase in the average allele length (covariate-adjusted OR, 1.05; 95% CI, 1.01-1.10; Table 1).
We observed no effect modification by body mass index (Table 2), an androgen-related characteristic. However, the association between AR allele length and ovarian cancer risk differed significantly by polycystic ovarian syndrome (P value for the interaction = 0.01) and fertility status (P for the interaction = 0.05). Among women with no reported infertility, those who possessed two alleles with ≥22 CAG repeats had a significantly increased risk of ovarian cancer (covariate-adjusted OR, 1.48; 95% CI, 1.11-1.98) compared with women with both alleles containing <22 repeats; in contrast, among infertile women, those with two alleles containing ≥22 CAG repeats had a nonsignificantly decreased risk of ovarian cancer (covariate-adjusted OR, 0.77; 95% CI, 0.42-1.41). Similarly, with the exception of religion, most ovarian cancer risk factors evaluated as potential effect modifiers did not significantly influence the association between CAG length and ovarian cancer risk (P value for the interaction = 0.04; Table 2). Jewish women with only one allele with ≥22 CAG repeats had a nonsignificant reduction in risk of ovarian cancer (covariate-adjusted OR, 0.36; 95% CI, 0.13-1.04), whereas non-Jewish women possessing one allele with ≥22 CAG repeats had a nonsignificant increase in ovarian cancer risk (covariate-adjusted OR, 1.12; 95% CI, 0.89-1.43). In contrast, women who possessed two alleles with ≥22 CAG repeats had a similar ovarian cancer risk in Jewish and non-Jewish women (Table 2). Although the androgen receptor influences progesterone receptor transcription (44), we observed no significant interaction between AR CAG repeat length and possession of the progesterone receptor PROGINS variant, which has been previously associated with ovarian cancer risk in our population (Table 2; ref. 43). However, the association between possession of two AR alleles with ≥22 CAG repeats and ovarian cancer risk was slightly stronger in women who possessed the PROGINS variant compared with women who did not.
Table 2.
Association between AR CAG repeats and ovarian cancer stratified by androgen-related variables or ovarian cancer risk factors
| Alleles with ≥22 CAG repeats | Cases, n (%) | Controls, n (%) | Adjusted OR* (95% CI) | Test for interaction† | |
|---|---|---|---|---|---|
| Androgen-related variables | |||||
| Body mass index‡ | P = 0.49 | ||||
| ≤24.4 | 0 | 97 (23) | 130 (27) | 1.00 | |
| 1 | 207 (48) | 241 (50) | 1.19 (0.85-1.66) | ||
| 2 | 126 (29) | 112 (23) | 1.58 (1.08-2.30) | ||
| >24.4 | 0 | 115 (24) | 119 (24) | 1.00 | |
| 1 | 224 (47) | 244 (50) | 0.96 (0.69-1.32) | ||
| 2 | 135 (28) | 126 (26) | 1.10 (0.76-1.58) | ||
| Fertility status | P = 0.05 | ||||
| No reported infertility | 0 | 161 (22) | 209 (27) | 1.00 | |
| 1 | 357 (48) | 397 (50) | 1.19 (0.92-1.54) | ||
| 2 | 218 (30) | 190 (24) | 1.48 (1.11-1.98) | ||
| Infertile | 0 | 51 (30) | 40 (22) | 1.00 | |
| 1 | 75 (44) | 91 (51) | 0.67 (0.39-1.15) | ||
| 2 | 43 (25) | 49 (27) | 0.77 (0.42-1.41) | ||
| Polycystic ovarian syndrome§ | P = 0.01 | ||||
| No | 0 | 194 (23) | 240 (25) | 1.00 | |
| 1 | 413 (48) | 479 (50) | 1.07(0.85-1.34) | ||
| 2 | 253 (29) | 231 (24) | 1.36 (1.04-1.76) | ||
| Yes | 0 | 18 (40) | 9 (35) | 1.00 | |
| 1 | 19 (42) | 9 (35) | 0.98 (0.31-3.12) | ||
| 2 | 8 (18) | 8 (31) | 0.45 (0.12-1.70) | ||
| Ovarian cancer risk factors | |||||
| Family history of breast or ovarian cancer in mother or sister | P = 0.11 | ||||
| No | 0 | 181 (25) | 209 (25) | 1.00 | |
| 1 | 356 (48) | 429 (51) | 0.97 (0.75-1.24) | ||
| 2 | 201 (27) | 205 (24) | 1.15 (0.86-1.52) | ||
| Yes | 0 | 31 (19) | 40 (30) | 1.00 | |
| 1 | 76 (46) | 59 (44) | 1.81 (0.99-3.30) | ||
| 2 | 60 (36) | 34 (26) | 2.64 (1.37-5.10) | ||
| Oral contraceptive use | P = 0.09 | ||||
| <3 months or never | 0 | 121 (25) | 88 (23) | 1.00 | |
| 1 | 226 (48) | 194 (51) | 0.87 (0.62-1.22) | ||
| 2 | 128 (27) | 99 (26) | 0.99 (0.67-1.46) | ||
| 3+ months | 0 | 91 (21) | 161 (27) | 1.00 | |
| 1 | 206 (48) | 294 (49) | 1.28 (0.93-1.76) | ||
| 2 | 133 (31) | 140 (24) | 1.66 (1.16-2.37) | ||
| Religion|| | P = 0.04 | ||||
| Jewish | 0 | 15 (18) | 7 (11) | 1.00 | |
| 1 | 26 (31) | 33 (53) | 0.36 (0.13-1.04) | ||
| 2 | 43 (51) | 22 (35) | 1.08 (0.37-3.15) | ||
| Non-Jewish | 0 | 197 (24) | 242 (26) | 1.00 | |
| 1 | 406 (49) | 455 (50) | 1.12 (0.89-1.43) | ||
| 2 | 218 (27) | 217 (24) | 1.27 (0.97-1.67) | ||
| Progesterone receptor polymorphism | |||||
| PROGINS genotype¶ | P = 0.52 | ||||
| Wild type | 0 | 146 (23) | 153 (26) | 1.00 | |
| 1 | 306 (49) | 304 (51) | 1.11 (0.83-1.47) | ||
| 2 | 170 (27) | 141 (24) | 1.30 (0.93-1.80) | ||
| Variant (PROGINS carrier) | 0 | 53 (22) | 81 (25) | 1.00 | |
| 1 | 104 (43) | 158 (49) | 0.95 (0.61-1.47) | ||
| 2 | 84 (35) | 81 (25) | 1.47 (0.92-2.37) | ||
NOTE: Restricted to Caucasians.
Adjusted for age (continuous), center (Massachusetts or New Hampshire), oral contraceptive use (<3 months or never, ≥3 months), parity (0, 1, 2+), family history of breast or ovarian cancer in mother or sister; stratification variables were excluded from the multivariate analysis.
Likelihood ratio test for interaction.
Those missing body mass index (n = 5 ) were excluded from analyses stratified by body mass index.
Adjusted for age and study center; multivariate model not possible due to small number of participants with polycystic ovarian syndrome.
Family history was excluded from models stratified by religion.
Those missing PROGINS genotype (n = 100) were excluded from analyses stratified by PROGINS genotype.
Because different ovarian cancer histologies may have distinct pathways to disease, AR’s association with ovarian cancer may vary by histologic category. Compared with carrying two alleles with <22 CAG repeats, carriage of two alleles with ≥22 CAG repeats was associated with a significantly increased risk of clear cell (covariate-adjusted OR, 2.00; 95% CI, 1.09-3.68); a nonsignificant increase in risk of serous invasive (covariate-adjusted O R, 1.32; 95% CI, 0.93-1.88) or mucinous (covariate-adjusted OR, 1.75; 95% CI, 0.98-3.10); and no association with serous borderline, endome-trioid, or undifferentiated ovarian cancer (data not shown).
Furthermore, we observed no association between CAG length and age at onset of ovarian cancer. The proportion of subjects who carried an allele with ≥22 CAG repeats did not differ significantly between cases diagnosed at ≤40 years of age and those diagnosed at >40 years (76% versus 79%).
Table 3 summarizes findings of our haplotype analysis. Six htSNPs spanning three haplotype blocks were genotyped in 987 cases and 1,034 controls. Genotypes that could not be obtained due to genotyping failure were considered missing data; consequently, the number of available genotypes varies by polymorphism [SNP 1 (n = 1,986), SNP 2 (n = 1,969), SNP 3 (n = 1,935), SNP 4 (n = 1,985), SNP 5 (n = 1,963), SNP 6 (n = 1,954)]. Of the four possible haplotypes for AR block 1, three occurred with >5% frequency and none of these were significantly associated with ovarian cancer risk. Of eight possible haplotypes in AR haplotype block 2, four occurred with >5% frequency and none were associated with ovarian cancer risk. Because AR haplotype block 3 consists of only one SNP, there were two unambiguous haplotypes and these were not associated with disease (Table 3).
Table 3.
AR haplotype blocks and ovarian cancer risk
| Frequency
|
|||||||
|---|---|---|---|---|---|---|---|
| Haplotype ID | htSNPs* | Cases | Controls | Crude OR† (95% CI) | Adjusted OR‡ (95% CI) | ||
| AR BLOCK 1 | SNP 1 | SNP 2 | |||||
| 1a | 0 | 0 | 0.84 | 0.85 | 1.00 | 1.00 | |
| 1b | 0 | 1 | 0.08 | 0.09 | 0.90 (0.57-1.43) | 0.86 (0.53-1.38) | |
| 1c | 1 | 1 | 0.07 | 0.07 | 1.28 (0.77-2.13) | 1.20 (0.71-2.03) | |
| AR BLOCK 2 | SNP 3 | SNP 4 | SNP 5 | ||||
| 2a | 0 | 0 | 0 | 0.78 | 0.78 | 1.00 | 1.00 |
| 2b | 0 | 1 | 0 | 0.06 | 0.07 | 1.01 (0.60-1.69) | 1.10 (0.64-1.86) |
| 2c | 1 | 0 | 0 | 0.08 | 0.09 | 0.91 (0.57-1.45) | 0.85 (0.52-1.38) |
| 2d | 1 | 0 | 1 | 0.07 | 0.07 | 1.21 (0.72-2.03) | 1.17 (0.69-1.98) |
| AR BLOCK 3 | SNP 6 | ||||||
| 3a | 0 | 0.87 | 0.87 | 1.00 | 1.00 | ||
| 3b | 1 | 0.13 | 0.13 | 1.02 (0.78-1.33) | 1.05 (0.79-1.38) | ||
NOTE: Restricted to Caucasians.
0, wild-type allele; 1, variant allele.
Adjusted for the age (continuous) and center (Massachusetts or New Hampshire).
Adjusted for age (continuous), center (Massachusetts or New Hampshire), oral contraceptive use (<3 months or never, >3 months), parity (0, 1, 2+ livebirths), family history of breast or ovarian cancer in mother or sister.
Linkage disequilibria of 0.8 or more were observed between all six htSNPs (Supplemental Table S2). Because we observed a high degree of linkage across the entire gene, we also evaluated haplotypes estimated from all six htSNPs. Out of 64 possible haplotypes, 5 occurred with >5% frequency. There was no global association between these five haplotypes and ovarian cancer risk, and none were individually associated with ovarian cancer risk (Table 4). Linkage disequilibrium analyses revealed that the AR CAG repeat is significantly associated with five of the six AR htSNPs (data not shown).
Table 4.
AR long-range haplotypes and ovarian cancer risk
| htSNPs* |
Frequency
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Haplotype | 1 | 2 | 3 | 4 | 5 | 6 | Cases | Controls | Crude OR† (95% CI) | Adjusted OR‡ (95% CI) |
| A | 0 | 0 | 0 | 0 | 0 | 0 | 0.72 | 0.72 | 1.00 | 1.00 |
| B | 0 | 0 | 0 | 0 | 0 | 1 | 0.06 | 0.06 | 1.10 (0.62-1.95) | 1.19 (0.66-2.14) |
| C | 0 | 0 | 0 | 1 | 0 | 0 | 0.06 | 0.07 | 1.01 (0.60-1.69) | 1.11 (0.65-1.90) |
| D | 0 | 1 | 1 | 0 | 0 | 1 | 0.07 | 0.07 | 1.02 (0.62-1.69) | 0.98 (0.59-1.64) |
| E | 1 | 1 | 1 | 0 | 1 | 1 | 0.06 | 0.06 | 1.19 (0.70-2.04) | 1.14 (0.65-1.97) |
NOTE: Restricted to Caucasians. Global test, χ2 = 0.50, P = 0.97.
0, wild-type allele; 1, variant allele.
Adjusted for the age (continuous) and center (Massachusetts or New Hampshire).
Adjusted for age (continuous), center (Massachusetts or New Hampshire), oral contraceptive use (<3 months or never, >3 months), parity (0, 1, 2+ livebirths), family history of breast or ovarian cancer in mother or sister.
Discussion
To our knowledge, this is the first study to evaluate the association between AR haplotypes and ovarian cancer risk, and, to date, the largest study to evaluate theAR CAG repeat polymorphism in relation to ovarian cancer risk. In our analysis of the AR CAG repeat polymorphism, we observed an increase in ovarian cancer risk with a greater number of CAG repeats. We estimated AR haplotypes using htSNPs identified by the National Cancer Institute Cohort Consortium and observed five AR haplotypes occurring with >5% frequency in our study population. There was no association between these haplotypes and ovarian cancer risk.
Our findings regarding the AR CAG trinucleotide repeats and ovarian cancer risk are largely in line with other studies addressing this association. In a population-based case-control study with 319 cases and 853 controls, Spurdle and colleagues observed a larger proportion of cases carrying two alleles with ≥22 CAG repeats than controls, similar to our own findings, but the association was not significant (OR, 1.18, 95% CI, 0.78, 1.78; ref. 31). In a smaller hospital case-control study based in Italy, Santarosa and colleagues found a stronger association between carriage of two alleles with ≥22 AR CAG repeats (OR, 3.45; 95% CI, 1.42-8.34) than we observed in our own population (OR, 1.31; 95% CI, 1.01-1.69; ref. 30). Variation in the strength of the association observed in studies may be partly attributable to population differences because 24% of the controls in our study carried two alleles with ≥22 AR CAG repeats whereas 26% of the controls in Spurdle’s study and only 18% of the controls in Santarosa’s study carried two alleles with ≥22 AR CAG repeats. Because AR CAG repeat length is known to vary by ethnicity (14, 42), differences due to nationality or ancestral background could contribute to allele length differences in these populations. In addition, observed associations in our study were of marginal significance. Therefore, these results could potentially be due to chance.
The association between AR CAG length and ovarian cancer has also been evaluated among high-risk populations (26–29). In two studies of BRCA1/2 carriers, no association was observed between AR CAG length and ovarian cancer risk (26, 27). However, carrying fewer AR CAG repeats was correlated with a younger age of ovarian cancer diagnosis among those carrying a BRCA1/2 mutation or of Ashkenazi Jewish descent (26, 28). In contrast, Menin and colleagues observed no association between AR CAG length and age of diagnosis among hereditary cases of ovarian cancer (29). In our data, we observed no association between AR CAG repeat number and age of onset and no significant interaction between CAG length and family history. However, we did observe a significant interaction between CAG length and religion. Non-Jewish women had an increased risk of ovarian cancer with longer CAG length whereas among Jewish women there was no association between CAG length and ovarian cancer risk.
For the androgen-related variables, we observed an increased risk of ovarian cancer with longer CAG length among women with no reported infertility or polycystic ovarian syndrome. Because both infertility and polycystic ovarian syndrome are associated with higher androgen levels, these women may have a disruption in the androgen/androgen receptor pathway that renders CAG length irrelevant with respect to their ovarian cancer risk.
Studies of breast and endometrial cancer also suggest that longer AR alleles may contribute to carcinogenesis in women. Some breast cancer studies report an increased risk of breast cancer with a greater number of AR CAG repeats (22, 45), whereas others report no association (23, 46). Similarly, a greater number of AR CAG repeats were associated with increased endometrial cancer risk in two small case-control studies (24, 25). Consistent with our findings, these studies suggest that longer AR alleles may contribute to cancer risk.
Although, collectively, the results of these epidemiologic studies suggest that longer AR CAG alleles increase the risk of ovarian cancer, the biological pathway is unclear. What is known to date is that each additional AR CAG repeat results in a 1.7% decrease in transactivational activity (14). In addition, increased AR CAG length negatively influences interaction with coactivators involved in the stabilization of the transcriptional complex and in the functional activity of the receptor, resulting in decreased expression of androgen-responsive target genes (14, 47). Androgen receptor knock-out mice show that androgens exert their physiologic effects through the androgen receptor, and gene expression assays comparing the ovaries of AR knock-out mice to those of normal mice have identified target genes in the ovary including follicle stimulating hormone receptor, insulin-like growth factor-I receptor, progesterone receptor, and p21 (44, 48). Female mice lacking the AR gene show that AR influences many parts of the female reproductive system including oocyte production, progesterone production, and endometrial growth, but the consequences for the ovarian epithelium, the presumed precursor tissue of epithelial ovarian cancer, remains unclear (44). Given the wide range of target genes and physiologic effects of the androgen receptor, predicting the role of AR target gene activation in ovarian carcinogenesis is difficult. We observed that long AR alleles are associated with increased ovarian cancer risk. Because increased AR CAG repeat length is associated with decreased activation of androgen target genes, our findings seem in conflict with the theory that androgens increase ovarian cancer risk. However, not all androgens act through the androgen receptor (some are converted to estrogens) and the androgen receptor regulates a variety of androgen target genes; thus, our results are not necessarily at odds with this theory.
A unique misclassification issue may affect the study of the androgen receptor due to its location on the X chromosome. At an early stage of development, most likely the late blastula stage, one of the two X chromosomes carried in each cell is inactivated (49). Whether the maternal or paternal X chromosome is inactivated varies from cell to cell, however, all the cells that descend from the initial inactivated cell will retain the same X chromosome inactivation (49). Consequently, genotypes of AR polymorphisms would not necessarily reflect the active AR allele because one of the AR alleles is inactivated in each cell. Only allele expression in homozygous genotypes can be determined unambiguously because their alleles are the same and inactivation of either allele will have the same result. For example, in our study, the OR comparing women with two alleles ≥22 AR CAG repeats to women carrying two alleles with <22 AR CAG repeats will not suffer from this misclassification. Some have suggested that X chromosome inactivation may not be random with respect to ovarian cancer (50) and that the longer allele is more often expressed (30). Interestingly, our results suggest that the shorter allele may be more important.
Although the haplotypes theoretically provide a more comprehensive approach to evaluating genetic variation due to consideration of polymorphisms in combination, it may miss an association that truly exists if the disease-causing locus is associated with multiple haplotypes and therefore its effect is attenuated. In our study, an increasing number of CAG repeats is associated with ovarian cancer risk and CAG length is associated with all but one of the htSNPs. Therefore, the effect of the CAG repeat may be divided among several haplotypes, resulting in an attenuated association for each of those haplotypes. Thus, the effect of the CAG repeat is not captured by the haplotype analysis.
Limitations of our study include generalizability and potential biases. Because the study population is primarily Caucasian women and our analyses are restricted to Caucasians, our results are not generalizable to other ethnicities. Alternatively, the homogeneity of our population is an advantage because it reduces the possibility for confounding by ethnicity. Due to our case-control design, we must also consider the possibility of selection bias. Ovarian cancer is an aggressive disease; 9% of our cases died before enrollment. If a particular AR genotype or haplotype imparted some survival benefit, it would be overrepresented among our cases, resulting in a misleading association between genotype and disease.
In conclusion, our study suggests an increased risk of ovarian cancer with increasing number of CAG repeats in exon 1, but no association between AR haplotypes and ovarian cancer risk. Further studies are needed to identify AR target genes that contribute to ovarian cancer risk.
Acknowledgments
Grant support: Supported in part by the National Cancer Institute and the NIH, and Department of Health and Human Services (grants CA10607-01 and CA054419-10). I. De Vivo is an American Cancer Society Research Scholar Recipient (RSG-00-061-01-CCE).
We thank Hardeep Ranu, Craig Labadie, Maura Regan, Pati Soule, Alicia Whittington, and Allison Vitonis for their technical assistance, and the participants of this study and their physicians who made this research possible.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
References
- 1.Whittemore AS, Harris R, Itnyre J. Characteristics relating to ovarian cancer risk: collaborative analysis of 12 US case-control studies. II. Invasive epithelial ovarian cancers in white women. Collaborative Ovarian Cancer Group Am J Epidemiol. 1992;136:1184–203. doi: 10.1093/oxfordjournals.aje.a116427. [DOI] [PubMed] [Google Scholar]
- 2.Schildkraut JM, Schwingl PJ, Bastos E, Evanoff A, Hughes C. Epithelial ovarian cancer risk among women with polycystic ovary syndrome. Obstet Gynecol. 1996;88:554–9. doi: 10.1016/0029-7844(96)00226-8. [DOI] [PubMed] [Google Scholar]
- 3.Mink PJ, Folsom AR, Sellers TA, Kushi LH. Physical activity, waist-to-hip ratio, and other risk factors for ovarian cancer: a follow-up study of older women. Epidemiology. 1996;7:38–45. doi: 10.1097/00001648-199601000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Helzlsouer KJ, Alberg AJ, Gordon GB, et al. Serum gonadotropins and steroid hormones and the development of ovarian cancer. JAMA. 1995;274:1926–30. [PubMed] [Google Scholar]
- 5.Vassilomanolakis M, Koumakis G, Barbounis V, et al. A phase II study of flutamide in ovarian cancer. Oncology. 1997;54:199–202. doi: 10.1159/000227688. [DOI] [PubMed] [Google Scholar]
- 6.Tumolo S, Rao BR, van der Burg ME, et al. Phase II trial of flutamide in advanced ovarian cancer: an EORTC Gynaecological Cancer Cooperative Group study. Eur J Cancer. 1994;30A:911–4. doi: 10.1016/0959-8049(94)90112-0. [DOI] [PubMed] [Google Scholar]
- 7.Edmondson RJ, Monaghan JM, Davies BR. The human ovarian surface epithelium is an androgen responsive tissue. Br J Cancer. 2002;86:879–85. doi: 10.1038/sj.bjc.6600154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silva EG, Tornos C, Fritsche HA, Jr, et al. The induction of benign epithelial neoplasms of the ovaries of guinea pigs by testosterone stimulation: a potential animal model. Mod Pathol. 1997;10:879–83. [PubMed] [Google Scholar]
- 9.Thompson MA, Adelson MD. Aging and development of ovarian epithelial carcinoma: the relevance of changes in ovarian stromal androgen production. Adv Exp Med Biol. 1993;330:155–65. doi: 10.1007/978-1-4615-2926-2_12. [DOI] [PubMed] [Google Scholar]
- 10.Lau KM, Mok SC, Ho SM. Expression of human estrogen receptor-α and -β, progesterone receptor, and androgen receptor mRNA in normal and malignant ovarian epithelial cells. Proc Natl Acad Sci U S A. 1999;96:5722–7. doi: 10.1073/pnas.96.10.5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuhnel R, de Graaff J, Rao BR, Stolk JG. Androgen receptor predominance in human ovarian carcinoma. J Steroid Biochem. 1987;26:393–7. doi: 10.1016/0022-4731(87)90106-3. [DOI] [PubMed] [Google Scholar]
- 12.Chadha S, Rao BR, Slotman BJ, van Vroonhoven CC, van der Kwast TH. An immunohistochemical evaluation of androgen and progesterone receptors in ovarian tumors. Hum Pathol. 1993;24:90–5. doi: 10.1016/0046-8177(93)90067-q. [DOI] [PubMed] [Google Scholar]
- 13.van Doorn HC, Burger CW, van der Valk P, Bonfrer HM. Oestrogen, progesterone, and androgen receptors in ovarian neoplasia: correlation between immunohistochemical and biochemical receptor analyses. J Clin Pathol. 2000;53:201–5. doi: 10.1136/jcp.53.3.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buchanan G, Yang M, Cheong A, et al. Structural and functional consequences of glutamine tract variation in the androgen receptor. Hum Mol Genet. 2004;13:1677–92. doi: 10.1093/hmg/ddh181. [DOI] [PubMed] [Google Scholar]
- 15.Kazemi-Esfarjani P, Trifiro MA, Pinsky L. Evidence for a repressive function of the long polyglutamine tract in the human androgen receptor: possible pathogenetic relevance for the (CAG)n-expanded neuronopathies. Hum Mol Genet. 1995;4:523–7. doi: 10.1093/hmg/4.4.523. [DOI] [PubMed] [Google Scholar]
- 16.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–9. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 17.Giovannucci E, Platz EA, Stampfer MJ, et al. The CAG repeat within the androgen receptor gene and benign prostatic hyperplasia. Urology. 1999;53:121–5. doi: 10.1016/s0090-4295(98)00468-3. [DOI] [PubMed] [Google Scholar]
- 18.Tut TG, Ghadessy FJ, Trifiro MA, Pinsky L, Yong EL. Long polyglutamine tracts in the androgen receptor are associated with reduced trans-activation, impaired sperm production, and male infertility. J Clin Endocrinol Metab. 1997;82:3777–82. doi: 10.1210/jcem.82.11.4385. [DOI] [PubMed] [Google Scholar]
- 19.Legro RS, Shahbahrami B, Lobo RA, Kovacs BW. Size polymorphisms of the androgen receptor among female Hispanics and correlation with androgenic characteristics. Obstet Gynecol. 1994;83:701–6. [PubMed] [Google Scholar]
- 20.Mifsud A, Ramirez S, Yong EL. Androgen receptor gene CAG trinucleotide repeats in anovulatory infertility and polycystic ovaries. J Clin Endocrinol Metab. 2000;85:3484–8. doi: 10.1210/jcem.85.9.6832. [DOI] [PubMed] [Google Scholar]
- 21.Nelson KA, Witte JS. Androgen receptor CAG repeats and prostate cancer. Am J Epidemiol. 2002;155:883–90. doi: 10.1093/aje/155.10.883. [DOI] [PubMed] [Google Scholar]
- 22.Suter NM, Malone KE, Daling JR, Doody DR, Ostrander EA. Androgen receptor (CAG)n and (GGC)n polymorphisms and breast cancer risk in a population-based case-control study of young women. Cancer Epidemiol Biomarkers Prev. 2003;12:127–35. [PubMed] [Google Scholar]
- 23.Haiman CA, Brown M, Hankinson SE, et al. The androgen receptor CAG repeat polymorphism and risk of breast cancer in the Nurses’ Health Study. Cancer Res. 2002;62:1045–9. [PubMed] [Google Scholar]
- 24.Sasaki M, Sakuragi N, Dahiya R. The CAG repeats in exon 1 of the androgen receptor gene are significantly longer in endometrial cancer patients. Biochem Biophys Res Commun. 2003;305:1105–8. doi: 10.1016/s0006-291x(03)00883-0. [DOI] [PubMed] [Google Scholar]
- 25.Yaron M, Levy T, Chetrit A, et al. The polymorphic CAG repeat in the androgen receptor gene in Jewish Israeli women with endometrial carcinoma. Cancer. 2001;92:1190–4. doi: 10.1002/1097-0142(20010901)92:5<1190::aid-cncr1437>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 26.Dagan E, Friedman E, Paperna T, Carmi N, Gershoni-Baruch R. Androgen receptor CAG repeat length in Jewish Israeli women who are BRCA1/2 mutation carriers: association with breast/ovarian cancer phenotype. Eur J Hum Genet. 2002;10:724–8. doi: 10.1038/sj.ejhg.5200880. [DOI] [PubMed] [Google Scholar]
- 27.Kadouri L, Easton DF, Edwards S, et al. CAG and GGC repeat polymorphisms in the androgen receptor gene and breast cancer susceptibility in carriers and non-carriers. Br J Cancer. 2001;85:36–40. doi: 10.1054/bjoc.2001.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levine DA, Boyd J. The androgen receptor and genetic susceptibility to ovarian cancer: results from a case series. Cancer Res. 2001;61:908–11. [PubMed] [Google Scholar]
- 29.Menin C, Banna GL, De Salvo G, et al. Lack association between androgen receptor CAG polymorphism and familial breast/ovarian cancer. Cancer Lett. 2001;168:31–6. doi: 10.1016/s0304-3835(01)00473-6. [DOI] [PubMed] [Google Scholar]
- 30.Santarosa M, Bidoli E, Gallo A, et al. Polymorphic CAG repeat length within the androgen receptor gene: identification of a subgroup of patients with increased risk of ovarian cancer. Oncol Rep. 2002;9:639–44. [PubMed] [Google Scholar]
- 31.Spurdle AB, Webb PM, Chen X, et al. Androgen receptor exon 1 CAG repeat length and risk of ovarian cancer. Int J Cancer. 2000;87:637–43. [PubMed] [Google Scholar]
- 32.Belsham DD, Pereira F, Greenberg CR, Liao Wrogemann K. Leu-676-Pro mutation of the androgen receptor causes complete androgen insensitivity syndrome in a large Hutterite kindred. Hum Mutat. 1995;5:28–33. doi: 10.1002/humu.1380050104. [DOI] [PubMed] [Google Scholar]
- 33.Chung HW, Kim SC, Kim HL. Frame-shift mutation in hormone binding domain of human androgen receptor gene causes complete androgen insensitivity. Mol Cells. 1998;8:741–5. [PubMed] [Google Scholar]
- 34.Zhang K, Calabrese P, Nordborg M, Sun F. Haplotype block structure and its applications to association studies: power and study designs. Am J Hum Genet. 2002;71:1386–94. doi: 10.1086/344780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–7. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 36.Cramer DW, Harlow BL, Titus-Ernstoff L, et al. Over-the-counter analgesics and risk of ovarian cancer. Lancet. 1998;351:104–7. doi: 10.1016/S0140-6736(97)08064-1. [DOI] [PubMed] [Google Scholar]
- 37.Stram DO, Haiman CA, Hirschhorn JN, et al. Choosing haplotype-tagging SNPS based on unphased genotype data using a preliminary sample of unrelated subjects with an example from the Multiethnic Cohort Study. Hum Hered. 2003;55:27–36. doi: 10.1159/000071807. [DOI] [PubMed] [Google Scholar]
- 38.Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–9. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 39.Excoffier L, Slatkin M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol. 1995;12:921–7. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- 40.Zaykin DV, Westfall PH, Young SS, et al. Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered. 2002;53:79–91. doi: 10.1159/000057986. [DOI] [PubMed] [Google Scholar]
- 41.Lewontin RC. The interaction of selection and linkage. II. Optimum models Genetics. 1964;50:757–82. doi: 10.1093/genetics/50.4.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edwards A, Hammond HA, Jin L, Caskey CT, Chakraborty R. Genetic variation at five trimeric and tetrameric tandem repeat loci in four human population groups. Genomics. 1992;12:241–53. doi: 10.1016/0888-7543(92)90371-x. [DOI] [PubMed] [Google Scholar]
- 43.Terry KL, De Vivo I, Titus-Ernstoff L, Sluss PM, Cramer DW. Genetic variation in the progesterone receptor and ovarian cancer risk. Am J Epidemiol. 2005;16:442–51. doi: 10.1093/aje/kwi064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu YC, Wang PH, Yeh S, et al. Subfertility and defective folliculogenesis in female mice lacking androgen receptor. Proc Natl Acad Sci U S A. 2004;101:11209–14. doi: 10.1073/pnas.0404372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elhaji YA, Gottlieb B, Lumbroso R, et al. The polymorphic CAG repeat of the androgen receptor gene: a potential role in breast cancer in women over 40. Breast Cancer Res Treat. 2001;70:109–16. doi: 10.1023/a:1012942910375. [DOI] [PubMed] [Google Scholar]
- 46.Dunning AM, McBride S, Gregory J, et al. No association between androgen or vitamin D receptor gene polymorphisms and risk of breast cancer. Carcinogenesis. 1999;20:2131–5. doi: 10.1093/carcin/20.11.2131. [DOI] [PubMed] [Google Scholar]
- 47.Irvine RA, Ma H, Yu MC, et al. Inhibition of p160-mediated coactivation with increasing androgen receptor polyglutamine length. Hum Mol Genet. 2000;9:267–74. doi: 10.1093/hmg/9.2.267. [DOI] [PubMed] [Google Scholar]
- 48.Yeh S, Tsai MY, Xu Q, et al. Generation and characterization of androgen receptor knock-out (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. Proc Natl Acad Sci U S A. 2002;99:13498–503. doi: 10.1073/pnas.212474399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stratchan T, Read AP. Human Molecular Genetics. 2nd ed. New York: Wiley-Liss; 1999. p. 576.
- 50.Buller RE, Sood AK, Lallas T, Buekers T, Skilling JS. Association between nonrandom X-chromosome inactivation and BRCA1 mutation in germline DNA of patients with ovarian cancer. J Natl Cancer Inst. 1999;91:339–46. doi: 10.1093/jnci/91.4.339. [DOI] [PubMed] [Google Scholar]