

Abstract
Uteroglobin-related proteins 1 and 2 (UGRP1 and -2) are thought to play important roles in inflammation and immunologic responses in the lung. In this study we demonstrate that IL-4 and IL-13 enhance Ugrp2 gene expression in the mouse transformed Clara cell line, mtCC, in a time- and dose-dependent manner. Addition of actinomycin D abrogated the IL-4- and IL-13-induced increase of Ugrp2 expression, demonstrating that this increase occurs at the transcriptional level. When mtCC cells were pretreated with IFN-γ before the addition of IL-4 or IL-13, IL-4- and 13-induced Ugrp2 mRNA increase was markedly decreased. IL-4 and IL-13 induced phosphorylation of STAT6 in mtCC cells, which binds to the proximal STAT-binding element (SBE) in the Ugrp2 gene promoter, leading to transcriptional activation of this gene. Mutations of the proximal SBE abrogated the binding of activated STAT6 to this site and the IL-4-induced increase in Ugrp2 gene promoter activity. IFN-γ-activated STAT1 binds to the same SBE in the Ugrp2 gene promoter to which STAT6 binds and decreases the binding of STAT6 to this site. Furthermore, an IL-4-induced increase in Ugrp2 expression was not observed in primary cultures of lung cells derived from STAT6-deficient mice. These results indicate that Ugrp2 expression is enhanced by IL-4 and IL-13 through STAT6 binding to the proximal SBE located in the Ugrp2 gene promoter.
Uteroglobin-related protein 2 (UGRP2),5 also called high in normal-1 (HIN-1) or secretoglobin 3A1, was identified as a candidate tumor suppressor gene that is silenced by methylation in the majority of breast carcinomas (1). Subsequently, it was found as a homologous gene to UGRP1, which is a downstream target gene for T/EBP/NKX2.1 homeodomain transcription factor (2). Mouse UGRP2 shows 33% amino acid sequence identity to mouse UGRP1 (2). Based on amino acid sequences, both UGRP1 and UGRP2 belong to a gene superfamily of the uteroglobin/Clara cell secretory proteins (UG/CCSP), officially termed secretoglobins (3-5).
In humans, the highest level of UGRP2 expression is detected in trachea, followed by salivary gland, lung, and mammary gland (3). In contrast, Ugrp2 expression in mice is predominantly found in trachea and lung (3, 5), with low levels of expression reported in other tissues, such as heart, stomach, small intestine, and mammary gland (5). Ugrp2 mRNA is detected on embryonic day 17.5 during mouse development in the tracheal epithelium and later also in the submucosal glands of the proximal trachea and bronchi (4, 5). Ugrp2 mRNA expression is up-regulated by retinoic acid, which induces mucinous differentiation in primary normal human bronchial epithelial cells (5). It was suggested that UGRP2 may have a role in the acquisition or maintenance of the terminally differentiated proximal airway epithelial phenotype (5).
The mouse Ugrp2 gene is localized at region 11A5-B1, which is homologous to human chromosome 5q35 (3). The human chromosome region 5q31-q34 was reported to contain a number of genes associated with inflammatory cytokines, such as IL-3, IL-4, IL-5, IL-13, and CSF-2. Furthermore, UG/CCSP, a prototypical protein of the secretoglobin gene superfamily to which UGRP2 is distantly related, exhibits several anti-inflammatory properties, including inhibition of phospholipase A2 (6), IFN-γ production (7), and neutrophil inflammation and chemotaxic response (8). These facts together with the site of UGRP2 expression suggest that UGRP2 may play a role in the regulation of inflammation and immunologic responses. However, little is known about the physiological function of UGRP2 in the lung.
IL-4, a Th2 cytokine, is a pleiotropic lymphokine produced by Ag-activated T cells, which acts on hemopoietic, endothelial, dendritic, osteoblastic, and fibroblastic cells and plays a particularly important role in allergic sensitization (9). IL-4 has a role in B cell class switching to IgE production (10). Together with IL-3, IL-4 functions as a growth factor for mast cells (11) and is able to up-regulate VCAM-1 expression, leading to preferential migration of eosinophils into tissues (12).
IL-4 activates gene expression by signaling through either type 1 or type 2 IL-4R complexes on the surface of IL-4-responsive target cells. Binding of IL-4 to the IL-4R α-chain induces subsequent heterodimerization with either the common γ-chain (γc) (13, 14) or the γ-chain-like receptor, IL-13Rα1 (15-17). This dimerization activates one or more members of the receptor-associated JAKs that initiates the phosphorylation cascade, subsequently resulting in the activation of secondary effector molecules, including STAT6 (18-21). Activated STAT6 then homodimerizes and translocates to the nucleus, where it binds with high affinity to STAT-binding elements (SBE) in the promoters of various IL-4-responsive genes (18, 22-29). In recent years it was found that IL-13 induces the expression of the same repertoire of genes that are inducible by IL-4 in monocytes (30).
The aim of the present study was to investigate the mechanisms governing the regulation of Ugrp2 expression by the Th2 cytokines, IL-4 and IL-13. We demonstrate that both IL-4 and IL-13 stimulate the expression of Ugrp2 through a mechanism dependent on the transcription factor STAT6 that is able to bind to one of the SBEs present in the Ugrp2 gene promoter and up-regulates the promoter activity. Furthermore, we show that IFN-γ markedly reduces the IL-4- and IL-13-induced increase in Ugrp2 mRNA expression.
Materials and Methods
Materials
Pregnant BALB/c mice were purchased from Charles River Laboratories, and STAT6-deficient mice were bred onto the BALB/c background by Dr. M. J. Grusby (Harvard Medical School, Boston, MA) (31). We obtained the breeding pairs from Dr. W. E. Paul (National Institute of Arthritis and Infectious Diseases, National Institutes of Health, Bethesda, MD). Recombinant murine IL-4, IL-13, and IFN-γ were purchased from R&D Systems. Actinomycin D was purchased from Sigma-Aldrich. Abs against murine STAT1, STAT6, and phospho-STAT6 were purchased from Santa Cruz Biotechnology. All animal studies were conducted after approval by the institutional animal care and use committee.
Cell culture
Mouse transformed Clara (mtCC) cells (32) were maintained in DMEM (BioSource International) supplemented with 10% FBS (Gemini Bio-Products) and antibiotic-antimycotic containing 100 U/ml penicillin G sodium, 100 μg/ml streptomycin sulfate, and 250 ng/ml amphotericin B (Invitrogen Life Technologies).
Lung primary culture
Mouse embryonic lung primary culture was prepared using the method previously described (33). In brief, embryos were obtained by dissection of pregnant females on embryonic day 17.5. Fetal lungs were dispersed by 10 U/ml collagenase (Sigma-Aldrich) and 1 U/ml dispase (dispase I; Roche) and were cultured in DMEM/Ham's F-12 medium (BioSource International) supplemented with 10% FBS (Gemini Bio-Products).
Isolation and culture of bone marrow-derived macrophages
Monolayers of mouse bone marrow-derived macrophage were generated according to standard procedures. Briefly, bone marrow aspirates from the tibias and femurs of wild-type mice were flushed with sterile DMEM to dislodge the cells. The harvested cells were then cultured in DMEM containing 10% FBS, 2 mM l-glutamine, antibiotics, and 100 ng/ml recombinant human M-CSF. The cultures were incubated at 37°C under a 5% CO2 atmosphere for 5-7 days.
RT-PCR
To prevent genomic DNA contamination, total RNA was treated with RNase-free DNase I (Ambion). For cDNA synthesis, total RNA was first incubated at 70°C for 10 min, then chilled on ice. The cDNA synthesis reaction was conducted in a final volume of 20 μl containing 1 μg of RNA, 4 μl of 5× first-strand synthesis buffer, 1 μl of a mixture of four deoxynucleotide triphosphates (10 mM each), 2 μl of 0.1 M DTT, and 1 μl of 500 pg/μl N6 random hexamers or oligo(dT)20 (Invitrogen Life Technologies). After incubation at 37°C for 2 min, 200 U of SuperScript II reverse transcriptase (Invitrogen Life Technologies) was added, and incubation was continued at 37°C for 60 min. Cells were then subjected to PCR using Advantage 2 Taq DNA polymerase (BD Biosciences) under the following conditions: denaturation at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s, 20 cycles for 18S and GAPDH, and 30 cycles for IL-2Rα, IL-4Rα, IL-9Rα, IL-10R1, IL-13Rα1, and γc. For IL-5Rα, IL-10R2, and common β-chain receptor (βc), annealing temperatures of 50, 58, and 55°C were used instead of 60°C, and the reaction was run for 35, 30, and 35 cycles, respectively. Oligonucleotide primers used for RT-PCR were as follows: mouse 18S, 5′-CGGCTACCACATCCAAG GAA-3′ and 5′-ATTGGAGCTGGAATTACCGC-3′; mouse GAPDH, 5′-ACCA CAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCT GTA-3′; mouse IL-2Rα, 5′-AGGTTTCCGAAGACTAAAGG-3′ and 5′-CCAC GAAATGATAGATTCTC-3′; mouse IL-4Rα, 5′-ATCTGCGTG CTTGCTGGTTCT-3′ and 5′-CTGGTATCTGTCTGATTGGACCG-3′; mouse IL-5Rα, 5′-CATGCTGGTTTCCCAGGAC-3′ and 5′-TTTCCCA CATAAATAGGTTG-3′; mouse IL-9Rα, 5′-AGTCTCGGTCCGAGAG CAAG-3′ and 5′-GTCTCCTGGGCAGGTACTGT-3′; mouse IL-10R1, 5′-GTCTGAGAGCACCTACTATG-3′ and 5′-TTGTAAACTCGGAG ATCCTT-3′; mouse IL-10R2, 5′-GAGACGTGGACCTTGAAGAA-3′ and 5′-ACGGAGACTATGAGGATGAT-3′; mouse IL-13Rα1, 5′-CAT CTTCTCCTCAAAAATGGTGCC-3′ and 5′-GGATTATGACTGCCAC TGCGAC-3′; mouse γc, 5′-GTTCTGAGCCTCAGGCAACC-3′ and 5′-CAGATTGCTGAGTGTTAGAT-3′; and mouse βc, 5′-GCTACATG GCACTGGTGGCT-3′ and 5′-GCTTATAAGCCACCTCAAAC-3′.
Northern blot analysis
Total RNAs from mtCC cells (5 μg) or lung primary culture cells (20 μg) were electrophoresed on 1% agarose gels containing 0.22 M formaldehyde and blotted onto GeneScreen Plus nylon membranes (PerkinElmer). Filters were serially hybridized with mouse Ugrp2 and 18S cDNA probes. Hybridization was performed in Perfect Hybridization Solution (Sigma-Aldrich) at 68°C overnight. The membrane was washed twice with 2× SSC containing 0.1% SDS at 68°C for 30 min, once with 2× SSC at 68°C for 30 min, followed by exposure to a Storm PhosphorImager screen (Amersham Biosciences). Signals were visualized and quantitated using ImageQuant software (Amersham Biosciences).
Western blot analysis
The mtCC or lung primary culture cells were washed with PBS and lysed in radio-immuno-protein-assay buffer (20 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, 2 mM EDTA, 10 mM sodium fluoride, and 1 mM sodium orthovanadate) with Protease Inhibitor Cocktail Tablets (Complete Mini; Roche). The protein lysates were mixed with SDS sample loading buffer containing 2-ME, electrophoresed on 10% SDS-polyacrylamide gels containing 0.1 or 0.8% bis-(N,N′-methylene-bis-acrylamide), and then electrotransferred to an Immobilon-P membrane (Millipore). Membranes were blocked with TBS containing Tween 20 (TBS-Tween; 20 mM Tris-HCl, 150 mM NaCl, and 0.1% Tween 20) and 5% skin milk, and were incubated with the first Ab in TBS-Tween. Membranes were washed with TBS-Tween, then incubated with HRP-conjugated secondary Ab (Santa Cruz Biotechnology). Protein bands were detected using Western Lighting Chemiluminescence Reagent Plus (PerkinElmer) and were exposed to scientific imaging film (Eastman Kodak).
Construction of mouse UGRP2-luciferase promoter plasmids
A BamHI fragment containing 3.2 kb of the 5′-flanking sequence of the mouse Ugrp2 genomic DNA was subcloned into pBluescript II, and PCR was performed with T7 primer (5′-GTAATACGACTCACTATAGGGC-3′) and a Ugrp2 gene-specific primer (5′-TGGTAGAATGAGTCTCAG-3′; +52/+35 based on the transcription start site as +1) (3). The PCR product was subcloned into pCR2.1 (Invitrogen Life Technologies), and an SpeI-XhoI fragment from this plasmid was inserted into NheI-XhoI site of the luciferase reporter vector pGL3-basic (Promega) to generate pGL3-3185 plasmid. A BglII fragment containing 420 bp of the 5′-flanking sequence of the mouse Ugrp2 genomic DNA was subcloned into pBluescript II, and PCR was performed with T3 primer (5′-AATTAACCCTCACTAAA GGG-3′) and a Ugrp2 gene-specific primer (+52/+35). The PCR product was subcloned into pCR2.1, and an SmaI-XhoI fragment from this plasmid was inserted into the SmaI-XhoI site of pGL3-basic to generate pGL3-411 plasmid. This construct was further digested with KpnI and NheI for preparation of deletion plasmids using Exonuclease III (New England Biolabs) and S1 nuclease (Invitrogen Life Technologies). Four deletion constructs thus obtained (pGL3-65, -154, -212, and -264) were sequenced to determine the exact sequences. To generate the pGL3-1158 construct, a -1158/+52 bp fragment was amplified by PCR using the primers -1158 mUgrp2 (5′-CGCCCTTCAGAGGAAGAGGCT-3′) and GLprimer2 located in the pGL3 basic as a reverse primer (5′-CTTTATGTTTTTGGCGTCTTCC-3′) and subcloned into pGL3-basic. Mutations were introduced into STAT binding elements (SBEs), -1143/-1154 (5′-CTTCAGAGGAAG-3′) and -201/-209 (5′-TTCCTGGAA-3′; putative STAT6 binding sequences are underlined), in the Ugrp2 gene promoter using PCR with primers 5′-CGC CATGCAGAGTACGAGGCT-3′ and 5′-CCGTGGTACCTGCAAGAAA GG-3′ (mutations are in italics), respectively. All plasmids were confirmed by nucleotide sequencing.
Luciferase reporter assay
The mtCC cells were seeded in 24-well tissue culture plates, grown to 90% confluence, and transfected with pGL3 reporter plasmids containing various lengths of the mouse Ugrp2 gene promoter using Lipofectamine 2000 (Invitrogen Life Technologies). As an internal control, plasmid phRL-TK containing the Renilla luciferase gene (Promega) was cotransfected. Cells were cultured in 0.5% FBS-containing medium in the presence or the absence of cytokine for 48 h, then lysed with passive lysis buffer (Promega). Luciferase activity was measured according to instructions in the technical manual for the Dual-Luciferase Reporter Assay System (Promega).
EMSA
Nuclear protein extracts were prepared from cytokine-treated cells using a modification of the original method described by Schreiber et al. (34). Double-stranded oligonucleotide (21 nt), prepared based on the DNA sequences present in the mouse Ugrp2 gene promoter, was used as a probe in gel-shift assays. This oligonucleotide contains a SBE that can bind to STAT6 and STAT1. Binding reactions were performed at room temperature for 30 min by incubating radiolabeled probe DNA (>105 cpm) with 5 μg of nuclear extracts prepared from IL-4-, IL-13-, IFN-γ-, or IL-4/IL-13- and IFN-γ-stimulated cells (30 min at 37°C) in the presence of 2 μg of polydeoxyinosinic-polydeoxycytidylic acid poly(dI:dC) (Amersham Biosciences), 10 mM DTT, 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, and 50% glycerol in a final volume of 20 μl. After incubation, 8 μl of each mixture was electrophoresed on nondenaturing 6% polyacrylamide gels (Novex; Invitrogen Life Technologies) using 0.25× TBE electrophoresis buffer containing 22 mM Tris-HCl (pH 8.0), 22 mM borate, and 0.5 mM EDTA. Gels were then dried and visualized by autoradiography.
Results
IL-4 and IL-13 enhance Ugrp2 mRNA expression
The induction of Ugrp2 mRNA by selected Th1 (IL-2) and Th2 (IL-4, IL-5, IL-9, IL-10, and IL-13) cytokines was examined in mtCC cells that were generated from tumor cells derived from transgenic mice expressing the SV40 large T Ag gene under the control of the Ug/Ccsp promoter (32). This cell line constitutively expresses modest levels of Ugrp2 mRNAs (Fig. 1A). After 12-h incubation in DMEM containing reduced serum (0.5%), cells were treated with 20 ng/ml IL-2, IL-4, IL-5, IL-9, IL-10, and IL-13 for 12 h. Among them, IL-4 and IL-13 were found to enhance the levels of Ugrp2 expression (Fig. 1A). Because cytokine signaling starts with the binding of cytokines to their specific cell surface receptor complexes (35), the presence of receptors for each cytokine was examined in mtCC cells using RT-PCR (Fig. 1B). The results suggest that no induction of Ugrp2 gene expression by IL-2, IL-5, and IL-9 in mtCC cells is due to the lack of at least one receptor component in each receptor complex. In contrast, the Ugrp2 gene was induced by IL-4 and IL-13 in the absence of the γc because mtCC cells express IL-13Rα1. This indicates that IL-4 and IL-13 activate Ugrp2 gene expression in mtCC cells by signaling through type 2 IL-4R complexes (15-17).
FIGURE 1.

Induction of Ugrp2 mRNA by cytokines and analysis of their receptors. A, The mtCC cells were treated with 20 ng/ml selected cytokines (IL-2, IL-4, IL-5, IL-9, IL-10, and IL-13) for 12 h. Total cellular RNAs were extracted, and mRNAs for Ugrp2 and 18S were examined by Northern blot analysis. B, RT-PCR analysis of IL-2Rα, IL-4Rα, IL-5Rα, IL-9Rα, IL-10R1, IL-10R2, IL-13Rα1, γc, and βc using RNAs prepared from mtCC cells, and adult mouse lung and spleen as controls. The sizes of the PCR products are 260 bp (IL-2Rα), 432 bp (IL-4Rα), 416 bp (IL-5Rα), 317 bp (IL-9Rα), 360 bp (IL-10R1), 272 bp (IL-10R2), 385 bp (IL-13Rα1), 266 bp (γc), 504 bp (βc), 452 bp (GAPDH), and 191 bp (18S).
Increase in Ugrp2 expression occurred at the transcriptional level in a time- and dose-dependent manner
Ugrp2 mRNA levels were increased in the presence of as little as 1 ng/ml IL-4 or IL-13 and were additionally increased in a dose-dependent manner, with the level reaching a plateau at ∼10 ng/ml (Fig. 2A). The time course for Ugrp2 mRNA induction by IL-4 and IL-13 was determined with a fixed concentration of 20 ng/ml IL-4 and IL-13 and at various time points between 3 and 24 h. The increase in Ugrp2 expression was detected 3 h after the addition of IL-4 and IL-13, and the expression level was already close to the maximum (Fig. 2B). To examine whether the increase in Ugrp2 mRNA expression by treatment with IL-4 and IL-13 occurs at the transcriptional level, mtCC cells were pretreated with the RNA synthesis inhibitor, actinomycin D, followed by treatment with IL-4 and IL-13 (Fig. 2C). Actinomycin D abrogated the IL-4- and IL-13-induced increase in Ugrp2 mRNA expression, suggesting that the enhancement of Ugrp2 gene expression by IL-4 and IL-13 in mtCC cells is mediated at the transcriptional level.
FIGURE 2.
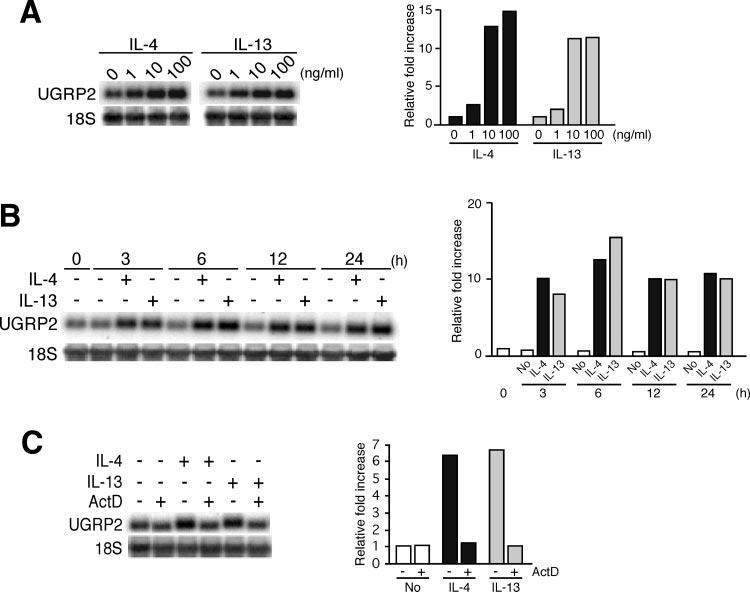
Dose- and time-dependent increase in Ugrp2 mRNA by IL-4 and IL-13 occurs at the transcriptional level. A, Dose effects of IL-4 and IL-13. The mtCC cells were treated with 1, 10, or 100 ng/ml IL-4 or IL-13 for 12 h. B, Time-course analysis of the effects of IL-4 and IL-13. The mtCC cells were treated with 20 ng/ml IL-4 or IL-13 for 3, 6, 12, or 24 h. C, IL-4 and IL-13 induced Ugrp2 mRNA at the transcriptional level. The mtCC cells were pretreated with 5 μg/ml actinomycin D (ActD) for 1 h, followed by treatment with 20 ng/ml IL-4 or IL-13 for 6 h. The expressions of Ugrp2 and 18S were detected by Northern blot analysis, and signals were quantitated using ImageQuant software. The relative fold increase in Ugrp2 mRNA levels after normalizing to those of 18S is plotted on the right in each section. Experiments were repeated twice, and each time a robust increase (A and B) or a decrease (C) in Ugrp2 mRNA expression was obtained when cells were treated with 10 ng/ml IL-4/IL-13 (A), 3 h after initiation of treatment with IL-4/IL-13 (B), and with ActD treatment together with IL-4/IL-13 (C).
Activation of STAT6 by IL-4 and IL-13 in mtCC cells
IL-4 and IL-13 bind to cell surface receptor complexes (15-17), leading to the activation of STAT6 (18-21) that is capable of binding to SBEs in the promoters of various IL-4- and IL-13-responsive genes (18, 22, 23). To determine whether STAT6 is involved in the IL-4- and IL-13-induced increase in Ugrp2 gene expression, the nuclear protein extracts prepared from IL-4- and IL-13-stimulated mtCC cells were analyzed by Western blotting using anti-phospho (Y)-STAT6 and anti-STAT6 Abs (Fig. 3). Because mtCC cells express IL-4Rα and IL-13Rα1, but not γc, these results also demonstrate that STAT6 is activated by IL-4 and IL-13 by signaling through the type 2 IL-4R complexes.
FIGURE 3.

STAT6 is activated by IL-4 and IL-13 in mtCC cells. The mtCC cells were treated with 20 ng/ml IL-4 and IL-13 for 30 min. The level of activated STAT6 was measured by Western blotting using a rabbit polyclonal anti-phospho-STAT6 (Tyr641) Ab.
IFN-γ inhibits IL-4- and IL-13-induced increases in Ugrp2 mRNA
IFN-γ is a major product of activated Th1 cells, whereas IL-4 and IL-13 are primarily produced by Th2 cells. IL-4 and IFN-γ exhibit mutual antagonism in many systems. For example, IFN-γ can markedly inhibit IL-4- and IL-13-stimulated expression of IL-1RI and IL-1RII (36), 15-lipoxygenase (29), and FcεRIIb (CD23) genes (37). When mtCC cells were treated with IFN-γ, the constitutive level of Ugrp2 expression was reduced (Fig. 4). Furthermore, IFN-γ pretreatment before the addition of IL-4 or IL-13 markedly reduced the IL-4- and IL-13-induced increase in Ugrp2 mRNA levels. These results demonstrate an effect of IFN-γ on the regulation of Ugrp2 gene expression by IL-4 similar to those seen with other IL-4-regulated genes. It is also notable that costimulation of cells with IL-4 and IL-13 did not further increase the level of Ugrp2 expression above that achieved with IL-4 or IL-13 treatment alone, suggesting that IL-4 and IL-13 signal through the same receptor complex.
FIGURE 4.

IFN-γ inhibits the increase in Ugrp2 mRNA caused by IL-4 and IL-13. The mtCC cells were pretreated with 20 ng/ml IFN-γ for 1 h, followed by treatment with 20 ng/ml IL-4 or IL-13 for 6 h. The expressions of Ugrp2 and 18S were detected by Northern blot analysis.
Identification of IL-4-responsive elements in mouse Ugrp2 gene promoter
A transcription start site of the mouse Ugrp2 gene was previously determined by 5′RACE analysis (3). Several Ugrp2 promoter-luciferase reporter plasmids were constructed and analyzed by transient transfection with or without IL-4 using mtCC cells that constitutively express Ugrp2. The reporter activity of the Ugrp2 gene promoter without IL-4 gradually increased up to -412 bp of the promoter sequence. With the construct -1158, the baseline activity decreased to a level similar to that of the construct -212, suggesting the possible presence of an element that represses the promoter activity in between -412 and -1158 bp of the Ugrp2 gene promoter (Fig. 5A). In the presence of IL-4, the reporter activity of the construct -212 was ∼2-fold higher than that with no IL-4, whereas the promoter activity of the -154 construct was similar regardless of the addition of IL-4 (Fig. 5A). All other constructs containing upstream sequences up to -264, -412, -1158, and -3185 bp exhibited ∼2-fold higher reporter activities with IL-4 compared without IL-4, suggesting that an element(s) responsible for the IL-4-induced increase in Ugrp2 gene promoter activity resides between -154 and -212 bp from the transcription start site of the Ugrp2 gene. Within this region, one putative SBE was found with the consensus sequence of TTC(N3)GAA (5′-TTCCTGGAA-3′) located at position -209 to -201 bp (proximal SBE) relative to the transcription start site of the gene. To confirm that the -209 to -201 bp SBE is responsible for the IL-4-induced increase in Ugrp2 promoter activity, mutations were introduced to modify two nucleotides (5′-TTCCTGGAA-3′ to 5′-TACCTGCAA-3′; putative STAT6 binding sequences are in italics, and mutations are underlined) in this SBE. The construct -212M1 carrying these mutated nucleotides exhibited almost complete suppression of the IL-4-induced increase in promoter activity (Fig. 5B), suggesting that the proximal SBE plays an important role in the induction of Ugrp2 promoter activity by IL-4. Furthermore, within 1.2 kb of the mouse Ugrp2 gene promoter, another putative SBE, 5′-CTTCAGAG GAAG-3′ (TTC(N4)GAA), was found at -1154 to -1143 bp (distal SBE) relative to the transcription start site. Mutations were also introduced into this distal SBE by modifying four nucleotides (5′-CTTCAGAGGAAG-3′ to 5′-ATG CAGAGTACG-3′). When the construct -1158M2 with these mutated nucleotides was subjected to transfection analysis, IL-4-induced reporter activity similar to that of a construct without the mutation (-1158 construct) was obtained, whereas construct -1158M1, which has the proximal SBE mutated, but with intact distal SBE, exhibited similar promoter activities both with and without IL-4, suggesting that the distal SBE is dispensable for IL-4-induced expression of the Ugrp2 gene. These results demonstrate that the proximal SBE, -209 to -201 bp relative to the transcription start site, is important for the induction of mouse Ugrp2 gene promoter activity by IL-4.
FIGURE 5.

Determination of the IL-4-responsive region in the mouse Ugrp2 gene promoter. A, Transfection analysis of a series of luciferase reporter plasmids containing various lengths (-3185, -1158, -412, -246, -212, -154, and -65/+52 bp) of the mouse Ugrp2 gene promoter with or without IL-4. Putative distal (5′-TTCAGAGGAA-3′; -1154 to -1143 bp) and proximal SBEs (5′-TTCCTGGAA-3′; -212 to -154 bp) are indicated by the illustration on the left. Putative SBEs are characterized by a palindromic sequence shown in bold. Luciferase assays were performed in mtCC cells with (■) or without ([unk]) stimulation with 50 ng/ml IL-4. Results shown are the mean ± SE from three independent experiments. A significant difference was obtained in the values with and without IL-4 treatment: *, p < 0.05; **, p < 0.01. B, Mutation analysis. The putative SBE sites and mutated sequences (underlined) are shown in bold. The mutation of proximal SBE abrogates the IL-4-induced promoter activities of -212 and -1158 constructs.
Binding of STAT6 to proximal SBE in mouse Ugrp2 gene promoter
To confirm whether STAT6 binds to the proximal SBE (-209 to -201 bp), EMSA was conducted using a 32P end-labeled, double-stranded oligonucleotide of -215 to -195 bp of the mouse Ugrp2 gene promoter (Fig. 6). Nuclear extracts prepared from IL-4-treated macrophages produced a shifted band that was supershifted by the addition of STAT6 Ab, but not STAT1 Ab (Fig. 6, lanes 2, 3, and 6). IL-13 produced the same shifted band corresponding to STAT6; however, it was less potent than IL-4 (Fig. 6, lanes 12 and 15). Interestingly, a DNA-protein complex corresponding to STAT1 was obtained when IFN-γ-treated nuclear extracts were used, which was supershifted by the addition of Ab for STAT1, but not STAT6 (Fig. 6, lanes 4, 5, 7, and 13). Introduction of mutations into the proximal SBE resulted in complete loss of both STAT6- and STAT1-specific bands (Fig. 6, lanes 9 and 10). These results demonstrate that both IL-4-activated STAT6 and IFN-γ-activated STAT1 can bind to the proximal SBE in the Ugrp2 gene promoter. When the cells were cotreated with IFN-γ and IL-4 or IL-13, the binding of STAT1 to the Ugrp2 SBE decreased the binding of STAT6 to the same element due to the apparently higher affinity of STAT1 for this site (Fig. 6, lanes 14 and 16). These results may explain the inhibitory effects of IFN-γ on the IL-4 induced expression of the Ugrp2 gene.
FIGURE 6.

EMSAs for STAT6 binding site in the Ugrp2 gene promoter. A putative SBE is underlined. In the mutated SBE oligonucleotide (mut), mutations were introduced by changing two nucleotides (bold). Nuclear extract proteins (5 μg) from macrophages stimulated with 20 ng/ml IL-4 (lanes 2, 3, 6, 9, and 12), IL-13 (lane 15), IFN-γ (lanes 4, 5, 7, 10, and 13), or IL-4/13 and IFN-γ together (lanes 14 and 16) for 30 min at 37°C were incubated with labeled wild-type (WT; lanes 1-7 and 11-16) and mutant (mut; lanes 8-10) SBE oligonucleotide. For supershift assays, 2 μg of STAT6 (lanes 3 and 7) or STAT1 (lanes 5 and 6) Ab was added to probe-extract complexes. After incubations, DNA/protein complexes were resolved by 6% PAGE.
Requirement of STAT6 in the regulation of Ugrp2 gene expression by IL-4
To demonstrate the role of STAT6 in the increased expression of Ugrp2 by IL-4, Ugrp2 expression levels were compared between wild-type and STAT6-deficient mice using embryonic lung primary culture cells (Fig. 7). First, the lack of STAT6 expression was confirmed in these primary culture cells prepared from STAT6-deficient mice (Fig. 7A). The presence of IL-4Rα and IL-13Rα1 and the lack of γc expression were confirmed by RT-PCR in wild-type and STAT6-deficient mouse lung primary cells (Fig. 7B). Activation of STAT6 by IL-4 was shown in wild-type primary cells (Fig. 7C). Treatment of wild-type and STAT6-deficient lung primary cells with IL-4 clearly demonstrated that IL-4 enhanced Ugrp2 mRNA levels in wild-type, but not STAT6-deficient, primary cells (Fig. 7D), indicating that IL-4 enhancement of Ugrp2 gene expression is dependent on the STAT6 pathway.
FIGURE 7.

STAT6 is required for IL-4 up-regulation of Ugrp2 gene transcription. A, The presence of STAT6 in wild-type, but not STAT6-deficient (STAT6-/-), mouse embryonic lung primary culture cells. Expression of STAT6 was detected by Western blotting using a rabbit polyclonal anti-STAT6 Ab. B, Expression of IL-4Rα and IL-13Rα1 and absence of γc in lung primary culture cells. RT-PCR was conducted using RNAs prepared from embryonic lung primary culture cells of wild-type and STAT6-deficient (STAT6-/-) mice for IL-4Rα and IL-13Rα1, and from wild-type mice for γc. Spleen was used as a positive control for γc. The sizes of PCR products are 432 bp (IL-4Rα), 385 bp (IL-13Rα1), 191 bp (18S), 266 bp (γc), and 452 bp (GAPDH). C, STAT6 is activated by IL-4 in lung primary culture cells. Wild-type lung primary culture cells were treated with 50 ng/ml IL-4 for 30 min. The level of activated STAT6 was measured by Western blotting using a rabbit polyclonal anti-phospho-STAT6 (Tyr641) Ab. D, Northern blot analysis of total RNAs (20 μg/lane) prepared from wild-type or STAT6-/- mouse embryo lung primary culture cells treated with IL-4 (0, 1, 10, and 100 ng/ml) for 24 h. Hybridization was serially conducted using a probe for Ugrp2 and 18S.
Discussion
This study demonstrates that Ugrp2 expression is enhanced by treatment with IL-4 and IL-13 in mtCC cells that express endogenous Ugrp2. Other selected Th2 cytokines, such as IL-5, IL-9, and IL-10, and the Th1 cytokine IL-2 did not induce the expression of Ugrp2 mRNA in this cell line. The lack of expression of at least one of their specific receptor components appears to be responsible for the lack of induction by IL-2, IL-5, and IL-9, whereas both IL-10R1 and IL-10R2 are present, suggesting that the Ugrp2 gene promoter does not respond to IL-10 signaling. Although IL-4 signal transduction can take place through a pathway involving IL-4Rα and the γc, the absence of a γ-chain in mtCC cells and the lack of an additive effect of IL-4 and IL-13 on the induction of Ugrp2 gene expression suggest that the IL-4 or IL-13 signal goes through the heterodimeric type 2 IL-4R complex, leading to activation of STAT6. A binding site for STAT6 was identified at position -209 to -201 bp relative to the transcription start site of the Ugrp2 gene by transient transfection assays and EMSA, which is responsible for the IL-4-induced increase in Ugrp2 gene expression. Furthermore, experiments using STAT6-deficient lung primary culture cells demonstrated that STAT6 is necessary for the IL-4-induced increase in Ugrp2 gene expression.
RT-PCR analysis revealed the presence of both IL-10R1 and IL-10R2 in mtCC cells. IL-10R1 is known to be primarily expressed by hemopoietic cells, such as B cells, T cells, NK cells, monocytes, and macrophages, whereas IL-10R2 is constitutively expressed by most cells and tissues (38, 39). We previously demonstrated the presence of IL-10R1 in the human lung adenocarcinoma-derived cell line NCI-H441 (33). It appears that lung epithelial cells express IL-10R1.
Promoters of IL-4-inducible genes contain unique DNA sequences that constitute specific binding sites for STAT6. These DNA sequences, referred to as SBEs, are characterized by a palindromic sequence, 5′-TTC(N4)GAA-3′. The symmetrical GAA half-sites with a 4-bp spacer, called an N4 motif, distinguish the STAT6 site from binding sites for other STAT proteins, where the GAA half-sites usually have 3-bp spacers, called an N3 motif (22). However, genes whose promoters contain 3-base spacer SBEs may still be selectively activated by STAT6. For example, the N3 motif SBE present in the human FcεRIIb (CD23) gene promoter can be bound by multiple STATs, yet only STAT6 appears to transactivate this gene (40). In the case of the Ugrp2 gene, the proximal SBE, located ∼200 bp upstream of the transcription start site, belongs to an N3 motif SBE, binds to the IL-4-activated STAT6, and activates transcription. Interestingly, STAT1 activated by IFN-γ can also bind to this site. Clearly, the presence of a 3- or 4-base spacer does not by itself define the specificity of an SBE.
To confirm STAT6 binding to the proximal SBE of the Ugrp2 gene promoter, we used nuclear extracts prepared from IL-4-stimulated macrophages instead of mtCC or lung primary culture cells for EMSA, because the latter two nuclear extracts did not produce a clear band for a specific binding complex between STAT6 and an oligonucleotide containing the proximal SBE. The exact reason for this is not known. However, in the case of mtCC cells, STAT1 is constitutively activated in the absence of any stimulation and can bind to SBE in the Ug/Ccsp promoter (32). Thus, the binding of STAT6 to the Ugrp2 gene-proximal SBE may be masked by constitutively activated STAT1. In fact, nuclear extracts prepared from control mtCC cells without IL-4 treatment produced a prominent protein-DNA complex that corresponds to STAT1 (A. Yamada and S. Kimura, unpublished observation). Furthermore, IFN-γ-activated STAT1 appears to bind to the same SBE in the Ugrp2 gene promoter and reduce STAT6 binding when macrophage nuclear extracts were used.
In contrast to the stimulatory effects of IL-4 and IL-13 on Ugrp2 expression, IFN-γ was found to repress both basal and IL-4-stimulated Ugrp2 expression. IFN-γ is a well-known antagonist that inhibits the expression of several IL-4- and IL-13-inducible genes, including IGF-1 (41), FcεRIIb (CD23) (37), IgE (42), 15-lipoxygenase (29), IL-1R1 and IL-1RII (36), and IL-1R antagonist (43). One mechanism to explain the antagonistic effect of IFN-γ is that it induces the expression of suppressor of cytokine signaling-1, which inhibits IL-4-induced activation of JAK1 and STAT6 (44, 45). Alternatively, there could exist a competition between STAT1 and STAT6 for binding to the SBE (N3 site). This mechanism occurs in the promoter of the IFN regulatory factor-1 gene (46). This appears to be the case for the inhibitory effect of IFN-γ observed with IL-4- and IL-13-induced Ugrp2 gene expression. It should be noted, however, that suppression of basal Ugrp2 expression by IFN-γ can suppress induction of Ugrp2 gene expression independently of IL-4 or IL-13.
In lungs, the expression of the Ug/Ccsp gene, a prototypical member of the secretoglobin gene superfamily to which Ugrp2 belongs, is regulated by IL-4 and IFN-γ (32, 47-49). However, the mode of regulation of Ug/Ccsp expression by IL-4 and IFN-γ appears to differ from what we observed for Ugrp2 in mtCC cells. Thus, transgenic mice overexpressing IL-4 in the airways had decreased expression of Ug/Ccsp (47), whereas IFN-γ increased the expression of Ug/Ccsp both in vivo, by intratracheal administration of IFN-γ to mice, and in vitro, using mtCC cells (32, 49) and the human bronchial epithelial cell line BEAS-2B in the presence of IFN-γ (48). In the latter, no induction of Ug/Ccsp expression was observed by IL-4 and IL-13. Two mechanisms for IFN-γ stimulation of Ug/Ccsp expression have been described. First, IFN-γ activates STAT1, which directly binds to SBE in the distal promoter of the Ug/Ccsp gene, leading to activation of the gene. Secondly, IFN-γ-activated STAT1 stimulates transcription of IFN regulatory factor-1, which, in turn, activates transcription of hepatocyte nuclear factor 3β (HNF3β), an essential transcription factor for Ug/Ccsp gene expression (50). These findings indicate that the expressions of Ug/Ccsp and Ugrp2 are differentially regulated by IFN-γ. It is also known that UG/CCSP feedback inhibits IFN-γ expression and activity in lung (7). It is possible that UGRP2 mediates a similar feedback inhibitory function to control IL-4/IL-13 activity in lung. Although the physiological consequence of differential regulation by IL-4/IL-13 and IFN-γ between Ugrp2 and Ug/Ccsp genes is not known, a compensatory mechanism may account for this phenomenon. Similar differential regulation was reported between Ugrp1 and Ug/Ccsp genes in the lungs of Ug/Ccsp-knockout mice, in which increased Ugrp1 expression was observed (51).
Transcription analysis of the Ugrp2 promoter-reporter constructs showed that the construct -154 has ∼4-fold higher promoter activity than the -65 construct regardless of IL-4 treatment, and the basal promoter activity gradually increased up to -412 bp. Within the -412 bp of the Ugrp2 gene promoter, there are putative HNF3β (-71 to -61 bp) and C/EBP (-74 to -64 bp) binding sites detected by the transcription factor Search program (52). Many lung-specific genes are known to be regulated by HNF3β and C/EBP (50, 53). A basal increase in Ugrp2 gene promoter activity could be due to binding of these transcription factors to their binding sites. Furthermore, an element that represses Ugrp2 promoter activity appears to be located in between -412 and -1158 bp. We do not know what protein binds to this element that causes repression of promoter activity. Additional studies are required to define the transcription factors that positively and negatively regulate Ugrp2 gene expression.
Finally, Ugrp2, also called HIN-1, was found to be significantly down-regulated due to hypermethylation of the HIN-1 gene promoter in a majority of human primary breast carcinomas, including in situ and invasive ductal and lobular carcinomas and breast cancer cell lines (1). Reintroduction of HIN-1 by transfecting expression plasmid to various breast cancer cell lines inhibited the growth of cells (1). Based on these results, it was proposed that HIN-1 is a candidate tumor suppressor gene (1). In normal adult mice and humans, the expression of Ugrp2 was found at the highest level in the epithelial cells of trachea and lung (3, 5). Ugrp2 mRNA expression was induced by retinoic acid, which correlates with up-regulation of mucinous cell phenotypes when examined in primary normal human bronchial epithelial cells, suggesting that Ugrp2 may have a role in the acquisition or maintenance of the terminally differentiated proximal airway epithelial phenotype (5). In this regard, we previously reported that Ugrp2 expression can also be induced by epidermal growth factor and TGF-α, suggesting a possible involvement of Ugrp2 in epithelial development (54). Despite these observations, the precise physiological and functional significance of Ugrp2 expression in the airways is not fully understood. In the present study, Ugrp2 gene expression is up-regulated by the Th2 cytokines, IL-4 and IL-13, which play a major role in the development of asthma (55, 56). Cotreatment with IL-4 and epidermal growth factor together did not increase Ugrp2 gene expression above that induced by either cytokine alone (A. Yamada and S. Kimura, unpublished observation). It appears therefore that the expression of the Ugrp2 gene can be positively or negatively regulated by multiple cytokines. Overall, our current findings are consistent with a potential role for this gene in the pathogenesis of inflammatory lung diseases, such as asthma. Additional studies are required to define the pathophysiological role of elevated Ugrp2 expression in lung disease.
In conclusion, we have demonstrated that both IL-4 and IL-13 stimulate the expression of the Ugrp2 gene through the binding of STAT6 to an SBE present in the Ugrp2 gene promoter.
Acknowledgments
We thank Drs. R. Kurotani, K. Matsusue, A. Kamiya, and R. Kamijo for their help during the course of experiments, and Dr. F. Gonzalez for his critical review of the manuscript.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. A.Y. was partially supported by a postdoctoral fellowship from the Japanese Society for the Promotion of Science.
- UGRP2
- uteroglobin-related protein 2
- βc
- cytokine receptor common β-chain
- CCSP
- Clara cell secretory protein
- γc
- common γ-chain
- HIN-1
- high in normal-1
- mtCC
- mouse transformed Clara cell line
- SBE
- STAT-binding element
- UG
- uteroglobin
- HNF3β
- hepatocyte nuclear factor 3β
Disclosures
The authors have no financial conflict of interest.
References
- 1.Krop IE, Sgroi D, Porter DA, Lunetta KL, LeVangie R, Seth P, Kaelin CM, Rhei E, Bosenberg M, Schnitt S, et al. HIN-1, a putative cytokine highly expressed in normal but not cancerous mammary epithelial cells. Proc. Natl. Acad. Sci. USA. 2001;98:9796–9801. doi: 10.1073/pnas.171138398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niimi T, Keck-Waggoner CL, Popescu NC, Zhou Y, Levitt RC, Kimura S. UGRP1, a uteroglobin/Clara cell secretory protein-related protein, is a novel lung-enriched downstream target gene for the T/EBP/NKX2.1 homeodomain transcription factor. Mol. Endocrinol. 2001;15:2021–2036. doi: 10.1210/mend.15.11.0728. [DOI] [PubMed] [Google Scholar]
- 3.Niimi T, Copeland NG, Gilbert DJ, Jenkins NA, Srisodsai A, Zimonjic DB, Keck-Waggoner CL, Popescu NC, Kimura S. Cloning, expression, and chromosomal localization of the mouse gene (Scgb3a1, alias Ugrp2) that encodes a member of the novel uteroglobin-related protein gene family. Cytogenet. Genome Res. 2002;97:120–127. doi: 10.1159/000064067. [DOI] [PubMed] [Google Scholar]
- 4.Reynolds SD, Reynolds PR, Pryhuber GS, Finder JD, Stripp BR. Secretoglobins SCGB3A1 and SCGB3A2 define secretory cell subsets in mouse and human airways. Am. J. Respir. Crit. Care Med. 2002;166:1498–1509. doi: 10.1164/rccm.200204-285OC. [DOI] [PubMed] [Google Scholar]
- 5.Porter D, Lahti-Domenici J, Torres-Arzayus M, Chin L, Polyak K. Expression of high in normal-1 (HIN-1) and uteroglobin related protein-1 (UGRP-1) in adult and developing tissues. Mech. Dev. 2002;114:201–204. doi: 10.1016/s0925-4773(02)00056-4. [DOI] [PubMed] [Google Scholar]
- 6.Miele L, Cordella-Miele E, Facchiano A, Mukherjee AB. Novel anti-inflammatory peptides from the region of highest similarity between uteroglobin and lipocortin I. Nature. 1988;335:726–730. doi: 10.1038/335726a0. [DOI] [PubMed] [Google Scholar]
- 7.Dierynck I, Bernard A, Roels H, De Ley M. Potent inhibition of both human interferon-γ production and biologic activity by the Clara cell protein CC16. Am. J. Respir. Cell Mol. Biol. 1995;12:205–210. doi: 10.1165/ajrcmb.12.2.7865218. [DOI] [PubMed] [Google Scholar]
- 8.Andersson O, Nordlund-Moller L, Bronnegard M, Sirzea F, Ripe E, Lund J. Purification and level of expression in bronchoalveolar lavage of a human polychlorinated biphenyl (PCB)-binding protein: evidence for a structural and functional kinship to the multihormonally regulated protein uteroglobin. Am. J. Respir. Cell Mol. Biol. 1991;5:6–12. doi: 10.1165/ajrcmb/5.1.6. [DOI] [PubMed] [Google Scholar]
- 9.Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu. Rev. Immunol. 1999;17:255–281. doi: 10.1146/annurev.immunol.17.1.255. [DOI] [PubMed] [Google Scholar]
- 10.Finkelman FD, Katona IM, Urban JF, Jr., Holmes J, Ohara J, Tung AS, Sample JV, Paul WE. IL-4 is required to generate and sustain in vivo IgE responses. J. Immunol. 1988;141:2335–2341. [PubMed] [Google Scholar]
- 11.Madden KB, Urban JF, Jr., Ziltener HJ, Schrader JW, Finkelman FD, Katona IM. Antibodies to IL-3 and IL-4 suppress helminth-induced intestinal mastocytosis. J. Immunol. 1991;147:1387–1391. [PubMed] [Google Scholar]
- 12.Schleimer RP, Sterbinsky SA, Kaiser J, Bickel CA, Klunk DA, Tomioka K, Newman W, Luscinskas FW, Gimbrone MA, Jr., McIntyre BW, et al. IL-4 induces adherence of human eosinophils and basophils but not neutrophils to endothelium: association with expression of VCAM-1. J. Immunol. 1992;148:1086–1092. [PubMed] [Google Scholar]
- 13.Kondo M, Takeshita T, Ishii N, Nakamura M, Watanabe S, Arai K, Sugamura K. Sharing of the interleukin-2 (IL-2) receptor γ chain between receptors for IL-2 and IL-4. Science. 1993;262:1874–1877. doi: 10.1126/science.8266076. [DOI] [PubMed] [Google Scholar]
- 14.Russell SM, Keegan AD, Harada N, Nakamura Y, Noguchi M, Leland P, Friedmann MC, Miyajima A, Puri RK, Paul WE, et al. Interleukin-2 receptor γ chain: a functional component of the interleukin-4 receptor. Science. 1993;262:1880–1883. doi: 10.1126/science.8266078. [DOI] [PubMed] [Google Scholar]
- 15.Vita N, Lefort S, Laurent P, Caput D, Ferrara P. Characterization and comparison of the interleukin 13 receptor with the interleukin 4 receptor on several cell types. J. Biol. Chem. 1995;270:3512–3517. doi: 10.1074/jbc.270.8.3512. [DOI] [PubMed] [Google Scholar]
- 16.Obiri NI, Debinski W, Leonard WJ, Puri RK. Receptor for interleukin 13: interaction with interleukin 4 by a mechanism that does not involve the common γ chain shared by receptors for interleukins 2, 4, 7, 9, and 15. J. Biol. Chem. 1995;270:8797–8804. doi: 10.1074/jbc.270.15.8797. [DOI] [PubMed] [Google Scholar]
- 17.Callard RE, Matthews DJ, Hibbert L. IL-4 and IL-13 receptors: are they one and the same? Immunol. Today. 1996;17:108–110. doi: 10.1016/0167-5699(96)80600-1. [DOI] [PubMed] [Google Scholar]
- 18.Kotanides H, Reich NC. Requirement of tyrosine phosphorylation for rapid activation of a DNA binding factor by IL-4. Science. 1993;262:1265–1267. doi: 10.1126/science.7694370. [DOI] [PubMed] [Google Scholar]
- 19.Hou J, Schindler U, Henzel WJ, Ho TC, Brasseur M, McKnight SL. An interleukin-4-induced transcription factor: IL-4 Stat. Science. 1994;265:1701–1706. doi: 10.1126/science.8085155. [DOI] [PubMed] [Google Scholar]
- 20.Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, Chu C, Quelle FW, Nosaka T, Vignali DA, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630–633. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 21.Quelle FW, Shimoda K, Thierfelder W, Fischer C, Kim A, Ruben SM, Cleveland JL, Pierce JH, Keegan AD, Nelms K. Cloning of murine Stat6 and human Stat6, Stat proteins that are tyrosine phosphorylated in responses to IL-4 and IL-3 but are not required for mitogenesis. Mol. Cell. Biol. 1995;15:3336–3343. doi: 10.1128/mcb.15.6.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darnell JE., Jr. STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 23.Ihle JN. Cytokine receptor signalling. Nature. 1995;377:591–594. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- 24.te Velde AA, Klomp JP, Yard BA, de Vries JE, Figdor CG. Modulation of phenotypic and functional properties of human peripheral blood monocytes by IL-4. J. Immunol. 1988;140:1548–1554. [PubMed] [Google Scholar]
- 25.Vercelli D, Jabara HH, Lee BW, Woodland N, Geha RS, Leung DY. Human recombinant interleukin 4 induces FcεR2/CD23 on normal human monocytes. J. Exp. Med. 1988;167:1406–1416. doi: 10.1084/jem.167.4.1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M, Giri JG, Dower SK, Sims JE, Mantovani A. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science. 1993;261:472–475. doi: 10.1126/science.8332913. [DOI] [PubMed] [Google Scholar]
- 27.Fenton MJ, Buras JA, Donnelly RP. IL-4 reciprocally regulates IL-1 and IL-1 receptor antagonist expression in human monocytes. J. Immunol. 1992;149:1283–1288. [PubMed] [Google Scholar]
- 28.de Wit H, Hendriks DW, Halie MR, Vellenga E. Interleukin-4 receptor regulation in human monocytic cells. Blood. 1994;84:608–615. [PubMed] [Google Scholar]
- 29.Conrad DJ, Kuhn H, Mulkins M, Highland E, Sigal E. Specific inflammatory cytokines regulate the expression of human monocyte 15-lipoxygenase. Proc. Natl. Acad. Sci. USA. 1992;89:217–221. doi: 10.1073/pnas.89.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Waal Malefyt R, Figdor CG, Huijbens R, Mohan-Peterson S, Bennett B, Culpepper J, Dang W, Zurawski G, de Vries JE. Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes: comparison with IL-4 and modulation by IFN-γ or IL-10. J. Immunol. 1993;151:6370–6381. [PubMed] [Google Scholar]
- 31.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 32.Magdaleno SM, Wang G, Jackson KJ, Ray MK, Welty S, Costa RH, DeMayo FJ. Interferon-γ regulation of Clara cell gene expression: in vivo and in vitro. Am. J. Physiol. 1997;272:L1142–L1151. doi: 10.1152/ajplung.1997.272.6.L1142. [DOI] [PubMed] [Google Scholar]
- 33.Srisodsai A, Kurotani R, Chiba Y, Sheikh F, Young HA, Donnelly RP, Kimura S. Interleukin-10 induces uteroglobin-related protein (UGRP) 1 gene expression in lung epithelial cells through homeodomain transcription factor T/EBP/NKX2.1. J. Biol. Chem. 2004;279:54358–54368. doi: 10.1074/jbc.M405331200. [DOI] [PubMed] [Google Scholar]
- 34.Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts,’ prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heim MH. The Jak-STAT pathway: cytokine signalling from the receptor to the nucleus. J. Recept. Signal Transduct. Res. 1999;19:75–120. doi: 10.3109/10799899909036638. [DOI] [PubMed] [Google Scholar]
- 36.Dickensheets HL, Donnelly RP. IFN-γ and IL-10 inhibit induction of IL-1 receptor type I and type II gene expression by IL-4 and IL-13 in human monocytes. J. Immunol. 1997;159:6226–6233. [PubMed] [Google Scholar]
- 37.te Velde AA, Rousset F, Peronne C, De Vries JE, Figdor CG. IFN-α and IFN-γ have different regulatory effects on IL-4-induced membrane expression of FcεRIIb and release of soluble FcεRIIb by human monocytes. J. Immunol. 1990;144:3052–3059. [PubMed] [Google Scholar]
- 38.Donnelly RP, Dickensheets H, Finbloom DS. The interleukin-10 signal transduction pathway and regulation of gene expression in mononuclear phagocytes. J. Interferon Cytokine Res. 1999;19:563–573. doi: 10.1089/107999099313695. [DOI] [PubMed] [Google Scholar]
- 39.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 40.Kohler I, Rieber EP. Allergy-associated Iε and Fcε receptor II (CD23b) genes activated via binding of an interleukin-4-induced transcription factor to a novel responsive element. Eur. J. Immunol. 1993;23:3066–3071. doi: 10.1002/eji.1830231204. [DOI] [PubMed] [Google Scholar]
- 41.Wynes MW, Riches DW. Induction of macrophage insulin-like growth factor-I expression by the Th2 cytokines IL-4 and IL-13. J. Immunol. 2003;171:3550–3559. doi: 10.4049/jimmunol.171.7.3550. [DOI] [PubMed] [Google Scholar]
- 42.Pene J, Rousset F, Briere F, Chretien I, Bonnefoy JY, Spits H, Yokota T, Arai N, Arai K, Banchereau J, et al. IgE production by normal human lymphocytes is induced by interleukin 4 and suppressed by interferons γ and α and prostaglandin E2. Proc. Natl. Acad. Sci. USA. 1988;85:6880–6884. doi: 10.1073/pnas.85.18.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohmori Y, Smith MF, Jr., Hamilton TA. IL-4-induced expression of the IL-1 receptor antagonist gene is mediated by STAT6. J. Immunol. 1996;157:2058–2065. [PubMed] [Google Scholar]
- 44.Dickensheets HL, Venkataraman C, Schindler U, Donnelly RP. Interferons inhibit activation of STAT6 by interleukin 4 in human monocytes by inducing SOCS-1 gene expression. Proc. Natl. Acad. Sci. USA. 1999;96:10800–10805. doi: 10.1073/pnas.96.19.10800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Losman JA, Chen XP, Hilton D, Rothman P. Cutting edge: SOCS-1 is a potent inhibitor of IL-4 signal transduction. J. Immunol. 1999;162:3770–3774. [PMC free article] [PubMed] [Google Scholar]
- 46.Ohmori Y, Hamilton TA. IL-4-induced STAT6 suppresses IFN-γ-stimulated STAT1-dependent transcription in mouse macrophages. J. Immunol. 1997;159:5474–5482. [PubMed] [Google Scholar]
- 47.Jain-Vora S, Wert SE, Temann UA, Rankin JA, Whitsett JA. Interleukin-4 alters epithelial cell differentiation and surfactant homeostasis in the postnatal mouse lung. Am. J. Respir. Cell Mol. Biol. 1997;17:541–551. doi: 10.1165/ajrcmb.17.5.2883. [DOI] [PubMed] [Google Scholar]
- 48.Yao XL, Ikezono T, Cowan M, Logun C, Angus CW, Shelhamer JH. Interferon-γ stimulates human Clara cell secretory protein production by human airway epithelial cells. Am. J. Physiol. 1998;274:L864–L869. doi: 10.1152/ajplung.1998.274.5.L864. [DOI] [PubMed] [Google Scholar]
- 49.Ramsay PL, Luo Z, Magdaleno SM, Whitbourne SK, Cao X, Park MS, Welty SE, Yu-Lee LY, DeMayo FJ. Transcriptional regulation of CCSP by interferon-γ in vitro and in vivo. Am. J. Physiol. 2003;284:L108–L118. doi: 10.1152/ajplung.00186.2002. [DOI] [PubMed] [Google Scholar]
- 50.Chang A, Ramsay P, Zhao B, Park M, Magdaleno S, Reardon MJ, Welty S, DeMayo FJ. Physiological regulation of uteroglobin/CCSP expression. Ann. NY Acad. Sci. 2000;923:181–192. doi: 10.1111/j.1749-6632.2000.tb05529.x. [DOI] [PubMed] [Google Scholar]
- 51.Watson TM, Reynolds SD, Mango GW, Boe IM, Lund J, Stripp BR. Altered lung gene expression in CCSP-null mice suggests immunoregulatory roles for Clara cells. Am. J. Physiol. 2001;281:L1523–L1530. doi: 10.1152/ajplung.2001.281.6.L1523. [DOI] [PubMed] [Google Scholar]
- 52.Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, et al. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26:362–367. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cassel TN, Nord M. C/EBP transcription factors in the lung epithelium. Am. J. Physiol. 2003;285:L773–L781. doi: 10.1152/ajplung.00023.2003. [DOI] [PubMed] [Google Scholar]
- 54.Yamada A, Kimura S. Induction of uteroglobin-related protein 2 (Ugrp2) expression by EGF and TGFα. FEBS Lett. 2005;579:2221–2225. doi: 10.1016/j.febslet.2005.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corry DB, Kheradmand F. Biology and therapeutic potential of the interleukin-4/interleukin-13 signaling pathway in asthma. Am. J. Respir. Med. 2002;1:185–193. doi: 10.1007/BF03256608. [DOI] [PubMed] [Google Scholar]
- 56.Izuhara K, Arima K. Signal transduction of IL-13 and its role in the pathogenesis of bronchial asthma. Drug News Perspect. 2004;17:91–98. doi: 10.1358/dnp.2004.17.2.829041. [DOI] [PubMed] [Google Scholar]