

Abstract
NO production by macrophages in response to lipoteichoic acid (LTA) and a synthetic lipopeptide (Pam3CSK4) was investigated. LTA and Pam3CSK4 induced the production of both TNF-α and NO. Inhibitors of platelet-activating factor receptor (PAFR) blocked LTA- or Pam3CSK4-induced production of NO but not TNF-α. Jak2 tyrosine kinase blocked LTA-induced production of NO but not TNF-α. PAFR inhibition blocked phosphorylation of Jak2 and STAT1, a key factor for expressing inducible NO synthase. In addition, LTA did not induce IFN-β expression, and p38 mitogen-activated protein serine kinase was necessary for LTA-induced NO production but not for TNF-α production. These findings suggest that Gram-positive bacteria induce NO production using a PAFR signaling pathway to activate STAT1 via Jak2. This PAFR/Jak2/STAT1 signaling pathway resembles the IFN-β, type I IFNR/Jak/STAT1 pathway described for LPS. Consequently, Gram-positive and Gram-negative bacteria appear to have different but analogous mechanisms for NO production.
During bacterial infections, bacterial constituents such as LPS can induce the production of various host factors, which can, in turn, cause multiorgan dysfunction. This medical condition often results in intractable hypotension, which is a significant cause of death for patients in intensive care units (2). Host factors associated with multiorgan dysfunction include increased levels of proinflammatory cytokines such as TNF-α, IL-1β, IL-6, and IL-8 (3), and glycerophospholipids (4) such as platelet-activating factor (PAF).3 TNF-α has been labeled “the hub of the cytokine network” (3) because it is sufficient to cause septic symptoms in animals (5) and because Abs to TNF-α protect animals in sepsis models (6, 7). PAF is elevated in patients with septic shock (8) and can cause hypotension in animals (9). It exerts its inflammatory properties by binding to a member of the 7-transmembrane receptor family (4) which then stimulates cells through mostly, but not always (10), G proteins.
NO has been implicated as another key host factor in sepsis for several reasons. First, excess amounts of NO are produced during sepsis (11). Second, NO can activate the myosin phosphatase and potassium channels in arterial smooth muscle cells, thereby causing vasodilation and hypotension (12). Third, inhibitors of NO synthesis have been shown to be beneficial for patients with severe sepsis (13) or for animals with various experimental infections (14-16). Lastly, animals do not become hypotensive after an LPS exposure if they are deficient in inducible NO synthase (iNOS) (17), an enzyme critical to the excessive production of NO. Understanding the mechanisms for NO production during infections is an important goal for the prevention of sepsis. During Gram-negative sepsis, the LPS reacts with TLR4 and elicits the expression of IFN-β using a signaling pathway independent of the MyD88 gene (1, 18, 19). IFN-β then activates various transcription factors, including STAT1 (20-22) that is necessary for iNOS gene expression (23). Activation of STAT1 by IFN-β requires phosphorylation of both tyrosine and serine residues (21), with the phosphorylation being mediated by Tyk2/Jak kinases (20, 24), p38 MAPKs (25, 26), and various other kinases (27, 28).
The production of NO during Gram-positive bacterial infection is an enigma, even though half of the microbiologically confirmed cases of sepsis are due to infections by Gram-positive bacteria (29). Unlike Gram-negative bacteria, Gram-positive bacteria primarily stimulate innate immunity, not by TLR4 but by TLR2 (30). All TLR2 signaling is mediated by the MyD88 gene (19, 31), and various TLR2 ligands (such as peptidoglycan (PGN) and bacterial lipoproteins) have been reported not to elicit the production of IFN-β (1, 18) and NO (1, 32). Indeed, Gram-positive bacteria do not appear to elicit IFN-β production in mice (33) even though they can induce NO production (34). Because lipoteichoic acid (LTA) may induce NO production (34, 35), we have investigated NO production using LTA from pneumococci and staphylococci, which account for the majority of cases of Gram-positive sepsis (29).
Materials and Methods
Reagents and cells
Nω-nitro-l-arginine methyl ester hydrochloride (l-NAME), Nω-nitro-d-arginine methyl ester hydrochloride (d-NAME), ABT-491, and Escherichia coli LPS (055:B5) were obtained from Sigma-Aldrich. LPS was repurified by phenol extraction (36) before use. 2-Iminopiperidine hydrochloride, an iNOS-specific inhibitor (37), was purchased from Biotrend. Pertussis toxin, AG1478, AG490, SB203580, SP600125, and PD98059 were purchased from Calbiochem. PAF inhibitors (CV6209 and CV3988) and G-protein antagonist 2A were purchased from BIOMOL. All the reagents for RT-PCR were purchased from Promega, except for recombinant TaqDNA polymerase (rTaq) and dNTP, which were purchased from Takara Bio. Rabbit polyclonal Abs specific for p38, the phosphorylated forms of p38, Erk1/2, and stress-activated protein kinase (SAPK)/JNK were obtained from Cell Signaling Technology. Rabbit Abs to phosphorylated Jak2, STAT1 with phosphorylated serine at 727, and STAT1 with phosphorylated tyrosine at 701 along with HRP-conjugated anti-rabbit IgG were also obtained from Cell Signaling Technology. Mouse macrophage-like cell line RAW 264.7 (TIB-71) was purchased from the American Type Culture Collection.
Preparation of LTA
Highly purified and structurally intact pneumococcal LTA (PnLTA) and staphylococcal LTA (StLTA) were prepared from nonencapsulated pneumococci R36A and Staphylococcus aureus (ATCC 6538; American Type Culture Collection), respectively, by organic solvent extraction, Octyl-Sepharose and ion-exchange chromatography, as we have previously described (38-40). Our LTA preparations had <5 pg of endotoxin/mg LTA. Additional studies of the purity of our LTA preparations have been reported (40).
Culture of RAW 264.7 cells
RAW 264.7 cells were cultured with DMEM (Cellgro/Mediatech) supplemented with 10% FBS (HyClone), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a 5% CO2 humidified incubator. They were stimulated with various stimulants at 5 × 105 cells/ml.
Stimulation of bone marrow macrophages
C57BL/6 mice were obtained from The Jackson Laboratory, and TLR2-deficient mice on a C57BL/6 background were bred in our animal facility using the breeding pairs from Dr. S. Akira (Osaka, Japan) with an Institutional Review Board (IRB) approval. Bone marrow cells were harvested from the tibia of 6- to 8-wk-old C57BL/6- or the TLR2-deficient mice using an IRB-approved protocol. Bone marrow cells (106 cells/ml) were suspended in DMEM supplemented with 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 20 ng/ml M-CSF (R&D Systems), and were cultured for 7 days. The adherent cells were harvested with trypsin-EDTA, washed with PBS, and suspended in DMEM containing 3% FCS. One-hundred microliters of the cell suspension (2 × 105 cells/ml) was placed in a 96 microwell and stimulated with 10 μg/ml StLTA. After 2 days of stimulation, the culture supernatant was analyzed for NO production.
Determination of NO and TNF-α
Nitrite accumulation was determined to be an indicator of NO production in the culture media, as previously described (41). Briefly, the culture media obtained at the end of the culture were mixed with an equal volume of Griess reagent (1% sulfanilamide, 0.1% naphthylethylenediamine dihydrochloride, and 2% phosphoric acid) in a 96-well ELISA plate. The OD540 was determined with an ELISA plate reader. Nitrite production was determined by comparing the OD with the standard curve obtained with NaNO2. The amount of TNF-α in the culture supernatant was determined with a mouse TNF-α ELISA kit (Ready-SET-Go kit; eBioscience) following the manufacturer’s recommended protocol. The assay is a sandwich-type ELISA using an immobilized anti-TNF-α Ab and an enzyme-conjugated anti-TNF-α Ab.
RT-PCR
RT-PCR was performed as follows: total RNA was isolated using TRIzol reagent (Invitrogen Life Technologies), and RNA was reverse-transcribed into cDNA with random hexamers (Promega). For RT-PCR, the amplifications were performed in a total volume of 30 μl containing 0.5 U of TaqDNA polymerase and 10 pmol of primers specific for murine IFN-β (5′-TCCAAGAAAGGACGAACATTCG-3′, 5′TGAGGACATCTCCCACGTCAA-3′) (1); iNOS (5′-GGATAGGCAGAGATTGGAGG-3′, 5′-AATGAGGATGCAAGGCTGG-3′); TNF-α (5′-ATGAGCACAGAAAGCATGATC-3′, 5′-TACAGGCTTGTCACTCGAATT-3′); and β-actin (5′-GTGGGGCGCCCCAGGCACCA-3′ and 5′-CTCCTTAATGTCACGCACGATTTC-3′). The amplifications were performed for 25 cycles for β-actin and for 30 cycles for all others. Equal amounts of RT-PCR products were separated on an agarose gel (1%) and visualized by ethidium bromide staining with a gel documentation system (Gel Doc 2000; Life Science Research).
Western blotting
RAW 264.7 cells (5 × 105 cells/ml, 10 ml) were plated onto a 100-mm tissue-culture dish in serum-free DMEM supplemented with antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin) for 3 h, washed, and resuspended in fresh serum-free DMEM supplemented with antibiotics for 1 h. The cells were exposed to an antagonist (e.g., CV6209) for 1 h before being stimulated with 50 μg/ml PnLTA, 5 μg/ml StLTA, or 1 μg/ml LPS for 30 min (or the indicated time periods). Cells were then washed with PBS and lysed with RIPA buffer (Upstate Biotechnology), as recommended by the manufacturer. Twenty micrograms of the whole-cell lysate were separated by 10% SDS-PAGE and electrotransferred to a polyvinylidene difluoride membrane (Millipore). The membrane was incubated with a blocking buffer (5% BSA/1× TBS/0.1% Tween 20) at room temperature for 1 h and then was kept on ice overnight with the same buffer containing rabbit polyclonal Abs for the MAPKs (or STAT1). After washing three times with TBST (1× TBS/0.1% Tween 20), the membrane was incubated with HRP-conjugated anti-rabbit IgG in the blocking buffer at room temperature for 1 h. Then, after washing three times with TBST, the immunoreactive bands were detected with ECL reagents (Amersham Biosciences).
Results
PnLTA induces NO production by murine macrophages
Cultures of the murine macrophage cell line RAW 264.7 were stimulated with various concentrations (1-100 μg/ml) of highly purified PnLTA for various time periods. The highly purified PnLTA was prepared using ion-exchange chromatography as well as hydrophobic interaction chromatography (38). Our LTA preparations had <5 pg of endotoxin/mg LTA and had undetectable amounts of other contaminants (e.g., DNA or protein) (40). Maximal NO production required 48 h (Fig. 1A), while maximal TNF-α production was observed in only 15 h (Fig. 1B). Consequently, in all subsequent experiments, NO production was measured after 48 h of stimulation and TNF-α after 15 h.
FIGURE 1.

PnLTA induces NO and TNF-α production. A and B, Cultures of the murine macrophage cell line RAW 264.7 were stimulated with PnLTA for the indicated time periods. The culture supernatants were analyzed for (A) nitrite levels or for (B) TNF-α levels. All error bars in the figures indicate SD. C, The cells were also stimulated with PnLTA, StLTA, or E. coli LPS at the indicated concentrations for 48 h, and the supernatants were analyzed for NO2 levels.
To examine the ability of LTA from other Gram-positive bacteria to induce NO production, we stimulated RAW 264.7 cells with various concentrations of StLTA, PnLTA, and E. coli LPS and assessed NO production at 48 h (Fig. 1C). All three stimuli induced NO production but with different potencies. Half-maximal stimulations were achieved at ∼0.01 μg/ml LPS, 2 μg/ml StLTA, and 50 μg/ml PnLTA. This indicates that, while both LTAs can stimulate NO production, PnLTA is less potent than StLTA, which in turn are less potent than LPS.
LTA does not induce IFN-β in murine macrophages
It has been reported that LPS stimulates NO production by inducing the expression of an autocrine stimulator, IFN-β (1), but many TLR2 stimulants such as a lipopeptide (Pam3CSK4) do not induce IFN-β (1). When we examined IFN-β mRNA expression, LPS induced the expression of IFN-β mRNA (Fig. 2A), but PnLTA and StLTA did not. However, all three stimuli (LPS, StLTA, and PnLTA) stimulated the production of iNOS mRNA (Fig. 2A). Furthermore, the PnLTA-induced NO production was almost completely suppressed by l-NAME but not by D-NAME (Fig. 2B). Also, NO production by StLTA, PnLTA, and LPS could be inhibited by iminopiperidine (Fig. 2C). l-NAME and iminopiperidine are inhibitors of iNOS; l-NAME can inhibit various isoforms of NOS but iminopiperidine is an iNOS-specific inhibitor (37). Thus, NO production is dependent on iNOS, and LTA, unlike LPS, can induce iNOS expression in the absence of IFN-β induction.
FIGURE 2.

A, LTA induces mRNA synthesis for iNOS and TNF-α but not for IFN-β. RAW 264.7 cells were stimulated with PnLTA, StLTA, or E. coli LPS for 3 h at the indicated concentrations. RT-PCR products of IFN-β, iNOS, TNF-α, and β-actin mRNA were separated in an agarose gel and visualized by ethidium bromide staining. B, NO production by the cells stimulated with PnLTA can be suppressed with a NO synthase inhibitor (l-NAME, ■) but not with an inert control molecule (d-NAME, □). ***, Significant reduction (p < 0.001) in nitrite levels by inhibitors. C, NO production by the cells stimulated with PnLTA can be suppressed with 2-IPD (iminopiperidine), an iNOS-specific inhibitor.
PAFR and TLR2 are involved in NO production by LTA
PnLTA resembles PAF in structure (42), and StLTA has been reported to stimulate PAFR to induce mucin gene expression by epithelial cells (43). In view of these observations, we investigated the involvement of PAFR in LTA-induced NO production. The PAFR inhibitor CV6209 did not inhibit NO production induced by LPS and CV3988 slightly reduced NO production by LPS (Fig. 3, C and F); however, they almost completely (>90%) suppressed NO production induced by either pneumococcal or staphylococcal LTA (Fig. 3, A, B, D, and E). In addition, the two PAFR inhibitors did not inhibit TNF-α production induced by any of the three stimulators (Fig. 3).
FIGURE 3.
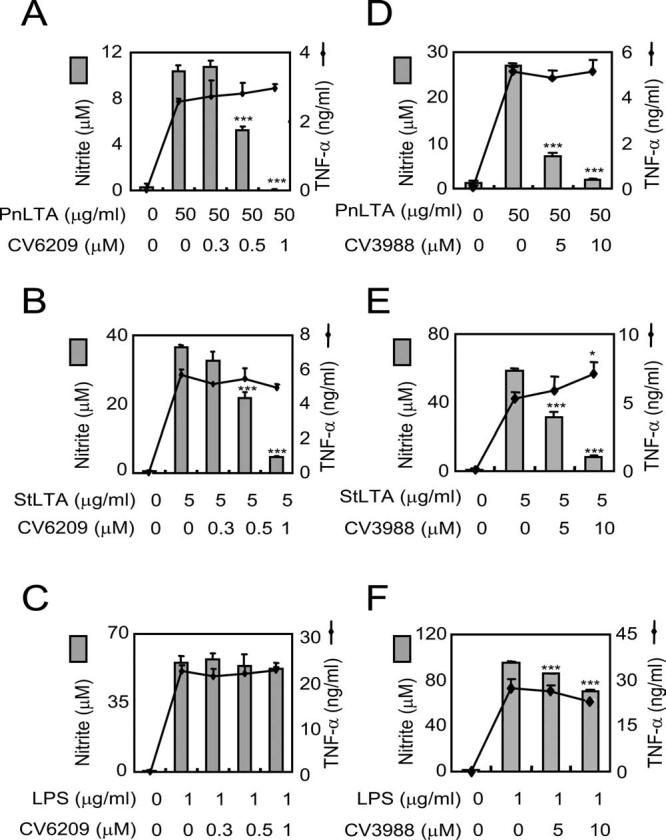
PAFR signaling is necessary for LTA induction of NO but not of TNF-α. RAW 264.7 cells were pretreated with varying concentrations of a PAFR antagonist for 1 h and then stimulated with (A and D) PnLTA, (B and E) StLTA, or (C and F) E. coli LPS for 16 or 48 h for assessment of TNF-α production or of NO production, respectively. PAFR antagonists were (A-C) CV6209 and (D-F) CV3988. At the end of stimulation, the amounts of nitrite (bar) and TNF-α (line) in the culture media were quantified. *, p < 0.05 and ***, p < 0.001 when the levels produced in the presence of an inhibitor are compared with those without an inhibitor.
Although we found PAFR involvement in LTA-induced NO production, LTA is a TLR2 ligand; therefore, we used TLR2-deficient mice to determine whether TLR2 is necessary for NO production. We obtained bone marrow macrophages from C57BL/6 mice or TLR2-deficient mice and stimulated the marrow cells with StLTA, PnLTA, and LPS in two separate experiments (Table I). StLTA was able to induce normal macrophages but not TLR2-deficient macrophages to produce NO (p = 0.02). Also, PnLTA induced NO production by normal macrophages but not byTLR2-/- mouse macrophages (p = 0.0025). In contrast, LPS induced NO production in both normal and TLR2-deficient macrophages (p = 0.39). These findings shown in Fig. 3 and Table I indicate that TLR2 and PAFR stimulations are necessary for LTA-induced NO production. Also, these findings indicate that NO production is not due to LPS contaminating our LTA preparations.
Table I.
Effect of TLR2 on NO production
| Stimulus | Mice |
p Valuea | |
|---|---|---|---|
| C57BL/6 mice | TLR2-/- mice | ||
| None | 0.31 (0.04)b | 0.27 (0.20) | 0.758 |
| StLTA (10 μg/ml) | 0.76 (0.19) | 0.24 (0.15) | 0.0217 |
| None | <0.49 (0.05) | <0.49 (0.00) | |
| PnLTA (50 μg/ml) | 3.70 (0.32) | <0.49 (0.00) | 0.00243 |
| LPS (1 μg/ml) | 13.89 (2.41) | 12.29 (1.31) | 0.387 |
p values were obtained with nonpaired t test by comparing the normal mouse group with the TLR2-deficient mouse group.
Average amount of nitrate (in micromoles) with standard deviation (in parenthesis) are shown. There are three samples per each test.
A synthetic lipopeptide, Pam3CSK4, induces NO production just as LTA does
To investigate whether another TLR2 ligand can induce NO production, we stimulated RAW 264.7 cells with Pam3CSK4, a chemically synthesized lipopeptide that mimics bacterial lipoprotein. Pam3CSK4 induced both TNF-α and NO production in a dose-dependent manner, with maximal responses seen at doses >40 ng/ml (Fig. 4A). To investigate the role of PAFR in this lipopeptide-induced NO production, RAW 264.7 cells were stimulated with Pam3CSK4 in the presence of one of two PAFR antagonists (CV6209 or ABT-491). ABT-491 was tested because it is not a PAF analog like CV6209 and its inhibition mechanism is different from CV6209 (44, 45). Production of NO, but not of TNF-α, was significantly reduced in the presence of CV6209 (Fig. 4B) or ABT-491 (Fig. 4C). The inhibition was achieved at inhibitor doses commonly used by others (44, 45). In summary, Pam3CSK4 stimulates RAW 264.7 cells to produce NO just as LTA does.
FIGURE 4.
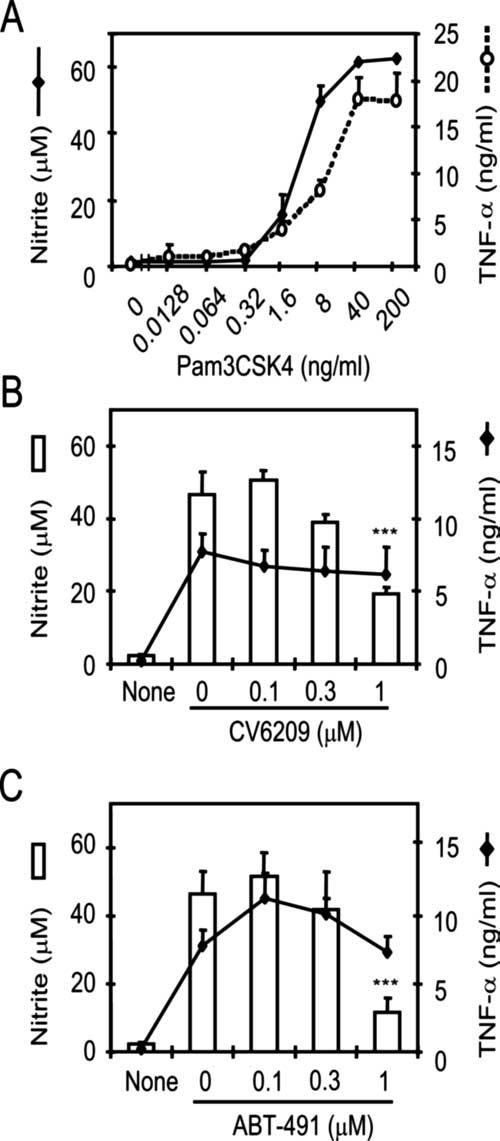
A synthetic lipopeptide, Pam3CSK4, induces NO production, and NO production requires PAFR signaling. A, RAW 264.7 cells were stimulated with Pam3CSK4 at the indicated concentrations for 16 or 48 h for assessment of TNF-α production (○) or of NO production (●), respectively. B and C, The cells were pretreated with varying concentrations of CV6209 or ABT-491 for 1 h and then stimulated with the Pam3CSK4 for 16 or 48 h for assessment of TNF-α production (lines) or of NO production (bars), respectively. ***, Significant (p < 0.001) reduction in NO production by an inhibitor.
A different pathway of PAFR stimulation is used for NO production by LTA
StLTA has been shown to directly stimulate epithelial cells to produce mucin via PAFR, with epidermal growth factor receptor (EGFR) and G proteins shown to be the signaling intermediates (43). Furthermore, G proteins have been shown to mediate most of the known PAFR-signaling pathways (4). Therefore, we investigated the involvement of EGFR and G proteins in NO production. A G-protein inhibitor (pertussis toxin) and an EGFR inhibitor (AG1478) failed to block LTA-induced NO or TNF-α production (Fig. 5). In addition, two different G-protein inhibitors (GDP-βS and G-protein antagonist 2A) did not block NO or TNF-α production (data not shown). These findings suggest that LTA stimulates murine macrophages to produce NO using a PAFR signaling pathway that is different from the one previously suggested for inducing mucin expression (43).
FIGURE 5.
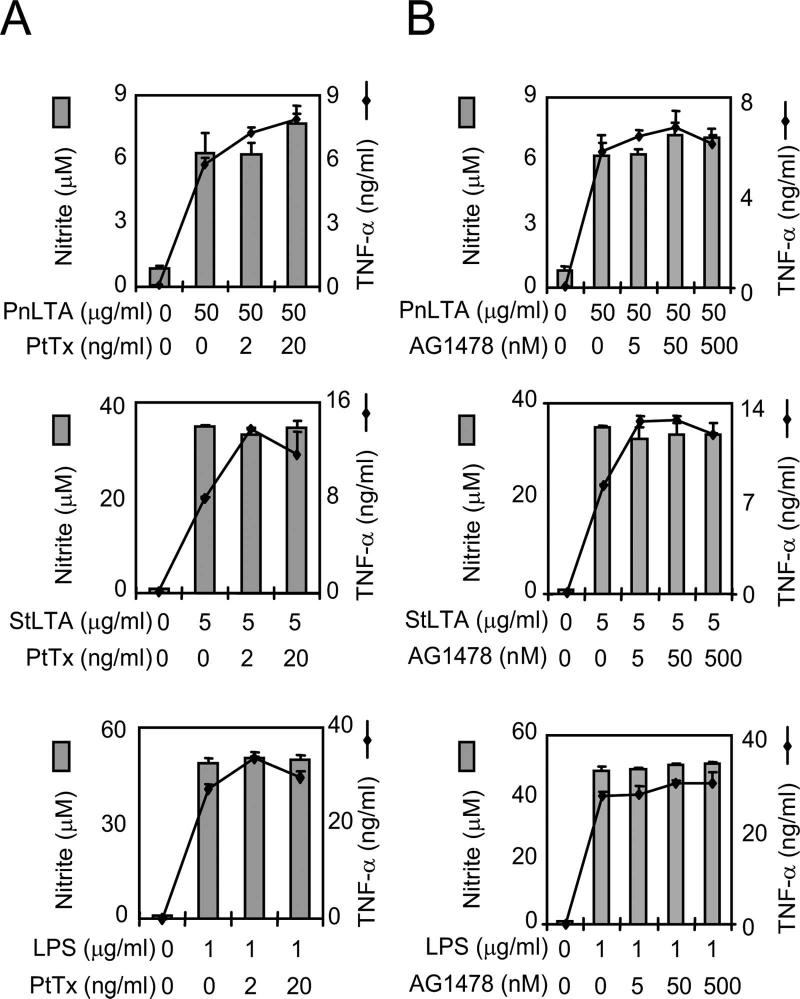
LTA signaling for NO or TNF-α production does not involve G proteins or EGFR. RAW 264.7 cells were incubated with (A) pertussis toxin or (B) AG1478 for 1 h and then stimulated with StLTA, PnLTA, or E. coli LPS for 48 h for the NO assay and 15 h for the TNF-α assay. At the end of the culture period, the media were analyzed for the levels of TNF-α (lines) and nitrite (bars).
Jak2 and p38 MAPK antagonists block production of NO but not of TNF-α
Without involving G proteins, PAFR has been shown to be able to activate Jak2 and Tyk2 tyrosine kinases directly and phosphorylate STAT1 (10, 46), which is a transcription factor important in iNOSgene expression (23). Thus, we examined the effect of a Jak2 inhibitor (AG490) on TNF-α and NO production. The inhibitor suppressed LTA-induced NO production without suppressing TNF-α production (Fig. 6A). Similarly, it suppressed LPS-induced NO production without affecting TNF-α production (Fig. 6A). The suppression of NO production appears to be more marked for LTA than for LPS.
FIGURE 6.
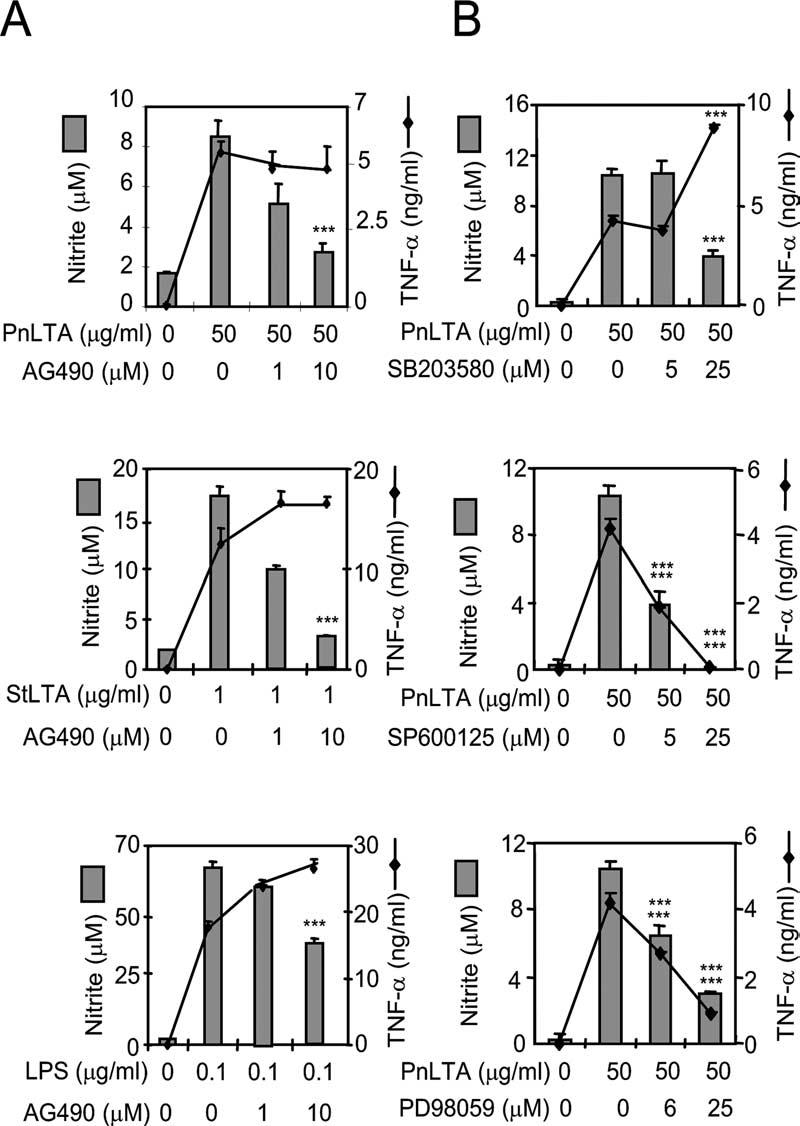
Intracellular signal transduction for LTA-induced NO vs TNF-α production. A, RAW 264.7 cells were stimulated with PnLTA (50 μg/ml), StLTA (1 μg/ml), or E. coli LPS (0.1 μg/ml) in the presence of varying concentration of a Jak2 inhibitor (AG490). Culture supernatants were harvested after 16 h for the TNF-α determination or after 48 h for the nitrite determination. B, RAW 264.7 cells were pretreated with specific inhibitors for p38 kinase (SB203580), SAPK/JNK (SP600125), or Erk (PD98059) for 1 h and then stimulated with PnLTA (50 μg/ml) for 16 h for the TNF-α determination or 48 h for the NO determination. At the end of the culture period, the media were analyzed for TNF-α and nitrite levels. ***, A significant (p < 0.001) change in TNF-α or nitrite levels due to an inhibitor.
Because p38 MAPKs are important in autologous PAF production (47) and phosphorylating the serine residue of STAT1 (25, 26), we examined MAPKs for their involvement in NO or TNF-α production. RAW 264.7 cells were stimulated with PnLTA in the presence of inhibitors for p38, JNK, and Erk MAPK pathways (SB203580, SP600125, and PD98059, respectively). All three inhibitors could suppress NO production (>75%), whereas the inhibitors of JNK and Erk inhibited TNF-α production (Fig. 6B). Thus, p38 MAPK is involved in only the signaling pathway used for iNOS gene expression.
PAFR inhibitor blocks LTA-induced phosphorylation of STAT1 and Jak2
To further confirm the involvement of Jak2 and STAT1 downstream of PAFR, we examined the effect of a PAFR inhibitor on Jak2 and STAT1 phosphorylation. STAT1 from unstimulated RAW 264.7 cells was not phosphorylated (Fig. 7A, lane 1). When the cells were stimulated with either PnLTA (10 μg/ml) or StLTA (1 μg/ml), STAT1 was phosphorylated within 2 h at both serine (residue 727) and tyrosine (residue 701) (Fig. 7A, lanes 2 and 3). However, when the cells were stimulated with LTA in the presence of a PAFR antagonist (CV6209), tyrosine phosphorylation was almost completely abolished and serine phosphorylation was significantly reduced (Fig. 7A, lanes 4 and 5). This variation was observed even though comparable amounts of protein were in each lane, as shown by the even staining for “Total p38” (Fig. 7). Similar to STAT1, Jak2 from unstimulated cells was only weakly phosphorylated (Fig. 7B, lane 1), and StLTA (1 μg/ml) stimulation increased Jak2 phosphorylation (Fig. 7B, lanes 2-4). Jak2 phosphorylation was most intense after a 30-min incubation and became less afterward. However, when a PAF antagonist (CV6209) was present, LTA stimulation did not increase the Jak2 phosphorylation for any of the incubation periods (Fig. 7B, lanes 5-7). Thus, LTA-induced phosphorylation of Jak2 and STAT1, especially at the STAT1 tyrosine residue, depends on PAFR signaling.
FIGURE 7.
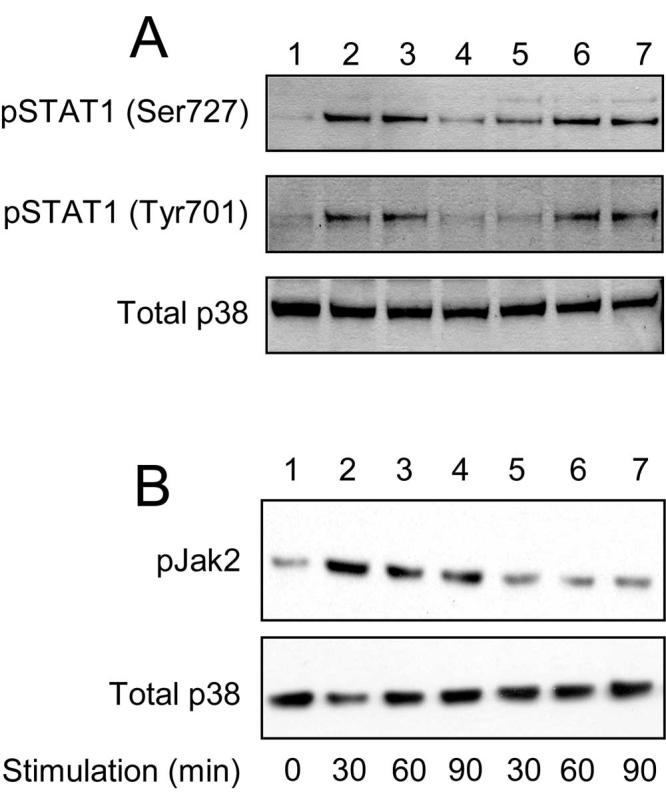
Effect of PAFR inhibitor on LTA-induced phosphorylation of STAT1 and Jak2. A, RAW 264.7 cells were treated with (lane 1) none, (lanes 2, 4, and 6) PnLTA, or (lanes 3, 5 and 7) StLTA. Cells were treated with (lanes 4 and 5) a PAFR inhibitor or with (lanes 6 and 7) DMSO alone (0.01%). The PAFR inhibitor is CV6209 (1 μM) in DMSO (0.01%). Phosphorylated serine at position 727 is shown at the top, phosphorylated tyrosine at position 701 in the middle, and the “Total p38” at the bottom. B, RAW 264.7 cells were treated with (lanes 1) none, (lanes 2-4) StLTA (5 μg/ml), or (lanes 5-7) both StLTA (5 μg/ml) and CV6209 (1 μM) for the indicated time periods. Jak2 phosphorylation is shown at the top, and the “Total p38” is at the bottom.
Discussion
Because TLR2 ligands do not induce the expression of IFN-β that is important in LPS-induced NO production (1), it is possible that biologically active contaminants present among TLR2 ligands rather than the TLR2 ligands themselves are responsible for the observed induction of NO by TLR2 ligands. For example, LTA was widely reported to induce NO production (34, 48, 49), but many studies used a commercially available LTA preparation (34, 35, 48, 49), a preparation that is now known to be contaminated with LPS (50). We show here that highly purified pneumococcal and staphylococcal LTAs as well as Pam3CSK4 can induce NO production. Because chemically synthesized Pam3CSK4 was used, it should be free from biological contaminants. Also, our LTA preparations had undetectable amounts of LPS, did not induce IFN-β production, and induced NO production only when TLR2 was available. Taken together, our data indicate that some TLR2 ligands can induce NO production without inducing IFN-β production.
Our studies with TLR2-deficient mice also show that TLR2 stimulation is essential for LTA-induced NO production. The TLR2 stimulation should provide NF-κB activation, which was shown to be required for iNOS gene expression (48, 49, 51). In addition, we found that several PAFR inhibitors can block NO production, but not TNF-α production, in response to both Pam3CSK4 and LTA. These findings indicate that PAFR stimulation provides additional signaling necessary for iNOS gene expression. To investigate this possibility, we examined involvement of PAFR on the activation of STAT1, which is another transcription factor required for iNOS gene expression (51) and is often limiting (23). LTA stimulation results in rapid phosphorylation of Jak2 and STAT1, and inhibition of PAFR prevents their phosphorylation. Thus, our data suggest that LTA and Pam3CSK4 need both PAFR and TLR2 to elicit NO production and that PAFR stimulation activates STAT1 via Jak2.
It is likely that PAFR directly activates Jak2 without involving G proteins because LTA-induced NO production is blocked by a Jak2 inhibitor but not by several G-protein inhibitors. This possibility is greatly enhanced since recent studies of PAF-stimulated human monocytes have shown that PAFR can directly activate Jak2 without involving G proteins and that the activated Jak2 then leads to STAT activation (10, 46). Nevertheless, our studies are primarily based on chemical inhibitors of enzymes or receptors, which may have limitations in their specificities. Also, STAT1 activation requires phosphorylation at tyrosine (residue 701) and serine (residue 727), and serine kinase needs to be identified. p38 MAPKs are potential candidates because they participate in LPS stimulations (25, 26). Thus, further studies are needed to elucidate the mechanism of PAFR-mediated STAT1 activation.
The PAFR pathway may be analogous to the IFN-β, type I IFNR pathway described in responses to LPS and may be widely used in inflammatory reactions to Gram-positive bacterial infections. For instance, this pathway may be involved in NO production during Gram-positive bacterial infections, which do not elicit IFN-β responses. Indeed, we observed that culture supernatants of various Gram-positive bacteria can induce NO production and that the NO production can be inhibited with various PAFR antagonists (our unpublished data). Also, this PAFR pathway may be involved in the expression of various inflammation-associated molecules (e.g., GARG16, CXCL10, and CXCL12), which were shown with LPS-stimulated cells to depend on endogenous IFN-β and STAT1 activation (52, 53). Since LPS stimulation has been shown to also induce PAF production (54, 55), the PAFR pathway may be operative in Gram-negative bacterial infections as well. However, the PAFR pathway may be more prominent in Gram-positive bacterial infections because it is overshadowed by the type I IFNR pathway in Gram-negative infections.
LTA has been proposed as a PAF analog that directly stimulates PAFR (43). However, for reasons described below, we believe that endogenously produced PAF is responsible for the PAFR stimulations despite our failure to detect increases in endogenous PAF (detection limit, 800 ng/ml) after LTA stimulations (our unpublished data). First, endogenous PAF is easily produced in response to various inflammatory stimuli, such as to TNF-α during an anaphylactic shock (56) or to LPS (54, 55). This may occur because stimulation of TLR2 or TLR4 activates p38 MAPKs, which can activate cytoplasmic phospholipase A2 and acetyltransferase, the two key enzymes necessary for endogenous PAF synthesis during inflammation (47, 57). Second, although Lemjabbar et al. (43) reported StLTA to be a PAF analog, they did not investigate the production of endogenous PAF and their findings can be explained with endogenously produced PAF. Lastly, involvement of endogenous PAF can easily explain the fact that NO production can be induced not only with LTA but also with Pam3CSK4.
Even though PAF is a potent inflammatory molecule, its role in innate immunity has not been emphasized thus far. PAF has been shown to influence dendritic cell maturation (58, 59) and should influence functioning of many types of cells. For instance, the effect of PAF on epithelial cells or endothelial cells should be relevant because PAFR appears to be involved in pneumococcal adhesion (60) or transcytosis (61). Also, PAFR has been shown to be important in nasopharyngeal carriage of pneumococci (60) or transcytosis of pneumococci through brain microvascular endothelial cells (61). A recent study showed that PAFR influences inflammation seen during pneumococcal pneumonia (62). In view of these findings, the role of PAF and PAFR in innate immunity and bacterial infections, particularly in Gram-positive infections, should be further investigated.
Acknowledgments
We thank Dr. William Benjamin, Jr., for a careful reading of the manuscript.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This work was partially supported by a National Institutes of Health Grant (AI-31473). S.H.H. was supported by a fellowship from the International Vaccine Institute (Seoul, Korea).
- PAF
- platelet activating factor
- iNOS
- inducible NO synthase
- LTA
- lipoteichoic acid
- l-NAME
- Nω-nitro-l-arginine methyl ester hydrochloride
- d-NAME
- Nω-nitro-d-arginine methyl ester hydrochloride
- SAPK
- stress-activated protein kinase
- PnLTA
- pneumococcal LTA
- StLTA
- staphylococcal LTA
- EGFR
- epidermal growth factor receptor.
The authors have no financial conflict of interest.
References
- Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-β-induced STAT1α/β-dependent gene expression in macrophages. Nat. Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- Bochud PY, Calandra T. Pathogenesis of sepsis: new concepts and implications for future treatment. Br. Med. J. 2003;326:262. doi: 10.1136/bmj.326.7383.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaillon JM, Adib-Conquy M, Fitting C, Adrie C, Payen D. Cytokine cascade in sepsis. Scand. J. Infect. Dis. 2003;35:535–544. doi: 10.1080/00365540310015935. [DOI] [PubMed] [Google Scholar]
- McManus LM, Pinckard RN. PAF, a putative mediator of oral inflammation. Crit. Rev. Oral Biol. Med. 2000;11:240–258. doi: 10.1177/10454411000110020701. [DOI] [PubMed] [Google Scholar]
- Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, Hariri RJ, Fahey TJ, Zentella A, Albert JD, et al. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–474. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF, Cerami A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662–664. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from the lethal effect of endotoxin. Science. 1985;229:869–871. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- Sorensen J, Kald B, Tagesson C, Lindahl M. Platelet-activating factor and phospholipase A2 in patients with septic shock and trauma. Int. Care Med. 1994;20:555–561. doi: 10.1007/BF01705721. [DOI] [PubMed] [Google Scholar]
- Bessin P, Bonnet J, Apffel D, Soulard C, Desgroux L, Pelas I, Benveniste J. Acute circulatory collapse caused by platelet-activating factor (PAF-acether) in dogs. Eur. J. Pharmacol. 1983;86:403–413. doi: 10.1016/0014-2999(83)90190-5. [DOI] [PubMed] [Google Scholar]
- Lukashova V, Asselin C, Krolewski JJ, Rola-Pleszczynski M, Stankova J. G-protein-independent activation of Tyk2 by the platelet-activating factor receptor. J. Biol. Chem. 2001;276:24113–24121. doi: 10.1074/jbc.M100720200. [DOI] [PubMed] [Google Scholar]
- Ochoa JB, Udekwu AO, Billiar TR, Curran RD, Cerra FB, Simmons RL, Peitzman AB. Nitrogen oxide levels in patients after trauma and during sepsis. Ann. Surg. 1991;214:621–626. doi: 10.1097/00000658-199111000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N. Engl. J. Med. 2001;345:588–595. doi: 10.1056/NEJMra002709. [DOI] [PubMed] [Google Scholar]
- Bakker J, Grover R, McLuckie A, Holzapfel L, Andersson J, Lodato R, Watson D, Grossman S, Donaldson J, Takala J. Administration of the nitric oxide synthase inhibitor NG-methyl-l-arginine hydrochloride (546C88) by intravenous infusion for up to 72 hours can promote the resolution of shock in patients with severe sepsis: results of a randomized, double-blind, placebo-controlled multicenter study (study no. 144-002) Crit. Care Med. 2004;32:1–12. doi: 10.1097/01.CCM.0000105118.66983.19. [DOI] [PubMed] [Google Scholar]
- Bergeron Y, Ouellet N, Simard M, Olivier M, Bergeron MG. Immunomodulation of pneumococcal pulmonary infection with N(G)-monomethyl-l-arginine. Antimicrob. Agents Chemother. 1999;43:2283–2290. doi: 10.1128/aac.43.9.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teale DM, Atkinson AM. Inhibition of nitric oxide synthesis improves survival in a murine peritonitis model of sepsis that is not cured by antibiotics alone. J. Antimicrob. Chemother. 1992;30:839–842. doi: 10.1093/jac/30.6.839. [DOI] [PubMed] [Google Scholar]
- Tsai WC, Strieter RM, Zisman DA, Wilkowski JM, Bucknell KA, Chen GH, Standiford TJ. Nitric oxide is required for effective innate immunity against Klebsiella pneumoniae. Infect. Immun. 1997;65:1870–1875. doi: 10.1128/iai.65.5.1870-1875.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 2001;167:5887–5894. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]
- Barton GM, Medzhitov R. Linking Toll-like receptors to IFN-α/β expression. Nat. Immunol. 2003;4:432–433. doi: 10.1038/ni0503-432. [DOI] [PubMed] [Google Scholar]
- Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu. Rev. Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- Darnell JE., Jr. STATs and gene regulation. Science. 1997;277:1630. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- Decker T, Stockinger S, Karaghiosoff M, Muller M, Kovarik P. IFNs and STATs in innate immunity to microorganisms. J. Clin. Invest. 2002;109:1271–1277. doi: 10.1172/JCI15770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Morrison DC, Parmely TJ, Russell SW, Murphy WJ. An interferon-γ-activated site (GAS) is necessary for full expression of the mouse iNOS gene in response to interferon-γ and lipopolysaccharide. J. Biol. Chem. 1997;272:1226–1230. doi: 10.1074/jbc.272.2.1226. [DOI] [PubMed] [Google Scholar]
- Karaghiosoff M, Neubauer H, Lassnig C, Kovarik P, Schindler H, Pircher H, McCoy B, Bogdan C, Decker T, Brem G, Pfeffer K, Muller M. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity. 2000;13:549–560. doi: 10.1016/s1074-7613(00)00054-6. [DOI] [PubMed] [Google Scholar]
- Kovarik P, Stoiber D, Eyers PA, Menghini R, Neininger A, Gaestel M, Cohen P, Decker T. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-γ uses a different signaling pathway. Proc. Natl. Acad. Sci. USA. 1999;96:13956–13961. doi: 10.1073/pnas.96.24.13956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SH, Jones BW, Toshchakov V, Vogel SN, Fenton MJ. Toll-like receptors 2 and 4 activate STAT1 serine phosphorylation by distinct mechanisms in macrophages. J. Biol. Chem. 2003;278:22506–22512. doi: 10.1074/jbc.M208633200. [DOI] [PubMed] [Google Scholar]
- Uddin S, Sassano A, Deb DK, Verma A, Majchrzak B, Rahman A, Malik AB, Fish EN, Platanias LC. Protein kinase C-δ (PKC-δ) is activated by type I interferons and mediates phosphorylation of Stat1 on serine 727. J. Biol. Chem. 2002;277:14408–14416. doi: 10.1074/jbc.M109671200. [DOI] [PubMed] [Google Scholar]
- Takauji R, Iho S, Takatsuka H, Yamamoto S, Takahashi T, Kitagawa H, Iwasaki H, Iida R, Yokochi T, Matsuki T. CpG-DNA-induced IFN-α production involves p38 MAPK-dependent STAT1 phosphorylation in human plasmacytoid dendritic cell precursors. J. Leukocyte Biol. 2002;72:1011–1019. [PubMed] [Google Scholar]
- Cohen J, Abraham E. Microbiologic findings and correlations with serum tumor necrosis factor-α in patients with severe sepsis and septic shock. J. Infect. Dis. 1999;180:116–121. doi: 10.1086/314839. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Kaufmann A, Grote K, Kawai T, Hoshino K, Morr M, Muhlradt PF, Akira S. Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a Toll-like receptor 2- and MyD88-dependent signaling pathway. J. Immunol. 2000;164:554–557. doi: 10.4049/jimmunol.164.2.554. [DOI] [PubMed] [Google Scholar]
- Jones BW, Means TK, Heldwein KA, Keen MA, Hill PJ, Belisle JT, Fenton MJ. Different Toll-like receptor agonists induce distinct macrophage responses. J. Leukocyte Biol. 2001;69:1036–1044. [PubMed] [Google Scholar]
- Sing A, Merlin T, Knopf HP, Nielsen PJ, Loppnow H, Galanos C, Freudenberg MA. Bacterial induction of β interferon in mice is a function of the lipopolysaccharide component. Infect. Immun. 2000;68:1600–1607. doi: 10.1128/iai.68.3.1600-1607.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kengatharan KM, De Kimpe S, Robson C, Foster SJ, Thiemermann C. Mechanism of Gram-positive shock: identification of peptidoglycan and lipoteichoic acid moieties essential in the induction of nitric oxide synthase, shock, and multiple organ failure. J. Exp. Med. 1998;188:305–315. doi: 10.1084/jem.188.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalpke AH, Frey M, Morath S, Hartung T, Heeg K. Interaction of lipoteichoic acid and CpG-DNA during activation of innate immune cells. Immunobiology. 2002;206:392–407. doi: 10.1078/0171-2985-00189. [DOI] [PubMed] [Google Scholar]
- Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine Toll-like receptor 2. J. Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- Southan GJ, Szabo C, Connor MP, Salzman AL, Thiemermann C. Amidines are potent inhibitors of nitric oxide synthases: preferential inhibition of the inducible isoform. Eur. J. Pharmacol. 1995;291:311–318. doi: 10.1016/0922-4106(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Han SH, Kim JH, Martin M, Michalek SM, Nahm MH. Pneumococcal lipoteichoic acid (LTA) is not as potent as staphylococcal LTA in stimulating Toll-like receptor 2. Infect. Immun. 2003;71:5541–5548. doi: 10.1128/IAI.71.10.5541-5548.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morath S, Geyer A, Hartung T. Structure-function relationship of cytokine induction by lipoteichoic acid from Staphylococcus aureus. J. Exp. Med. 2001;193:393–397. doi: 10.1084/jem.193.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Seo H, Han SH, Lin J, Park MK, Sorensen UB, Nahm MH. Monoacyl lipoteichoic acid from pneumococci stimulates human cells but not mouse cells. Infect. Immun. 2005;73:834–840. doi: 10.1128/IAI.73.2.834-840.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- Cabellos C, MacIntyre DE, Forrest M, Burroughs M, Prasad S, Tuomanen E. Differing roles for platelet-activating factor during inflammation of the lung and subarachnoid space: the special case of Streptococcus pneumoniae. J. Clin. Invest. 1992;90:612–618. doi: 10.1172/JCI115900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemjabbar H, Basbaum C. Platelet-activating factor receptor and ADAM10 mediate responses to Staphylococcus aureus in epithelial cells. Nat. Med. 2002;8:41–46. doi: 10.1038/nm0102-41. [DOI] [PubMed] [Google Scholar]
- Adachi T, Aoki J, Manya H, Asou H, Arai H, Inoue K. PAF analogues capable of inhibiting PAF acetylhydrolase activity suppress migration of isolated rat cerebellar granule cells. Neurosci. Lett. 1997;235:133–136. doi: 10.1016/s0304-3940(97)00742-8. [DOI] [PubMed] [Google Scholar]
- Albert DH, Magoc TJ, Tapang P, Luo G, Morgan DW, Curtin M, Sheppard GS, Xu L, Heyman HR, Davidsen SK, et al. Pharmacology of ABT-491, a highly potent platelet-activating factor receptor antagonist. Eur. J. Pharmacol. 1997;325:69–80. doi: 10.1016/s0014-2999(97)00109-x. [DOI] [PubMed] [Google Scholar]
- Lukashova V, Chen Z, Duhe RJ, Rola-Pleszczynski M, Stankova J. Janus kinase 2 activation by the platelet-activating factor receptor (PAFR): roles of Tyk2 and PAFR C terminus. J. Immunol. 2003;171:3794–3800. doi: 10.4049/jimmunol.171.7.3794. [DOI] [PubMed] [Google Scholar]
- Nixon AB, O’Flaherty JT, Salyer JK, Wykle RL. Acetyl-CoA:1-O-alkyl-2-lyso-sn-glycero-3-phosphocholine acetyltransferase is directly activated by p38 kinase. J. Biol. Chem. 1999;274:5469–5473. doi: 10.1074/jbc.274.9.5469. [DOI] [PubMed] [Google Scholar]
- Kao SJ, Lei HC, Kuo CT, Chang MS, Chen BC, Chang YC, Chiu WT, Lin CH. Lipoteichoic acid induces nuclear factor-κB activation and nitric oxide synthase expression via phosphatidylinositol 3-kinase, Akt, and p38 MAPK in RAW 264.7 macrophages. Immunology. 2005;115:366–374. doi: 10.1111/j.1365-2567.2005.02160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien HF, Yeh KY, Jiang-Shieh YF, Wei IH, Chang CY, Chang ML, Wu CH. Signal transduction pathways of nitric oxide release in primary microglial culture challenged with Gram-positive bacterial constituent, lipoteichoic acid. Neuroscience. 2005;133:423–436. doi: 10.1016/j.neuroscience.2004.09.067. [DOI] [PubMed] [Google Scholar]
- Gao JJ, Xue Q, Zuvanich EG, Haghi KR, Morrison DC. Commercial preparations of lipoteichoic acid contain endotoxin that contributes to activation of mouse macrophages in vitro. Infect. Immun. 2001;69:751–757. doi: 10.1128/IAI.69.2.751-757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinert H, Schwarz PM, Forstermann U. Regulation of the expression of inducible nitric oxide synthase. Biol. Chem. 2003;384:1343–1364. doi: 10.1515/BC.2003.152. [DOI] [PubMed] [Google Scholar]
- Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S. Differential involvement of IFN-β in Toll-like receptor-stimulated dendritic cell activation. Int. Immunol. 2002;14:1225–1231. doi: 10.1093/intimm/dxf089. [DOI] [PubMed] [Google Scholar]
- Weighardt H, Jusek G, Mages J, Lang R, Hoebe K, Beutler B, Holzmann B. Identification of a TLR4- and TRIF-dependent activation program of dendritic cells. Eur. J. Immunol. 2004;34:558–564. doi: 10.1002/eji.200324714. [DOI] [PubMed] [Google Scholar]
- Svetlov SI, Liu H, Chao W, Olson MS. Regulation of platelet-activating factor (PAF) biosynthesis via coenzyme A-independent transacylase in the macrophage cell line IC-21 stimulated with lipopolysaccharide. Biochim. Bio-phys. Acta. 1997;1346:120–130. doi: 10.1016/s0005-2760(97)00022-2. [DOI] [PubMed] [Google Scholar]
- Tonks AJ, Tonks A, Morris RH, Jones KP, Jackson SK. Regulation of platelet-activating factor synthesis in human monocytes by di-palmitoyl phosphatidylcholine. J. Leukocyte Biol. 2003;74:95–101. doi: 10.1189/jlb.1202601. [DOI] [PubMed] [Google Scholar]
- Choi IW, Kim YS, Kim DK, Choi JH, Seo KH, Im SY, Kwon KS, Lee MS, Ha TY, Lee HK. Platelet-activating factor-mediated NF-κB dependency of a late anaphylactic reaction. J. Exp. Med. 2003;198:145–151. doi: 10.1084/jem.20022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker PR, Owen JS, Nixon AB, Thomas LN, Wooten R, Daniel LW, O’Flaherty JT, Wykle RL. Regulation of platelet-activating factor synthesis in human neutrophils by MAP kinases. Biochim. Biophys. Acta. 2002;1592:175–184. doi: 10.1016/s0167-4889(02)00314-2. [DOI] [PubMed] [Google Scholar]
- Al-Darmaki S, Schenkein HA, Tew JG, Barbour SE. Differential expression of platelet-activating factor acetylhydrolase in macrophages and monocyte-derived dendritic cells. J. Immunol. 2003;170:167–173. doi: 10.4049/jimmunol.170.1.167. [DOI] [PubMed] [Google Scholar]
- Dichmann S, Rheinen H, Panther E, Herouy Y, Czech W, Termeer C, Simon JC, Gebicke-Haerter PJ, Norgauer J. Downregulation of platelet-activating factor responsiveness during maturation of human dendritic cells. J. Cell. Physiol. 2000;185:394–400. doi: 10.1002/1097-4652(200012)185:3<394::AID-JCP9>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature. 1995;377:435–438. doi: 10.1038/377435a0. [DOI] [PubMed] [Google Scholar]
- Ring A, Weiser JN, Tuomanen EI. Pneumococcal trafficking across the blood-brain barrier: molecular analysis of a novel bidirectional pathway. J. Clin. Invest. 1998;102:347–360. doi: 10.1172/JCI2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijneveld AW, Weijer S, Florquin S, Speelman P, Shimizu T, Ishii S, van der Poll T. Improved host defense against pneumococcal pneumonia in platelet-activating factor receptor-deficient mice. J. Infect. Dis. 2004;189:711–716. doi: 10.1086/381392. [DOI] [PubMed] [Google Scholar]