

Abstract
Synaptic dysfunction is one of the earliest events in the pathogenesis of Alzheimer’s disease. However, the molecular mechanisms underlying synaptic defects in AD are largely unknown. We report here that Aβ, the main component of senile plaques, induced a significant decrease in dynamin 1, a protein that is essential for synaptic vesicle recycling, and hence, for memory formation and information processing. The Aβ-induced dynamin 1 decrease occurred in the absence of overt synaptic loss and was also observed in the Tg2576 mouse model of Alzheimer’s disease. In addition, our results provided evidence that the Aβ-induced decrease in dynamin 1 was likely the result of a calpain-mediated cleavage of dynamin 1 protein and possibly the down-regulation of dynamin 1 gene expression. These data suggest a mechanism to explain the early cognitive loss without a major decline in synapse number observed in Alzheimer’s disease, and propose a novel therapeutic target for Alzheimer’s disease intervention.
Senile plaques, neurofibrillary tangles, synapse loss, and gross neurodegeneration are common findings in the brain of AD1 patients (1–4). Numerous genetic, biochemical, and animal model studies have implicated the gradual build-up of Aβ, the main component of senile plaques, as the catalyst for AD. However, the mechanistic link between Aβ accumulation and the progressive cognitive impairment associated with this disease has not been elucidated. Synapse loss seems to be the best morphological correlate of the functional deficits observed in the mid to late-stages of AD (3,4). In contrast, patients in the earliest stages of the disease show no significant decline in synapse number (5). Based on these findings, it has been hypothesized that a stage of synaptic dysfunction might precede frank synapse loss, plaque accumulation, and tangle formation in AD (6,7). The mechanisms underlying such synaptic dysfunction remain unknown. It is tempting to speculate that proteins involved in synaptic vesicle biogenesis and/or recycling might play a critical role in AD. Data obtained recently seem to support this view. Thus, changes in the levels of a number of presynaptic proteins, including SNAP-25, syntaxin, and synaptotagmin, have been reported in AD (8). More recently, dynamin 1, a protein highly enriched in presynaptic terminals, has been shown to be significantly reduced in AD brains (9). Dynamin 1, a well-studied neuron-specific mechanochemical GTPase, pinches off synaptic vesicles freeing them from the membrane and allowing them to re-enter the synaptic vesicle pool to be refilled for future release (10,11). The essential role for dynamin 1 in vesicle scission and synaptic function has been best supported by studies of the Drosophila model shibire, a temperature-sensitive mutant of a dynamin ortholog (12,13). At restrictive temperatures these flies displayed a paralysis phenotype. This functional deficit was accompanied by the depletion of synaptic vesicles and the accumulation of invaginated pits at pre-synaptic membranes adjacent to the synaptic clefts. Collectively, these data suggested that stressors which induce dynamin 1 loss-of-function could result in the diminution of available synaptic vesicles leading to synaptic dysfunction.
In the present study we analyzed whether Aβ was one of such stressors using hippocampal neurons that develop in culture and an AD animal model system. Our results showed that Aβ induced a significant reduction in dynamin 1 levels that preceded synapse loss in both model systems. In cultured neurons, our data suggested that the Aβ-induced decrease in dynamin 1 was likely the result of a bi-modal mechanism that involved calpain-mediated proteolysis and down-regulation of dynamin 1 gene expression. On the other hand in the AD mouse model, Tg2576, the decrease in dynamin 1 was mainly the result of calpain activation. These mechanisms identify novel therapeutic targets to address the synaptic dysfunction preceding synapse loss and neurodegeneration in the context of AD.
EXPERIMENTAL PROCEDURES
Preparation of hippocampal cultures
Embryonic day (E) 18 rat embryos were used to prepare primary hippocampal cultures as previously described (14,15). Briefly, hippocampi were dissected and freed of meninges. The cells were dissociated by trypsinization followed by trituration with a fire-polished Pasteur pipette. For biochemical experiments, hippocampal neurons were plated at high density (500,000 cells/60-mm dish) in MEM with 10% horse serum (MEM10). After 2 hours, the medium was changed to glia-conditioned MEM containing N2 supplements plus ovalbumin (0.1%) and sodium pyruvate (0.1 mM) (16). For immunocytochemistry studies the neurons were plated onto poly-L-lysine-coated coverslips in MEM10. After 2 hours, the coverslips were transferred to dishes containing an astroglial monolayer and maintained in MEM containing N2 supplements plus ovalbumin (0.1%) and sodium pyruvate (0.1 mM). These cultures contain approximately 95% pyramidal neurons and 5% glial cells.
Aβ treatment
Synthetic Aβ 1–40 or Aβ1–42 (Sigma, St. Louis, MO) was dissolved in N2 medium at 0.1 mg/ml and incubated for 4 days at 37° C to preaggregate the peptide (17). This preaggregated Aβ was added to the culture medium at final concentrations ranging from 0.02 μM to 20 μM. For some experiments, the aggregated Aβ was centrifuged at 100,000 x g for 1 hour in order to separate the oligomeric (supernatant) from the fibrillar (pellet) forms of the peptide. The oligomeric fraction was obtained by removing the supernatant. The fibrillar fraction was obtained by resuspending the pellet in a volume of N2 medium equal to the supernatant. These fractions were added directly to the neurons at final Aβ concentrations calculated using the initial concentration of the monomeric form of the peptide. Monomeric Aβ preparations were prepared by dissolving the peptide in N2 medium immediately before use. Hippocampal neurons were grown in the presence of the peptide for up to 36 hours.
Protease inhibitors and in vitro cleavage
A series of cell-permeable inhibitors including the general caspase inhibitor Z-VAD-FMK (5 – 50 μM; Promega, Madison, WI) and calpain inhibitors ALLN (5 – 50 μM; Santa Cruz Biotechnology, Santa Cruz, CA), calpeptin (0.1 – 10 μM; Calbiochem, San Diego, CA), and MDL-28170 (0.1 – 10 μM; Calbiochem) were added to the medium of three weeks in culture hippocampal neurons 1 hour prior to, and for the duration of, the Aβ treatment. For experiments using purified caspase-3 (Sigma) and calpain (Calbiochem), whole cell lysates prepared from cultured hippocampal neurons or tissue from the hippocampal region were incubated with caspase-3 or calpain for 1 hour at 37° C. Both proteases were used at final concentrations ranging from 0.002 to 200 units.
Electrophoresis and immunoblotting
Whole cells extracts were prepared from hippocampal neurons that developed either in culture or in vivo. To prepare these fractions, hippocampal neurons kept in culture for 3 weeks were rinsed in PBS, scraped into Laemmli buffer, and homogenized in a boiling water bath for 10 minutes. Hippocampi were also dissected from SwAPP695 transgenic (Tg2576) mice and strain-matched, wild-type (WT) mice. The following numbers of mice were used for these experiments: 2-month-old WT (n=3), 2-month-old Tg2576 (n=3), 5-month-old WT (n=4), 5-month-old Tg2576 (n=6), 8-month-old WT (n=3), and 8-month-old Tg2576 (n=5). Hippocampi were mechanically homogenized in Laemmli buffer and boiled for 10 minutes. Samples were run on 7.5% SDS-polyacrylamide gels and transferred to Immobilon membranes (Millipore, Billerica, MA). Immunodetection was performed using the following antibodies: anti-α-tubulin (1:200,000; clone DM1A; Sigma Aldrich, St. Louis, MO), anti-dynamin 1 was raised against an immunogen corresponding to amino acids 633–647 (1:5,000; Affinity BioReagents, Golden, CO), anti-dynamin 1 (1:1,000; gift from Dr. Mark McNiven, Mayo Clinic, Rochester, MN), anti-dynamin 2 (1:1,000; Affinity BioReagents), anti-dynamin 3 (1:1,000; Affinity BioReagents), anti-synaptophysin (1:100,000; Santa Cruz Biotechnology), anti-class III β-tubulin (1:500; clone TUJ1) (18), and anti-spectrin (1:1,000; Chemicon, Temecula, CA). Secondary antibodies conjugated to horseradish peroxidase (Promega) followed by enhanced chemiluminescence reagents (Amersham Pharmacia Biotech, Piscataway, NJ) were used for the detection of proteins. Immunoreactive bands were detected and imaged using a ChemiDoc XRS (Bio-Rad, Hercules, CA). Densitometry of the images was performed using Quantity One software (Bio-Rad). Densitometric values were normalized using tubulin as internal controls.
Immunocytochemistry
Hippocampal neurons cultured for 3 weeks were fixed for 20 minutes with 4% paraformaldehyde in PBS containing 0.12 M sucrose. They were then permeabilized in 0.3% Triton X-100 in PBS for 5 minutes and rinsed twice in PBS. The cells were preincubated in 10% BSA in PBS for 1 hour at 37° C and exposed to the primary antibodies overnight at 4° C. The neurons were then rinsed in PBS and incubated with secondary antibodies for 1 hour at 37° C. The following primary antibodies were used for neuronal staining: polyclonal anti-dynamin 1 (1:1000; Affinity BioReagents) and monoclonal anti-synaptophysin (1:1000; Santa Cruz Biotechnology). The following secondary antibodies were used: anti-mouse IgG fluorescein-conjugated and anti-rabbit IgG rhodamine-conjugated (1:1000; Chemicon). To quantify immunoreactive spots, untreated and Aβ-treated cultured hippocampal neurons were stained using dynamin 1 and synaptophysin antibodies as described above. Five random fields were selected for quantification of dynamin 1 and synaptophysin immunoreactive spots, which was done at a set intensity using MetaMorph Image analysis software (Universal Imaging Corporation, Fryer Company Inc., Huntley, IL). For the staining of the Aβ peptide, the aggregated, fibrillar, and oligomeric fractions were dried on a slide, fixed with 4% paraformaldehyde, and rinsed twice in PBS. The fractions were preincubated in 10% BSA in PBS for 1 hour at 37° C and exposed to anti-Aβ(1:500; clone 6e10; Sigma Aldrich) overnight at 4° C. The fractions were then rinsed in PBS and incubated with anti-mouse IgG fluorescein-conjugated antibody for 1 hour at 37° C.
RT-PCR
To obtain total mRNA, cultured hippocampal neurons and hippocampi obtained from WT and Tg2576 mice were homogenized in TRIzol® Reagent (Life Technologies, Gaithersburg, MD). RNA was extracted by phenol/chloroform according to the TRIzol® Reagent manufacturer’s protocol. Reverse transcription was performed in 20 μl reactions containing 1 μg sample RNA, 2.5 U MuLV reverse transcriptase, 2.5 μM random hexamers, 1 U RNase inhibitor, 1 mM dATP, 1 mM dCTP, 1 mM dTTP, 1 mM dGTP, 5 mM MgCl2 solution, 2 μl l0X Buffer II (Perkin Elmer GeneAmp RNA PCR Core Kit, N808-0143). Tubes were incubated at 42° C for 15 min, at 99° C for 5 min, and then at 5°C for 5 min. Real-time RT-PCR was performed using 18s ribosomal-subunit primers (Applied Biosystems, Foster City, CA) as an endogenous control, and dynamin 1 specific primers (Applied Biosystems) as the target gene. Real-time RT-PCR reactions were performed in 20 μl reactions using TaqMan Universal PCR Master Mix (Applied Biosystems), 135 ng cDNA, and TaqMan® primers which included probes conjugated with FAM as the reporter dye. All real-time RT-PCR reactions were performed in triplicate and analyzed as relative quantification of dynamin 1 in Aβ-treated cultured hippocampal neurons vs. untreated cultured hippocampal neurons and Tg2576 hippocampi vs. WT hippocampi using the ABI 7900HT Detection System (Applied Biosystems).
RESULTS
Aβ induced a dynamin 1 reduction in cultured hippocampal neurons
To test whether the deposition of Aβ could cause a depletion of dynamin 1, we analyzed dynamin 1 protein levels in cultured hippocampal neurons treated with Aβ. We have chosen this model system because the hippocampus is one of the brain regions most affected in AD. In addition, synaptic integrity in the hippocampus is crucial to memory formation. Furthermore, we and others have previously shown that, when kept in culture for more than 3 weeks, hippocampal neurons reproduce the molecular composition and functional characteristics of mature neurons in vivo (17,19,20). The addition of Aβ1–40(20 μM) to the culture medium of these mature hippocampal neurons induced a significant decrease in dynamin 1 (100-kDa) as determined by Western blot analysis. Dynamin 2, a ubiquitous dynamin isoform, as well as synaptophysin, a pre-synaptic protein, were also decreased in Aβ-treated neurons (Fig. 1A). However, the loss of dynamin 1 and 2 was more extensive than that of synaptophysin. Synaptophysin is a synaptic vesicle protein with four transmembrane domains that anchor it to synaptic vesicles in the presynaptic compartment of nerve terminals (21). Synaptophysin levels correlate closely with synapse number and are commonly used to assay for loss of synapses (5,22). On the other hand, no changes in the levels of dynamin 3, a dynamin isoform also expressed in the nervous system, were detected in Aβ-treated neurons when compared to untreated controls. Interestingly, in addition to the changes in dynamin 1 levels described above, a second smaller dynamin 1 immunoreactive band (~90-kDa) was detected in the Aβ-treated neurons (Fig. 1A). Similar results were obtained when cultured hippocampal neurons were incubated with preaggregated Aβ1–42 (Fig 1B). All experiments described below were performed with Aβ1–40.
Figure 1. Aβ induced a decrease in dynamin levels in a dose- and time-dependent manner in cultured hippocampal neurons.
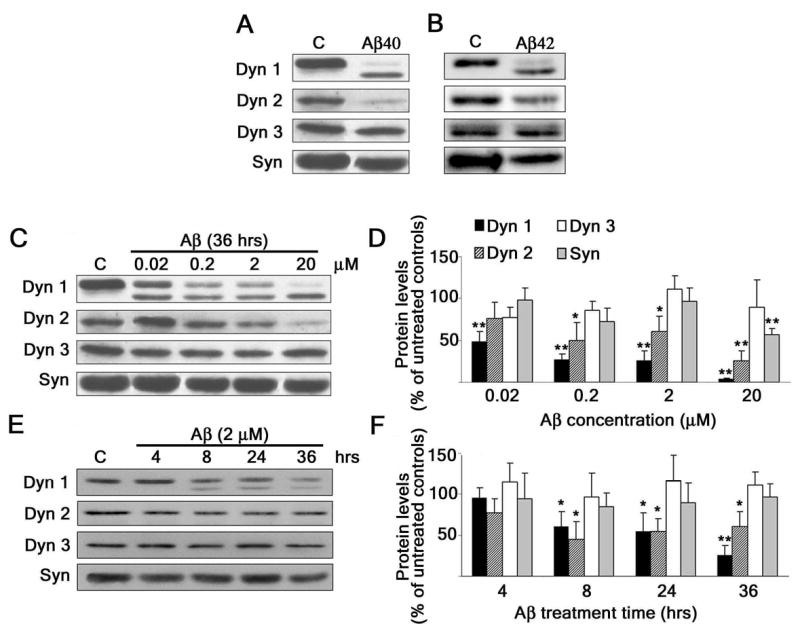
(A and B) Western blot analysis of dynamin 1 (Dyn 1), dynamin 2 (Dyn 2), dynamin 3 (Dyn 3), and synaptophysin (Syn) content in whole cell extracts prepared from 21-DIV hippocampal neurons cultured in the absence (c) or in the presence of Aβ1–40 or Aβ 1–42 (20 μM) for 36 hours. Note the decrease in Dyn 1 (100-kDa) and Dyn 2 immunoreactive bands and the appearance of a second Dyn 1 (~90-kDa) immunoreactive band in Aβ-treated neurons. (C and E) Western blot analysis of dynamin and synaptophysin content in whole cell extracts prepared from 21-DIV hippocampal neurons cultured with increasing concentrations of Aβ for 36 hours (C) or cultured in the presence of Aβ 2 (μM) for up to 36 hrs (E). (D and F) Quantification analysis of the dose-response (C) and time-course (E) effects of Aβ in cultured hippocampal neurons. The results were normalized using tubulin as internal controls. The values obtained in untreated controls were considered 100%. Values represent the mean ± SEM obtained from 6–8 independent experiments. Differs from untreated controls, *P < 0.05, **P < 0.01
To determine whether Aβ induced the decrease of dynamin levels in a dose-dependent manner, mature hippocampal neurons were incubated with Aβ at final concentrations ranging from 0.02 μM to 20 μM for 36 hours. The content of dynamin 1, 2, and 3, as well as synaptophysin, in whole cell lysates was determined by means of Western blot analysis (Fig. 1C and 1D). Our results showed a dose-dependent decrease in the levels of dynamin 1 and 2, but not dynamin 3, in Aβ-treated neurons as compared to untreated controls. However, synaptophysin levels were unchanged when cultured hippocampal neurons were incubated with Aβ at concentrations below 20 μM. These results suggested that synapse numbers were not affected at these lower concentrations. On the other hand, the appearance of the ~90-kDa dynamin 1 immunoreactive band was evident in cell extracts prepared from hippocampal neurons cultured in the presence of Aβ at all concentrations analyzed (Fig. 1C). Next, we determined whether Aβ affects dynamin levels in a time-dependent manner. For these experiments, hippocampal neurons kept in culture for 3 weeks were incubated with Aβ (2 μM) for up to 36 hours (Figs. 1E and 1F). The effect of Aβ on dynamin 1 and 2 levels was evident as early as 8 hours after the addition of the peptide. In contrast, no changes in dynamin 3 and synaptophysin levels were detected throughout the time period analyzed (Figs. 1C and 1E).
Since the aggregation state of Aβ has been proposed to play a critical role in its’ neurotoxic effects, we next determined which form of the Aβ peptide was causing these effects on dynamin 1. The preaggregated Aβ preparation likely contained both large, insoluble fibrillar forms and smaller, soluble oligomeric forms of the peptide. Therefore, we separated these two forms of Aβ by centrifugation. In order to determine if we had successfully separated the fibrillar Aβ from the oligomeric Aβ, we immunostained each fraction with an Aβ antibody. The aggregated, pre-centrifuged preparation, termed “mixed”, showed large globular immunoreactive aggregates along with smaller species (Fig. 2A). Following centrifugation, the oligomeric fraction showed numerous small spherical immunoreactive species, while the fibrillar fraction showed mainly large globular aggregates (Fig. 2A). To test whether these separate forms of Aβ had different effects on dynamin 1, we incubated cultured hippocampal neurons with the monomeric, mixed, oligomeric, and fibrillar forms of Aβ (2 μM each) for 24 hours. Western blot analysis showed a decrease in dynamin 1 levels only in whole cell extracts obtained from hippocampal neurons incubated with the mixed and oligomeric forms of Aβ (Figs. 2B and 2C). We also observed the appearance of the ~90-kDa band in the mixed and oligomeric preparations only (Fig. 2B).
Figure 2. Oligomeric forms of Aβ depleted dynamin 1 in cultured hippocampal neurons.
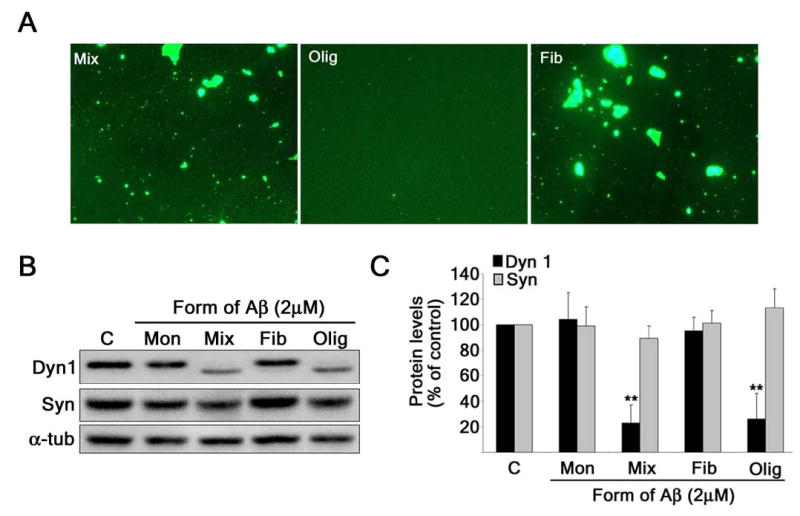
(A) Aβ peptide aggregated for 4 days at 37° C (Mix) was centrifuged (100, 000 x g) to separate the oligomers (Olig) from the fibrils (Fib) and stained with an anti-Aβ antibody. (B) Western blot analysis of dynamin 1 and synaptophysin in whole cell extracts prepared from 21-DIV hippocampal neurons cultured with 2 μM monomeric (Mon), mixed (Mix), fibrillar (Fib), or oligomeric (Olig) Aβ for 24 hours. (C) Quantification analysis of dynamin 1 and synaptophysin protein levels in cultured hippocampal neurons treated with these different forms of Aβ. The results were normalized using α-tubulin as internal controls. The values obtained in untreated controls were considered 100%. Values represent the mean ± SEM obtained from 3 independent experiments. Differs from untreated controls, *P < 0.05, **P < 0.01
Treatment with Aβ also altered the distribution of dynamin 1 in mature hippocampal neurons. In untreated neurons, dynamin 1 was highly enriched in the cell bodies. In addition, robust dynamin 1 punctate immunostaining was detected along the processes extended by these pyramidal neurons (Fig. 3A) Synaptophysin staining appeared in a typical punctuate pattern distributed along the processes (Fig. 3B). Most dynamin 1 immunoreactive spots co-localized with synaptophysin at synaptic sites (Figs. 3C and 3D). However, dynamin 1 immunoreactivity was also detected in extrasynaptic areas. The incubation of hippocampal neurons with Aβ (2 μM) for 24 hours affected neither the morphology nor the distribution of synaptophysin staining (Fig. 3F). In contrast, dynamin 1 immunoreactivity was greatly decreased throughout the neuritic network and was mainly restricted to the cell body and adjacent regions of the processes extended by Aβ-treated hippocampal neurons (Fig. 3E). As a consequence, synaptophysin immunoreactivity did not co-localize with that of dynamin 1 in distal neurites (Figs. 3G and 3H). Quantitative analysis of the dynamin 1 and synaptophysin immunoreactive puncta showed a similar number of spots detected by each antibody in untreated controls (726 ± 127 dynamin 1 and 651 ± 52 synaptophysin). On the other hand, a significant decrease in the number of dynamin 1 (204 ± 146), but not synaptophysin (706 ± 165) puncta, was detected in Aβ-treated neurons. These data suggested that Aβ severely depleted levels of dynamin 1 in the synapses present along the distal portion of the neurites extended by hippocampal neurons.
Figure 3. Fibrillar Aβ decreased dynamin 1 at synaptic sites in cultured hippocampal neurons.
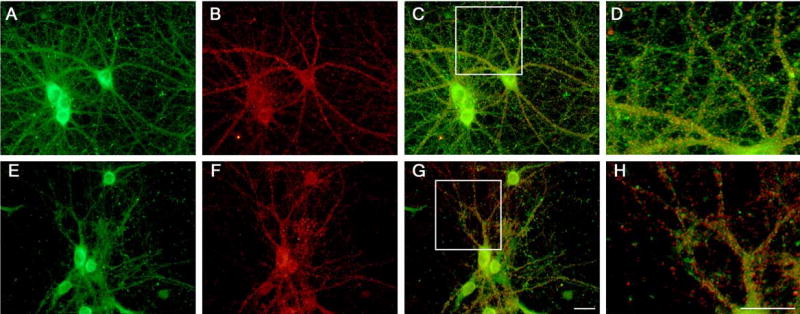
Hippocampal neurons kept in culture for 21 days were incubated in the absence (A – D) or presence (E –H) of Aβ (2 μM) for 24 hours, fixed, and co-stained with dynamin 1 (A, E) and synaptophysin (B, F) antibodies. Note the decrease of dynamin 1 staining in the distal portions of processes extended by Aβ-treated neurons (G, H) as compared to untreated controls (C, D). The scale bar for low magnification pictures (A, B, C, E, F, G) is shown in G, while the scale bar for the high magnification ones (D, H) is shown in H. Scale bars: 20 μm
Aβ induced dynamin 1 reduction by a bi-modal mechanism
We then determined to what extent post-translational degradation and/or down-regulation of the expression of dynamin 1 contributed to the Aβ-induced decrease in its protein levels observed in cultured hippocampal neurons. The results described previously showing the appearance of a second, lower molecular weight immunoreactive band in Aβ-treated neurons strongly suggested that a post-translational proteolytic event was involved in the decrease of dynamin 1 levels. Two proteases proposed to play a role in the pathogenesis of AD are caspase-3 and calpain (23–27). Therefore, we first tested the ability of these proteases to cleave dynamin 1 in vitro (Fig. 4A). Whole cell lysates prepared from untreated cultured hippocampal neurons were incubated with increasing amounts of recombinant caspase-3 or calpain. Caspase-3 failed to cleave dynamin 1 at all concentrations used in our in vitro assays. On the other hand, Western blot analysis showed a dose-dependent decrease of full-length dynamin 1 when incubated with calpain. In addition, recombinant calpain cleavage generated a second dynamin 1 band similar to the one detected when hippocampal neurons were incubated with Aβ (Fig. 4A). In order to further determine whether Aβ could induce the proteolysis of dynamin 1 in hippocampal neurons, we treated them with several cell-permeable inhibitors of caspase-3 or calpain prior to Aβ incubation (Fig. 4B). The broad-spectrum caspase inhibitor Z-VAD-FMK (VAD) did not prevent the dynamin 1 decrease induced by Aβ. Conversely, three different calpain inhibitors, ALLN, calpeptin, and MDL-28170 (MDL) did block the cleavage of dynamin 1 in a dose-dependent manner (Fig. 4B).
Figure 4. Aβ induced the reduction of dynamin 1 both at transcriptional and posttranscriptional levels in cultured hippocampal neurons.
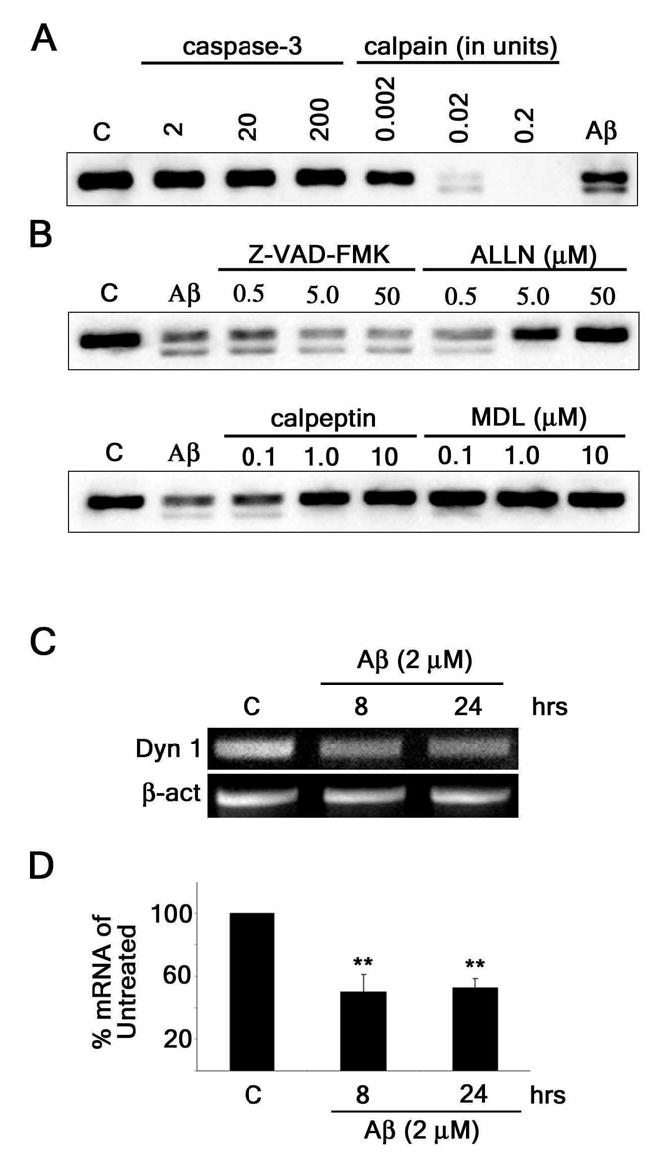
(A) Western blot analysis of dynamin 1 (Dyn 1) content in whole cell extracts obtained from 21-DIV hippocampal neurons incubated in the presence of increasing concentrations of recombinant caspase-3 or calpain. Note that calpain incubation produced a lower molecular weight dynamin 1 immunoreactive band (~90-kDa) similar to the one obtained when cultured hippocampal neurons were treated with Aβ (Aβ). (B) Western blot analysis of dynamin 1 content in whole cell extracts prepared from 21-DIV hippocampal neurons treated with increasing concentrations of various protease inhibitors for 1 hour prior to the addition of Aβ (2μM). Inhibitors included a general caspase inhibitor (VAD), a calpain/proteosome inhibitor (ALLN), and calpain inhibitors (calpeptin and MDL). Only inhibitors blocking calpain activity prevented cleavage of dynamin 1. (C) Conventional RT-PCR analysis of dynamin 1 mRNA bands in untreated and Aβ-treated (2 μM) cultured hippocampal neurons. β-actin was used as an internal control. (D) Real-time RT-PCR of dynamin 1 mRNA in untreated and Aβ-treated (2μM) cultured hippocampal neurons. Dynamin 1 signal was normalized using the 18s ribosomal gene as an endogenous control. Values obtained in untreated controls were set at 100%. Values represent the mean ± SEM obtained from 6 independent experiments. Differs from untreated controls, *P < 0.05, **P < 0.01
Next, we investigated the effect of Aβ treatment on dynamin 1 expression in cultured hippocampal neurons. Neurons were incubated with Aβ (2 μM) for 8 and 24 hours and their RNA was harvested for RT-PCR. Conventional RT-PCR bands showed a qualitative decrease in dynamin 1 mRNA from cultured hippocampal neurons that were incubated with Aβ for 8 and 24 hrs (Fig. 4C). Real-time RT-PCR showed a significant decrease of dynamin 1 mRNA from hippocampal neurons incubated in the presence of Aβ for 8 and 24 hours as compared to untreated controls (~50% and ~48% decrease, respectively) (Fig. 4D). Taken together, these data suggested that Aβ induced the decrease of dynamin 1 in cultured hippocampal neurons by a bi-modal process involving both calpain-mediated proteolysis and a decrease in dynamin 1 expression.
Decreased dynamin 1 levels were also detected in the hippocampus of Tg2576 mice
Finally, we studied whether endogenous Aβ had similar affects on dynamin 1 in the AD animal model Tg2576. Tg2576 transgenic mice harbor a double mutation in APP that enhances production of the Aβ peptide (28). These mice recapitulate many characteristics of AD pathology including Aβ plaque formation beginning at 10–12 months of age (28). On the other hand, cognitive deficits were detected as early as 4 months after birth in Tg2576 mice (29). Surprisingly, changes in the number of synapses were not detected even in 1-year-old Tg2576 mice (30,31). The cognitive impairments observed in these mice in the absence of pathology suggested a discrete mechanism affecting neuronal function. Dynamin 1 might play a key role in such a mechanism. To examine this possibility, we compared dynamin 1 and synaptophysin protein levels in whole cell extracts prepared from the hippocampi of 2-, 5-, and 8-month-old Tg2576 mice with those of strain-, and age-matched WT mice (Fig. 5). Dynamin 1 and synaptophysin levels were normalized using neuron-specific β-tubulin as an internal control. No differences in dynamin 1 protein levels were detected in the Tg2576 mice as compared to WT mice 2 months after birth (Figs. 5A and 5C). On the other hand, beginning at 5-months of age, Tg2576 mice showed a significant decline (~22%) in dynamin 1 levels. This declining trend in dynamin 1 levels continued at 8-months-of-age when Tg2576 mice show a greater yet (~36%) decrease in dynamin 1 levels when compared to WT mice. Importantly, no changes in synaptophysin protein levels were detected in the Tg2576 mice when compared to WT mice throughout the whole period analyzed (Figs. 5B and 5D). These data indicated that a decrease in dynamin 1 protein occurred independently from synapse loss both in neurons that developed in culture and in situ.
Figure 5. Dynamin 1 was decreased in the hippocampus of Tg2576 mice.
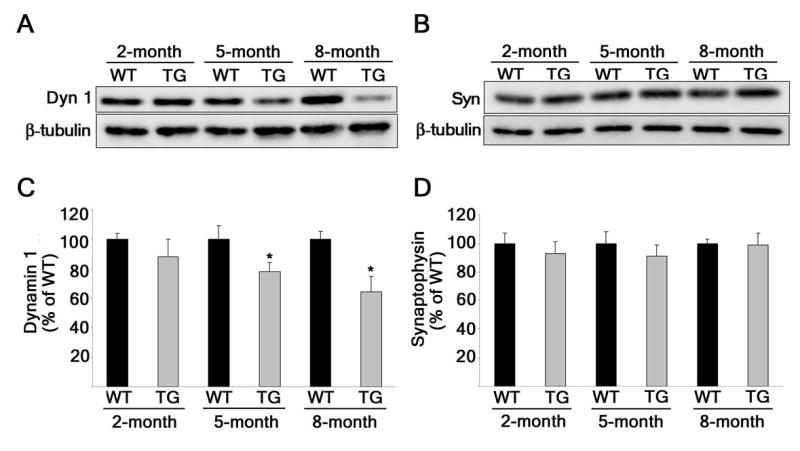
(A) Western blot analysis of dynamin 1 (Dyn 1) and class III β-tubulin (β-tubulin) in whole cell extracts prepared from the hippocampi of 2-, 5-, and 8-month-old Tg2576 (TG) mice and strain-, and age-matched WT mice. (B) Western blot analysis of synaptophysin (Syn) and class III β-tubulin (β-tubulin) in whole cell extracts prepared from the hippocampi of 2-, 5-, and 8-month-old Tg2576 mice and strain-, and age-matched WT. (C and D) Quantification of dynamin 1 from (A) and synaptophysin from (B), respectively. The results were normalized using class III β-tubulin as internal control. Values represent the mean ± SEM. Differs from WT which is set at 100%, *P < 0.05.
Decrease of dynamin 1 in Tg2576 mice was likely due to calpain activation
Aβ-induced calpain activation resulted in the cleavage of dynamin 1 and the appearance of a fragment of ~90 kDa in cultured hippocampal neurons. If calpain was abnormally activated in the hippocampus of Tg2576 mice, we would expect to see a dynamin 1 fragment band at approximately the same molecular weight (~90-kDa). No dynamin 1 cleaved bands were detected in the WT or 2-month-old Tg2576 mice (Fig. 6A). By contrast, cleaved products were detected directly below full length dynamin 1 in 5-, and 8-month-old Tg2576 mice. To test whether calpain was responsible for generating this faster migrating dynamin 1 band, we incubated hippocampal lysates obtained from 2-month-old Tg2576 mice with calpain for 1 hour. Western blot analysis showed the presence of a faster migrating dynamin 1 band in 2-month-old lysates incubated with calpain and untreated lysates from 8-month-old Tg2576 mice. On the other hand, no such cleaved fragment was detected in untreated lysates obtained from 2-month-old mice (Fig. 6B). To further assess calpain activation in the hippocampus of these mice, we analyzed the degradation of spectrin, a common calpain substrate (32). Activation of calpain results in cleavage of full length spectrin (240-kDa) to breakdown products of approximately 145- and 150-kDa apparent molecular weights (33). An increase in the ratio of 150/240-kDa spectrin bands was detected in Tg2576 mice when compared to strain-, and age-matched WT mice at 5 and 8 months after birth (Fig. 6C). Interestingly, calpain activation in these mice seems to correlate with the decrease in dynamin 1 and the appearance of the dynamin 1 fragment.
Figure 6. Decrease of dynamin 1 in Tg2576 mice was consistent with calpain activation.
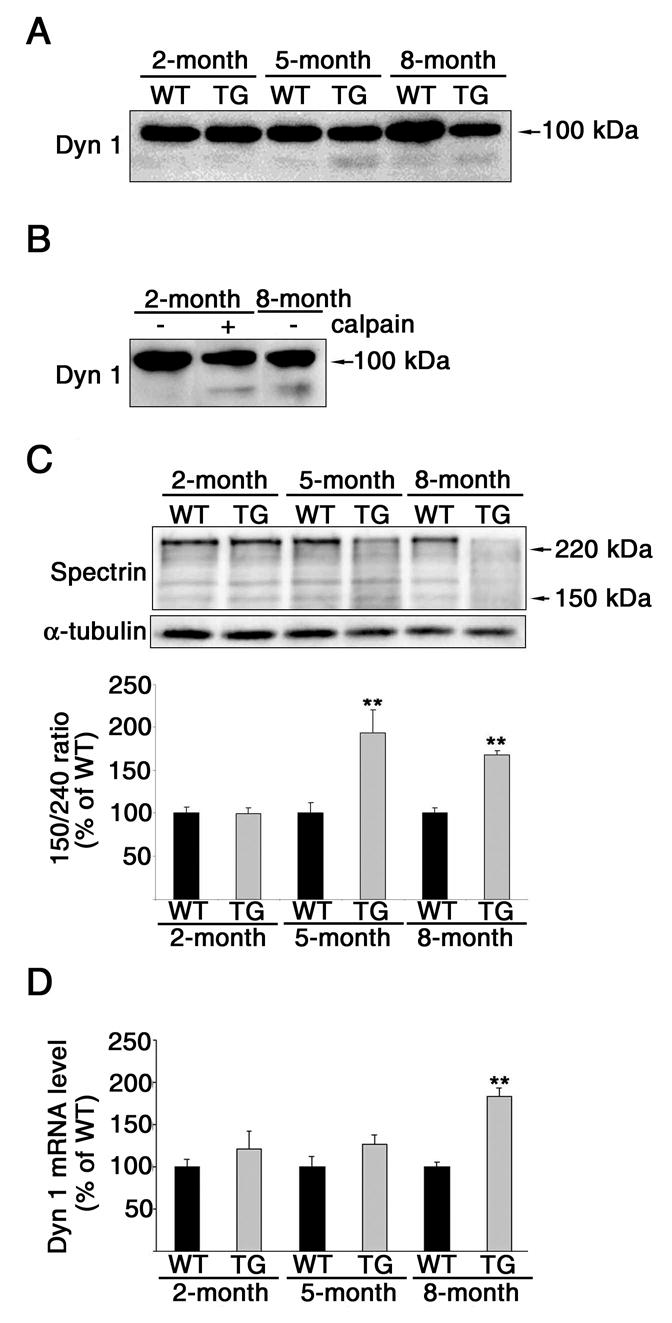
(A) Western blot analysis of dynamin 1 (Dyn 1) in whole cell extracts prepared from the hippocampi of 2-, 5-, and 8-month-old Tg2576 (TG) mice and strain-, and age-matched WT mice. Note the appearance of a lower molecular weight dynamin 1 band (~90kDa) in the 5- and 8-month-old Tg2576 mice. (B) Western blot analysis of dynamin 1 (Dyn1) in whole cell extracts prepared from the hippocampi of 2- and 8-month-old Tg2576 mice. The 2-month-old samples were either untreated (−) or incubated with 0.2 units of calpain (+) and run next to an untreated sample from an 8-month-old Tg2576 mouse. Note that calpain treatment in the 2-month-old sample produced a similar, faster migrating band as in the 8-month-old untreated sample. (C) Western blot analysis of spectrin in whole cell extracts prepared from the hippocampi of 2-, 5-, and 8-month-old Tg2576 mice and strain-, and age-matched WT mice. The antibody used recognized both full-length spectrin (240-kDa) and calpain-mediated cleavage products (150- and 145-kDa). Spectrin degradation was assessed by determining the 150/240-kDa ratio. α-tubulin was used as a loading control. (D) Real-time RT-PCR of dynamin 1 mRNA obtained from the hippocampi of 2-, 5-, and 8-month-old Tg2576 mice and strain-, and age-matched WT mice. Dynamin 1 signal was normalized using the 18s ribosomal gene as an endogenous control. Values represent the mean ± SEM. Differs from WT which is set at 100%, *P < 0.05, **P < 0.01.
We next studied dynamin 1 mRNA levels in the hippocampi of these transgenic mice. Relative quantification real-time PCR showed that at 2- and 5-months, Tg2576 mice have no significant change in dynamin 1 mRNA levels compared to strain-, and age-matched WT (Fig. 6D). A significant increase (~84%) in dynamin 1 mRNA expression was detected in 8-month-old Tg2576 mice when compared to WT mice.
DISCUSSION
The results presented herein indicated that Aβ induced a decrease of dynamin 1 in hippocampal neurons. In addition, our findings suggested that this Aβ-induced dynamin 1 decrease was the result of a bi-modal mechanism that involved calpain-mediated proteolysis and down-regulation of dynamin 1 gene expression in cultured hippocampal neurons. In Tg2576 mice, on the other hand, the decrease in dynamin 1 protein was likely due to calpain-mediated proteolysis. Given the importance of dynamin 1 to synaptic function, these results provide insights into the molecular mechanisms underlying the cognitive decline observed in the early stages of AD. Furthermore, they help to bridge an extensive gap between our knowledge of the mechanisms leading to Aβ accumulation in different brain areas and synaptic deficits in AD patients.
Synaptic dysfunction has been a tempting explanation for the mild cognitive deficits observed at early stages of the disease when no significant decline in the synapse number could be detected (5). However, the mechanisms underlying these functional deficits are not known. These results indicated that changes in dynamin 1 could mediate, at least in part, these deficits. Thus, low concentrations of Aβ (~10-fold lower than insoluble Aβ levels in AD brains (34)) significantly reduced dynamin 1 protein levels in a time- and dose-dependent manner in cultured hippocampal neurons. Importantly, when used at these concentrations, Aβ induced the reduction in dynamin 1 prior to synapse loss and/or neurite degeneration in cultured hippocampal neurons. Similar results were obtained when the levels of dynamin 1 were analyzed in the hippocampal region of Tg2576 mice, the most commonly used AD animal model. These mice, which have enhanced production of Aβ1–40 (5-fold) and Aβ1–42 (14-fold) in their hippocampus, showed a significant reduction in dynamin 1 protein levels in the absence of synapse loss as determined using synaptophysin as a synaptic marker.
Our results suggested that decreased dynamin 1 levels might lead to changes in synaptic vesicle recycling and the accumulation of fused vesicles at the membrane, thus affecting synaptic function in the complex neuronal network of the hippocampus. This network depends on flawless synaptic vesicle turnover in millions of synapses for memory acquisition following LTP (35,36). Our data support previous studies showing that Aβ-induced cognitive decline is a reversible or transient phenomenon in animal models (37,38). These studies have suggested that cognition decline due to Aβ is not due to frank synapse loss or neurodegeneration, rather a stage of synaptic dysfunction that does not cause permanent disruptions in neuronal networks important to learning and memory. It is tempting to speculate that this stage of synaptic dysfunction is mediated by a decrease in dynamin 1 leading to the depletion of releasable synaptic vesicles due to the inability to endocytose from the presynaptic membrane. Interestingly, it has been documented that the average synaptic area in the dentate gyrus and frontal cortex was significantly larger in AD brains than in age-matched controls (39,40). This particular ultrastructural abnormality could be a result of the accumulation of fused synaptic vesicles at the synapse. Similar ultrastructural features have been described in temperature-sensitive mutants of a dynamin ortholog in Drosophila (12,13).
This study also provided insights into the mechanisms leading to Aβ-induced decrease in dynamin 1 levels. Our in vitro and in vivo experiments suggested that calpain mediated dynamin 1 proteolysis in Aβ-treated hippocampal neurons. The activation of this protease in the presence of Aβ is in agreement with previous reports showing that AD patients had increased levels of active calpain in their central nervous system (23,24,27). Calpain has also been linked to the dysregulation of Cdk5 leading to aberrant phosphorylation of tau, another salient feature of AD (41). Furthermore, initial studies using calpain inhibitors in a mouse model of AD showed an encouraging recovery of cognitive function in transgenic mice that were treated with a calpain inhibitor at an early age (42). The mechanisms by which Aβ induces the activation of this protease have not been completely elucidated. Calpain is a cytosolic cysteine protease physiologically activated by intracellular increases in Ca2+ (43). Thus, one potential mechanism of activation could involve the disruption of Ca2+ homeostasis. The molecular mechanism by which Aβ causes calcium dyshomeostasis is yet to be fully identified. To this point, cases have been made for increased calcium influx through glutamate receptors, receptor-independent membrane permeabilization, and intracellular calcium leakage (44–46).
Our results obtained using cultured hippocampal neurons support the idea that Aβ also reduces dynamin 1 expression. This Aβ-induced decrease in dynamin 1 might involve the regulation of its transcription factor. Expression of the dynamin 1 gene is partly controlled by, and dependent on, the transcription factor Sp1 (47). Sp1 is activated by cAMP-dependent PKA-mediated phosphorylation (48). Therefore, disruptions in cAMP/PKA/Sp1 pathway could have significant effects on the level of dynamin 1 gene expression and synaptic function. Support for this hypothesis has been provided by a recent study showing that Aβ blocked PKA activity and LTP in cultured hippocampal neurons (49).
Surprisingly, dynamin 1 mRNA levels were increased in the hippocampus of 8-month-old Tg2576 mice. The results obtained using this AD animal model system are in agreement with a previous report showing an increase in dynamin 1 expression from brains of these mice (9). This apparent discrepancy between the effects of Aβ on the expression of dynamin 1 mRNA in neurons that develop either in culture or in situ could be due to several factors. First, compensatory mechanisms leading to an increase in dynamin 1 mRNA could be triggered by the chronic cleavage of this protein in Tg2576 mice. These mechanisms might not get into play under acute conditions leading to dynamin 1 cleavage like the ones observed in Aβ-treated cultured neurons. Alternatively, the increase in dynamin 1 mRNA observed in the hippocampal region of the Tg2576 mice could be the result of different responses of specific neuronal populations to Aβ. It is possible that granular cells from the dentate gyrus, absent from our cultures prepared from embryonic tissue, increase the transcription of dynamin 1 in response to the over expression of the double-mutated APP present in these transgenic animals. Regardless, it is worth noting that our results obtained using cultured hippocampal neurons closely resembled the decrease in dynamin 1 expression observed in the brains of AD patients (9).
Taken collectively, our results suggest that Aβ induces a decrease of dynamin 1 in hippocampal neurons. This effect on dynamin 1 is an early event that precedes synapse loss both in Aβ-treated cultured hippocampal neurons and in hippocampal neurons obtained from Tg2576 mice. These findings support a previous study that showed the decrease of dynamin 1 in human AD brains (9). In addition, these results present a molecular explanation for the cognitive decline in absence of synapse loss seen in the early stages of AD. The idea that synapses become dysfunctional before they are lost presents an attractive window for therapeutic intervention before irreversible neuronal damage is done. Thus, drugs that inhibit calpain and/or stimulate PKA, two attractive targets to prevent synaptic dysfunction, could become useful therapeutic tools for treating AD.
Acknowledgments
We thank Caryn Tournell, Kelsi Anderson, and Holly Oakley for excellent technical support. We thank Dr. Mark McNiven (Mayo Clinic, Rochester, MN) for the generous gift of a dynamin 1 antibody. We also thank Tracy O’Connor for help with real-time PCR. This study was supported by grants NIA/R01AG022560 to R.V., NIH/NS39080 and Alzheimer’s Association IIRG to A.F. B.L.K was supported by an NIA/AG20506 training grant and a fellowship from the American Foundation for Aging Research.
Footnotes
The abbreviations used are: AD, Alzheimer’s Disease; Aβ, beta amyloid; MEM, minimum essential medium; Z-VAD-FMK, carbobenzoxy-alyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone; ALLN, N-Acetyl-Leu-Leu-Nle-CHO; MDL-28170, Z-Val-Phe-CHO; SDS, Sodium dodecyl sulfate; RT-PCR, reverse transcription polymerase chain reaction APP, amyloid precursor protein; LTP, log term potentiation; PKA, protein kinase A; cAMP, Cyclic adenosine monophosphate; SEM, standard error of the mean; DIV, days in vitro
References
- 1.Glenner GG, Wong CW. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 2.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. J Biol Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- 3.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 4.DeKosky ST, Scheff SW. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 5.Tiraboschi P, Hansen LA, Alford M, Masliah E, Thal LJ, Corey-Bloom J. Neurology. 2000;55:1278–1283. doi: 10.1212/wnl.55.9.1278. [DOI] [PubMed] [Google Scholar]
- 6.Selkoe DJ. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 7.Yao PJ. Trends Neurosci. 2004;27:24–29. doi: 10.1016/j.tins.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Honer WG. Neurobiol Aging. 2003;24:1047–1062. doi: 10.1016/j.neurobiolaging.2003.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Yao PJ, Zhu M, Pyun EI, Brooks AI, Therianos S, Meyers VE, Coleman PD. Neurobiol Dis. 2003;12:97–109. doi: 10.1016/s0969-9961(02)00009-8. [DOI] [PubMed] [Google Scholar]
- 10.Clark SG, Shurland DL, Meyerowitz EM, Bargmann CI, van der Bliek AM. Proc Natl Acad Sci U S A. 1997;94:10438–10443. doi: 10.1073/pnas.94.19.10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damke H, Baba T, Warnock DE, Schmid SL. J Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koenig JH, Ikeda K. J Neurosci. 1989;9:3844–3860. doi: 10.1523/JNEUROSCI.09-11-03844.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Bliek AM, Meyerowitz EM. Nature. 1991;351:411–414. doi: 10.1038/351411a0. [DOI] [PubMed] [Google Scholar]
- 14.Ferreira A, Li L, Chin LS, Greengard P, Kosik KS. Mol Cell Neurosci. 1996;8:286–299. doi: 10.1006/mcne.1996.0064. [DOI] [PubMed] [Google Scholar]
- 15.Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- 16.Bottenstein JE, Sato GH. Proc Natl Acad Sci U S A. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferreira A, Lu Q, Orecchio L, Kosik KS. Mol Cell Neurosci. 1997;9:220–234. doi: 10.1006/mcne.1997.0615. [DOI] [PubMed] [Google Scholar]
- 18.Ferreira A, Caceres A. J Neurosci Res. 1992;32:516–529. doi: 10.1002/jnr.490320407. [DOI] [PubMed] [Google Scholar]
- 19.Bartlett WP, Banker GA. J Neurosci. 1984;4:1954–1965. doi: 10.1523/JNEUROSCI.04-08-01954.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartlett WP, Banker GA. J Neurosci. 1984;4:1944–1953. doi: 10.1523/JNEUROSCI.04-08-01944.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiedenmann B, Franke WW. Cell. 1985;41:1017–1028. doi: 10.1016/s0092-8674(85)80082-9. [DOI] [PubMed] [Google Scholar]
- 22.Sabbagh MN, Corey-Bloom J, Tiraboschi P, Thomas R, Masliah E, Thal LJ. Arch Neurol. 1999;56:1458–1461. doi: 10.1001/archneur.56.12.1458. [DOI] [PubMed] [Google Scholar]
- 23.Saito K, Elce JS, Hamos JE, Nixon RA. Proc Natl Acad Sci U S A. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsuji T, Shimohama S, Kimura J, Shimizu K. Neurosci Lett. 1998;248:109–112. doi: 10.1016/s0304-3940(98)00348-6. [DOI] [PubMed] [Google Scholar]
- 25.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 26.Gastard MC, Troncoso JC, Koliatsos VE. Ann Neurol. 2003;54:393–398. doi: 10.1002/ana.10680. [DOI] [PubMed] [Google Scholar]
- 27.Veeranna, Kaji T, Boland B, Odrljin T, Mohan P, Basavarajappa BS, Peterhoff C, Cataldo A, Rudnicki A, Amin N, Li BS, Pant HC, Hungund BL, Arancio O, Nixon RA. Am J Pathol. 2004;165:795–805. doi: 10.1016/S0002-9440(10)63342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 29.Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- 30.King DL, Arendash GW. Brain Res. 2002;926:58–68. doi: 10.1016/s0006-8993(01)03294-2. [DOI] [PubMed] [Google Scholar]
- 31.Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW. J Neurosci. 2002;22:3376–3385. doi: 10.1523/JNEUROSCI.22-09-03376.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siman R, Baudry M, Lynch G. Proc Natl Acad Sci U S A. 1984;81:3572–3576. doi: 10.1073/pnas.81.11.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bahr BA, Tiriveedhi S, Park GY, Lynch G. J Pharmacol Exp Ther. 1995;273:902–908. [PubMed] [Google Scholar]
- 34.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malenka RC. Cell. 1994;78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- 36.Ryan TA, Ziv NE, Smith SJ. Neuron. 1996;17:125–134. doi: 10.1016/s0896-6273(00)80286-x. [DOI] [PubMed] [Google Scholar]
- 37.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 38.Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 39.Bertoni-Freddari C, Fattoretti P, Pieroni M, Meier-Ruge W, Ulrich J. Pathol Res Pract. 1992;188:612–615. doi: 10.1016/S0344-0338(11)80066-X. [DOI] [PubMed] [Google Scholar]
- 40.Scheff SW, DeKosky ST, Price DA. Neurobiol Aging. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- 41.Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 42.Battaglia F, Trinchese F, Liu S, Walter S, Nixon RA, Arancio O. J Mol Neurosci. 2003;20:357–362. doi: 10.1385/JMN:20:3:357. [DOI] [PubMed] [Google Scholar]
- 43.Friedrich P. Biochem Biophys Res Commun. 2004;323:1131–1133. doi: 10.1016/j.bbrc.2004.08.194. [DOI] [PubMed] [Google Scholar]
- 44.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Demuro, A., Mina, E., Kayed, R., Milton, S. C., Parker, I., and Glabe, C. G. (2005) J Biol Chem [DOI] [PubMed]
- 46.Suen KC, Lin KF, Elyaman W, So KF, Chang RC, Hugon J. J Neurochem. 2003;87:1413–1426. doi: 10.1111/j.1471-4159.2003.02259.x. [DOI] [PubMed] [Google Scholar]
- 47.Yoo J, Jeong MJ, Kwon BM, Hur MW, Park YM, Han MY. J Biol Chem. 2002;277:11904–11909. doi: 10.1074/jbc.M111788200. [DOI] [PubMed] [Google Scholar]
- 48.Rohlff C, Ahmad S, Borellini F, Lei J, Glazer RI. J Biol Chem. 1997;272:21137–21141. doi: 10.1074/jbc.272.34.21137. [DOI] [PubMed] [Google Scholar]
- 49.Vitolo OV, Sant’Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M. Proc Natl Acad Sci U S A. 2002;99:13217–13221. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]