

Abstract
The mechanisms employed by nonenveloped animal viruses to penetrate the membranes of their host cells remain enigmatic. Membrane penetration by the nonenveloped mammalian reoviruses is believed to deliver a partially uncoated, but still large (∼70-nm), particle with active transcriptases for viral mRNA synthesis directly into the cytoplasm. This process is likely initiated by a particle form that resembles infectious subvirion particles (ISVPs), disassembly intermediates produced from virions by proteolytic uncoating. Consistent with that idea, ISVPs, but not virions, can induce disruption of membranes in vitro. Both activities ascribed to ISVP-like particles, membrane disruption in vitro and membrane penetration within cells, are linked to N-myristoylated outer-capsid protein μ1, present in 600 copies at the surfaces of ISVPs. To understand how μ1 fulfills its role as the reovirus penetration protein, we monitored changes in ISVPs during the permeabilization of red blood cells induced by these particles. Hemolysis was preceded by a major structural transition in ISVPs, characterized by conformational change in μ1 and elution of fibrous attachment protein σ1. The altered conformer of μ1 was required for hemolysis and was markedly hydrophobic. The structural transition in ISVPs was further accompanied by derepression of genome-dependent mRNA synthesis by the particle-associated transcriptases. We propose a model for reovirus entry in which (i) primed and triggered conformational changes, analogous to those in enveloped-virus fusion proteins, generate a hydrophobic μ1 conformer capable of inserting into and disrupting cell membranes and (ii) activation of the viral particles for membrane interaction and mRNA synthesis are concurrent events. Reoviruses provide an opportune system for defining the molecular details of membrane penetration by a large nonenveloped animal virus.
Before viruses can launch their replicative programs, they must first gain access to the interior of host cells where raw materials and machinery essential for viral multiplication are present. Enveloped animal viruses employ membrane fusion to cross the membrane barrier and enter the cytoplasm, and the viral surface proteins that mediate this process are termed fusion proteins. These well-studied proteins, exemplified by the influenza virus hemagglutinin (HA) and the tick-borne encephalitis virus (TBEV) E protein (reviewed in references 33 and 63), share several features. (i) They are oligomeric integral membrane proteins. (ii) They can adopt structurally distinct water-soluble and membrane-seeking states. (iii) They are primed, often by proteolytic cleavage, to undergo a conformational transition between these states. (iv) They are induced to undergo this transition by a triggering stimulus such as low pH or receptor binding. The primed and triggered conformational changes in the fusion proteins result in close apposition of the viral and cellular bilayers, membrane merger, and cytoplasmic delivery of the viral nucleocapsid(s).
Nonenveloped animal viruses, in contrast to their enveloped counterparts, cannot utilize membrane fusion to enter cells. The mechanisms employed by these viruses to cross the cellular membrane barrier are less well understood but may include the formation of small protein-lined pores through the bilayer analogous to those made by protein toxins such as colicin A (55) or larger disruptions in bilayer integrity analogous to those caused by membranolytic peptides such as magainin (59). Nonenveloped viruses generally contain a capsid protein or proteins that mediate membrane penetration. It has been proposed that these “penetration proteins” also undergo primed and triggered conformational transitions that allow them to interact with the cellular membrane during entry. For example, poliovirus virions incubated with receptor externalize the N-myristoylated VP4 peptide and expose amphipathic sequences of VP1 that can insert into liposomes (5, 26). Adenovirus (Ad) virions incubated at low pH are thought to undergo a conformational change in the penton base protein that renders the particles hydrophobic and capable of permeabilizing liposomes (7, 58). As a step toward understanding the membrane penetration mechanism of the nonenveloped mammalian orthoreoviruses (reoviruses), we undertook studies to identify and characterize entry-related changes in their capsid proteins.
Reovirus virions are 85-nm particles comprising the double-stranded RNA genome enclosed by two concentric icosahedral protein capsids (reviewed in reference 52) (Fig. 1A). The outer capsid mediates delivery of viral particles into the cytoplasm of host cells, where viral replication occurs. It has been proposed that protein μ1 (76 kDa, 200 trimers per particle), which appears to be found in virions mostly as autolytic fragments μ1N (4 kDa) and μ1C (72 kDa) (53), participates directly in membrane penetration during entry into cells (16, 35, 42, 43, 51, 53). σ3 (41 kDa, 600 monomers per particle), the major surface protein of virions, interacts closely with the trimers of μ1 (42, 61), thereby protecting them from the extracellular environment and regulating their activities. Protein σ1 (50 kDa, 12 trimers per particle) forms fibers that extend from the fivefold axes of virions and that mediate viral attachment to cellular receptors (4, 27, 41). The fourth outer-capsid protein, λ2 (144 kDa, 12 pentamers per particle), is involved in viral mRNA capping and outer-capsid assembly but is not known to participate directly in entry (22, 45).
FIG. 1.
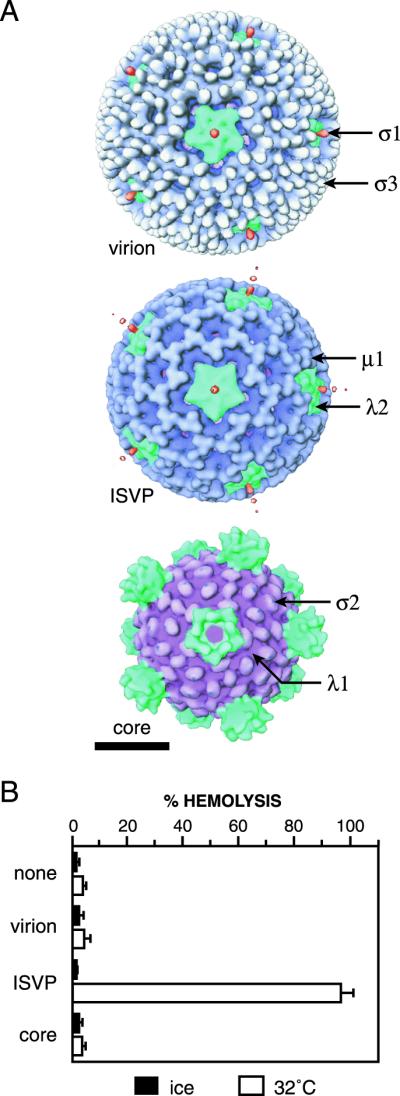
Reovirus particles and their capacities to mediate hemolysis. (A) Surface views of the reovirus virion, ISVP, and core obtained from transmission cryoelectron microscopy and three-dimensional image reconstruction as previously reported (22). Color coding of the capsid proteins was applied for clarity (50). Capsid proteins are labeled in a representative fashion. Bar, 20 nm. (B) Purified T3D virions, ISVPs, or cores (1013 particles/ml) or an equal volume of virion buffer lacking particles was incubated with bovine calf RBCs in virion buffer for 15 min on ice or at 32°C. The amount of hemolysis induced by each particle type is shown as the average ± standard deviation from three trials.
When virions are incubated with proteases in vitro, the outer capsid is sequentially disassembled to yield two subvirion particles: infectious subvirion particles (ISVPs) and cores (reviewed in reference 52) (Fig. 1A). ISVPs lack σ3 and contain protein μ1C as particle-bound fragments δ (59 kDa) and φ (13 kDa) (37, 51). Cores lack not only σ3 but also μ1 and its fragments as well as σ1 (37). Numerous observations suggest that subvirion particles resembling the ISVP and core play essential roles in reovirus infection. Proteolytic removal of σ3 from an infecting virion is thought to prime the particle for membrane penetration by freeing μ1 to interact with a cellular membrane (15, 42, 66) (also see below). Membrane penetration initiated by the ISVP-like particle is then believed to result in cytoplasmic delivery of the primary transcriptase particle. Like the core, this primary transcriptase particle is activated to synthesize the viral mRNAs for translation and packaging (38, 62).
One line of evidence that supports the involvement of an ISVP-like particle in membrane penetration is the capacity of ISVPs, but not virions or cores, to promote permeabilization of membranes in vitro. The target membranes include murine L929 cell membranes (10), human and bovine red blood cell (RBC) membranes (15, 16, 36), planar phospholipid bilayers (67), and liposomes (K. Chandran and M. L. Nibert, unpublished data). In some of these previous experiments, Cs+ ions were needed to promote the membrane interaction, for reasons that have remained largely mysterious but that may relate to the capacity of Cs+ to promote reovirus uncoating (9, 11, 57). The M2 gene (which encodes μ1) was identified as the genetic determinant of a viral strain difference in the ISVP-associated permeabilization of L929 cell membranes (43), suggesting that μ1 is a participant in this process. Also consistent with a role for μ1 in membrane permeabilization are observations that (i) μ1 is modified with a myristoyl (C14 fatty-acyl) group at its N terminus (53), (ii) viral particles containing mutant forms of μ1 show a reduced capacity to permeabilize membranes (35), and (iii) in vitro addition of μ1 to cores is sufficient to restore the membrane-permeabilizing activity of the particles (16). The crystal structure of the μ1 trimer in complex with three σ3 monomers was recently determined at 2.8-Å resolution and suggested additional aspects of the functions of μ1 in membrane penetration (42). Of particular note for the present study was an indication that large conformational changes in the μ1 trimer must be required for externalization of the N-myristoylated μ1N peptide, which, it was proposed, inserts into the cellular membrane as a part of the penetration mechanism (42, 53).
For a description of the molecular mechanism of reovirus membrane penetration that can take advantage of the recently determined μ1 crystal structure (42), more information about the biochemical and structural changes in μ1 and other viral proteins that accompany the membrane interactions is needed. In the present study, we determined that ISVPs undergo a major structural transition involving outer-capsid proteins μ1, σ1, and λ2 that precedes, and is required for, virus-induced membrane permeabilization and activation of the core-associated viral transcriptases. The nature of the structural changes in μ1 and σ1 and their roles in membrane permeabilization by viral particles were also investigated. Our results provide mechanistic insights into two key steps in reovirus entry: membrane penetration and generation of the primary transcriptase particles.
MATERIALS AND METHODS
Cells.
Spinner-adapted murine L929 cells were grown in Joklik's modified minimal essential medium (Irvine Scientific Co., Irvine, Calif.) supplemented to contain 2% fetal bovine serum and 2% bovine calf serum (HyClone Laboratories, Logan, Utah) in addition to 2 mM glutamine, 100 U of penicillin/ml, and 100 μg of streptomycin/ml (Irvine Scientific Co.).
Virions, ISVPs, and cores.
Virions of reoviruses T1L, T3D, and the T1L × T3D reassortants in Fig. 2 were obtained by the standard protocol (27) and stored in virion buffer (150 mM NaCl, 10 mM MgCl2, 10 mM Tris [pH 7.5]). Purified T1L ISVPs were obtained by the same protocol. Unless mentioned otherwise, nonpurified ISVPs were obtained by digesting virions at a concentration of 1013 particles/ml with Nα-p-tosyl-l-lysine chloromethyl ketone-treated chymotrypsin (200 μg/ml) for 8 to 20 min at 32°C. Digestion was stopped by addition of ethanolic phenylmethylsulfonyl fluoride (2 to 5 mM). Purified cores were obtained from virions as described previously (54). Particle concentrations were estimated on the basis of A260 (20).
FIG. 2.

Conformational state of the μ1 protein within hemolysis reactions and reassortant genetic analysis of the difference in hemolysis between T1L and T3D ISVPs. (A) Nonpurified ISVPs of strains T1L (circles), T3D (squares), E3 (diamonds), or EB144 (triangles) (8 × 1012 particles/ml) were incubated with RBCs in virion buffer for different times at 32°C. Only 0- and 20-min time points were taken for EB144 and E3 ISVPs. Each sample was divided into two equal aliquots. One aliquot was used to measure the amount of hemolysis. Results from two trials are shown. (B) The second aliquot was treated with trypsin for 30 min on ice, and digestion was stopped by addition of soybean trypsin inhibitor. Samples were subjected to SDS-PAGE, and δ, the major fragment of μ1 within ISVPs, was visualized by immunoblotting with μ1-specific monoclonal antibody 10H2. The positions of δ and an unidentified proteolytic fragment derived from δ (∗) are marked. (C) Reassortant strains chosen for this study represent all 16 possible combinations of T1L (L) and T3D (D) alleles for genome segments L2, M2, S1, and S4, which encode outer-capsid proteins λ2, μ1, σ1, and σ3, respectively. Strains are listed in order of decreasing amount of hemolysis. The allelic origin of genome segment M2 is in boldface. For determining the strain behaviors, nonpurified ISVPs (1013 particles/ml) were incubated with RBCs in virion buffer for 7 min at 32°C. Each sample was divided into two equal aliquots. One aliquot was used to measure the amount of hemolysis. Averages from five or six trials (T1L and T3D) and one or two trials (reassortant strains) are shown. The second aliquot was treated with trypsin for 20 min on ice, and digestion was stopped by addition of soybean trypsin inhibitor. Samples were subjected to SDS-PAGE, and the δ fragment of μ1 was visualized by Coomassie staining. Conformational (conf.) change: + or −, loss or retention of δ after trypsin treatment, respectively; nd, not determined.
SDS-PAGE and immunoblotting.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out on 10% acrylamide gels as described previously (51). Viral proteins were detected by staining with Coomassie brilliant blue R-250 (Sigma) or were transferred to a nitrocellulose membrane with the Mini-TransBlot kit (Bio-Rad, Hercules, Calif.) and visualized by immunoblotting. The μ1-specific mouse monoclonal antibody 10H2 (70) and T1L σ1-specific rabbit antisera raised against a glutathione S-transferase-σ1 fusion protein (to be described elsewhere) were employed at a 1:1,000 dilution as primary antibodies. Mouse- or rabbit-specific goat immunoglobulin Gs conjugated to alkaline phosphatase (Sigma) were used at a 1:2,000 dilution as secondary antibodies. Antibody binding was detected with colorimetric reagents p-nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolylphosphate p-toluidine salt (Bio-Rad) according to the manufacturer's instructions.
Hemolysis.
Citrated bovine calf RBCs (Colorado Serum Co., Denver, Colo.) were washed with phosphate-buffered saline supplemented with 2 mM MgCl2 (PBS-Mg2+) and suspended in PBS-Mg2+ at a stock concentration of 30% (vol/vol) just prior to use. All experiments were performed in a 4°C cold room to minimize variability in results. Viral particles were incubated with RBCs (3% [vol/vol]) in virion buffer or hemolysis reaction buffer (50 mM Tris-Cl [pH 7.5]) in a total volume of 20 to 100 μl. Na+ or Cs+ ions were added to hemolysis reaction mixtures as their chloride salts from stock solutions prepared in water. Reactions were initiated by transfer of samples to the experimental temperature (ice or 32°C) and terminated by their removal onto ice. After resting on ice for at least 5 min, cells within each sample were pelleted by centrifugation (300 × g for 5 min at 4°C), and 10 to 20 μl of the supernatant was diluted into virion buffer (100 μl total) in a 96-well microplate (Costar, Cambridge, Mass.). The amount of hemoglobin released from RBCs was quantitated by measuring A415 with a microplate reader (Molecular Devices, Sunnyvale, Calif.) and by using the equation percent hemolysis = {[A415 (sample) − A415 (blank)]/[A415 (detergent) − A415 (blank)]} × 100%, where the blank lacked viral particles but contained all other components of the reaction mixture and the detergent was either Triton X-100 (Sigma; 1% [vol/vol]) or Nonidet P-40 (Sigma; 0.5% [vol/vol]). Addition of either detergent to the hemolysis reaction mixture effected complete RBC lysis.
Proteolysis assay for conformational changes in viral proteins.
Viral particles were chilled on ice for at least 5 min and incubated with trypsin (100 μg/ml) for 20 to 60 min on ice. Reactions were stopped by addition of soybean trypsin inhibitor (300 μg/ml) and further incubation for 5 to 10 min on ice. Incubation of samples containing viral particles with trypsin for longer times (up to 2 h tested; data not shown) did not appear to alter the pattern of digestion observed. Following addition of Laemmli sample buffer, samples were disrupted by boiling for 2 to 5 min and were subjected to SDS-PAGE.
bis-ANS fluorescence.
High-purity bis-ANS (4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid, dipotassium salt) was obtained from Molecular Probes (Junction City, Oreg.). bis-ANS stock solutions (5 to 10 mM) were prepared in methanol and stored in the dark at 4°C. The concentration of bis-ANS in these solutions was measured from its molar extinction coefficient (∼24,000 M−1 cm−1) just prior to use. Viral particles were incubated with bis-ANS (25 μM) at the same reaction conditions used for hemolysis experiments (see above), except that RBCs were not added. Reactions were initiated by transfer of samples to the experimental temperature (ice or 32°C) and were terminated by their removal onto ice. After resting on ice for at least 5 min, 10 to 20 μl of each sample was diluted into virion buffer (100 μl total) in a 96-well black microplate designed for fluorescence applications (Costar). bis-ANS fluorescence was measured at an excitation wavelength (λex) of 405 nm and an emission wavelength (λem) of 485 nm with the Spectramax Gemini XS fluorescence microplate reader (Molecular Devices). An emission cutoff filter (λcut < 475 nm) was employed to minimize detection of scattered light from the excitation beam. bis-ANS fluorescence intensity (I) was calculated from Icorrected = Isample − Iblank, where the blank lacked viral particles but contained all other components of the reaction mixture.
Triton X-114 partitioning assay.
Werck-Reichhart and coworkers' (71) modification of Bordier's original Triton X-114 partitioning protocol (8) was employed. Viral particles were diluted to a volume of 1 ml with virion buffer, chilled for at least 5 min on ice, and incubated with ice-cold Triton X-114 (2% [vol/vol]; Sigma) for 1 h on ice with occasional mixing. Glycerol (40% [vol/vol]) was then added. Samples were incubated for 10 min at 37°C, followed by centrifugation (500 × g, 5 min, room temperature) to induce separation into detergent-poor and -rich phases. Inclusion of glycerol in the partitioning mixture allowed flotation, rather than sedimentation, of the detergent-rich phase (71), thereby removing the concern that the detergent-rich fraction contained particles merely due to pelleting during centrifugation. The bottom of each centrifuge tube was punctured with a needle, and both detergent-poor and -rich phases were harvested by collecting drops. The detergent-poor fraction was reextracted with fresh Triton X-114 and glycerol as described above. Detergent-rich fractions from both extractions were pooled, and the second detergent-poor fraction was discarded. Viral proteins within the detergent-poor and -rich fractions were precipitated with trichloroacetic acid (TCA) as follows. Samples were incubated with TCA (10% [wt/vol]) and purified carbonic anhydrase (5 μg) as a carrier protein for at least 1 h on ice. Precipitated material was concentrated by centrifugation (10,000 × g for 30 min at 4°C), washed twice with ice-cold acetone, and dried under reduced pressure. Dried pellets were solubilized in room-temperature virion buffer. Following addition of Laemmli sample buffer, samples were disrupted by boiling for 5 min. Viral proteins were resolved by SDS-PAGE.
RESULTS
T3D ISVPs, but not virions or cores, induce lysis of RBCs.
Previous work showed that only ISVPs can release 51Cr from preloaded L929 cells (10, 43), form ion-permeant channels in planar phospholipid bilayers (67), and lyse RBCs in the presence of Cs+ ions (15, 16, 36). To test whether certain reovirus particles can permeabilize RBC membranes in a Cs+-free buffer, we incubated purified T3D virions, ISVPs, or cores (Fig. 1A) with human RBCs either on ice or at 32°C and then measured the extent of hemolysis. None of the particles induced hemolysis upon incubation on ice (Fig. 1B), but, when incubated with RBCs at 32°C, T3D ISVPs lysed essentially all of the cells (Fig. 1B). In contrast, T3D virions and cores were inactive for hemolysis at these and all conditions tested (Fig. 1B; data not shown). Thus ISVPs, but not virions or cores, can induce permeabilization of different types of membranes, both in vitro and in cell culture, in the presence or absence of Cs+ ions. In addition, the failure of ISVPs to induce hemolysis at 4°C suggests that the permeabilization process is temperature dependent and may require changes in protein conformation and/or lipid mobility.
Genome segment M2, which encodes μ1, determines a difference in hemolytic capacity between T1L and T3D ISVPs.
To extend our findings to particles of other reovirus strains, we tested the capacity of T1L ISVPs to mediate hemolysis in a Cs+-free buffer. T1L ISVPs failed to induce RBC lysis upon incubation at the conditions used with T3D ISVPs above (data not shown; Fig. 2A). To determine whether this difference between T1L and T3D ISVPs arises from one or more genetic differences between the two strains, we analyzed the behavior of a panel of T1L × T3D reassortant strains in the hemolysis assay. The capacity of ISVPs to induce hemolysis segregated with the parental origin of their M2 genome segments (Fig. 2C). All reassortant ISVPs containing an M2 segment derived from T3D were positive for hemolysis, whereas all containing an M2 segment derived from T1L were negative. No other genome segment contributed significantly to the phenotypic difference between T1L and T3D ISVPs (Fig. 2C). Lucia-Jandris and coworkers (43) previously found that M2 also genetically controls the difference between T1L and T3D ISVPs in their capacity to release 51Cr from preloaded L929 cells (T1L ISVPs were inactive in that assay as well). The simplest hypothesis based on these results is that μ1, the protein encoded by M2 and the major surface protein of ISVPs (Fig. 1A), participates directly in permeabilization of both RBC and L929 cell membranes. A related hypothesis is that the μ1 proteins encoded by the T1L and T3D alleles of M2 must differ in their capacities to induce permeabilization at the assay conditions.
Protein μ1 changes conformation in association with hemolysis.
We used sensitivity to protease digestion to monitor conformational changes in viral particles during hemolysis reactions. T1L or T3D ISVPs were incubated with RBCs at 32°C for different times. At each time point, samples were removed to ice and divided into two aliquots. One aliquot was analyzed for RBC lysis. The other was incubated with trypsin for 60 min on ice. Viral proteins in the trypsin-treated samples were resolved by SDS-PAGE and detected by immunoblotting with protein-specific antibodies. Evidence for a change in protease sensitivity over time was obtained only in the case of T3D μ1 (Fig. 2B; data not shown for the other viral proteins, but see Fig. 3C). Specifically, the δ fragment of μ1 in T3D ISVPs became sensitive to trypsin cleavage after between 8 and 12 min of incubation at 32°C. This corresponded to the onset time of hemolysis induced by T3D ISVPs (Fig. 2A). When we repeated the experiment with T1L ISVPs, which did not mediate hemolysis under these conditions (Fig. 2A), we found that T1L μ1 remained resistant to cleavage by trypsin over the entire time course (Fig. 2B). Essentially identical results were obtained with several other proteases (data not shown). These findings indicate that protein μ1 acquires a protease-sensitive conformation when T3D ISVPs are incubated with RBCs at 32°C, but not at 4°C, and that this conformational change in μ1 occurs in temporal correlation with hemolysis.
FIG. 3.

Effect of Cs+ ions and RBCs on hemolysis and the conformational state of μ1. (A and B) Nonpurified ISVPs of strains T1L (circles) or T3D (squares) (4 × 1012 particles/ml) were incubated with RBCs in reaction buffer (50 mM Tris-Cl [pH 7.5]) containing CsCl (300 mM) for different times at 32°C. Measurement of the amount of hemolysis (A) and assessment of the protease sensitivity of δ (B) were carried out as described for Fig. 2A and B. Results from two trials are shown in panel A. (C) Samples were generated as described for panels A and B except that no RBCs were added. Samples were treated with trypsin for 30 min on ice, and digestion was stopped by addition of soybean trypsin inhibitor. Samples were subjected to SDS-PAGE, and the viral proteins were visualized by Coomassie staining.
We also tested ISVPs derived from T1L × T3D reassortant strains in the protease sensitivity assay. Reassortant ISVPs that scored positive for hemolysis (i.e., those that carry T3D M2) underwent μ1 conformational change in association with hemolysis, whereas reassortant ISVPs that scored negative for hemolysis (i.e., those that carry T1L M2) did not undergo μ1 conformational change detected by protease treatment (Fig. 2B and C). These results provide additional evidence that μ1 changes conformation in association with hemolysis. They also show that the difference between T1L and T3D ISVPs in the propensity of μ1 to change conformation is determined by one or more sequence differences between the T1L and T3D alleles of M2.
Rates of hemolysis and μ1 conformational change increase in parallel in the presence of Cs+ ions.
T3D ISVPs are proteolytically digested to cores more efficiently when incubated with K+, Rb+, or Cs+ ions in place of Na+ or Li+ ions (9, 11). Since differences among viral strains in the susceptibility of ISVPs to undergo proteolytic digestion to cores are genetically determined by M2, encoding μ1 (10), these results suggest that K+, Rb+, and Cs+ ions allow more efficient conversion of ISVPs to cores by increasing the capacity of μ1 to undergo protease digestion (12, 13). Based on these previous findings, we speculated that the propensity of T1L μ1 to acquire a protease-sensitive conformation when ISVPs are incubated with RBCs may be enhanced by inclusion of K+, Rb+, or Cs+ ions in the hemolysis reaction. Accordingly, we repeated the hemolysis experiments in a low-Na+ buffer supplemented with K+ or Cs+ ions. Consistent with our hypothesis, the μ1 protein in T1L ISVPs became protease sensitive upon incubation with RBCs at 32°C in the presence of K+ (data not shown) or Cs+ ions (Fig. 3B), indicating that T1L μ1 had indeed changed conformation. Moreover, T1L ISVPs induced hemolysis at these conditions (Fig. 3A), consistent with previous evidence that they have that capacity (15, 16, 36). In addition, the presence of Cs+ in the hemolysis reaction decreased the onset time of μ1 conformational change with T3D ISVPs (compare Fig. 3A and 2A), and for both T1L and T3D ISVPs, there was a correspondence between the onset times of μ1 conformational change and hemolysis (Fig. 3A). Thus, Cs+ ions accelerate in parallel both μ1 conformational change and hemolysis. The consistent temporal correlation between these phenomena argues that they are mechanistically linked.
RBCs are dispensable for μ1 conformational change.
In the experiments described thus far (Fig. 2 and 3), μ1 conformational change was observed in hemolysis reactions with RBCs. To test whether interaction of viral particles with one or more components of the RBC membrane are required for induction of μ1 conformational change, we repeated the experiments described above but omitted RBCs from the samples. Protein μ1 acquired a protease-sensitive conformation when either T1L or T3D ISVPs were incubated with Cs+ ions at 32°C in the absence of RBCs (Fig. 3C). Moreover, the kinetics of μ1 conformational change in the absence or presence of RBCs were essentially indistinguishable (Fig. 3C). RBCs also had little or no effect on the rate of μ1 conformational change in experiments carried out in the presence of Na+ ions (data not shown). Therefore, induction and maintenance of a protease-sensitive conformation of μ1 do not require interaction of viral particles with a component of RBC membranes.
μ1 conformational change is required for hemolysis.
We investigated the causal relationship between μ1 conformational change and hemolysis by attempting to decouple the two events. T1L ISVPs were incubated for different times at 32°C in the absence of RBCs and then removed to ice. RBCs were then added, and hemolysis reactions were allowed to proceed on ice (see Fig. 4A for a schematic diagram). The conformational status of μ1 after the incubation at 32°C was assessed by protease treatment of parallel samples on ice. The only samples in which hemolysis occurred at 4°C were those containing a protease-sensitive conformer of μ1 generated by preincubation of the ISVPs at 32°C (Fig. 4B and C). These results strongly suggest that μ1 conformational change is required for hemolysis. They also suggest that hemolysis is normally blocked at 4°C because the conformational change cannot occur at that temperature. The RBC lysis step(s) is, by comparison, less dependent on temperature: hemolysis mediated by samples containing a protease-sensitive conformer of μ1 was slowed from a few seconds at 32°C to ∼5 min at 4°C but occurred efficiently at both temperatures (data not shown).
FIG. 4.

Requirement of μ1 conformational change for hemolysis. (A) Schematic diagram for the experiment. Nonpurified T1L ISVPs (4 × 1012 particles/ml) were incubated in reaction buffer containing CsCl (300 mM) for different times at 32°C. After a 5-min incubation on ice, RBCs were added to each sample, and hemolysis reactions were allowed to proceed for 15 min on ice. (B) The amount of hemolysis in each sample was determined. Results from two trials are shown. (C) Same as panel B except that trypsin was added to samples instead of RBCs. Protease digestion was allowed to proceed for 60 min on ice and was stopped by addition of soybean trypsin inhibitor. Samples were subjected to SDS-PAGE, and the viral proteins were visualized by Coomassie staining.
A protease-sensitive conformer of μ1 is necessary for hemolysis.
Although the preceding experiments suggested that μ1 conformational change is required for hemolysis (Fig. 2 to 4), it remained possible that the conformationally altered μ1 protein per se is dispensable. For instance, hemolysis might be carried out by another viral protein activated by μ1 conformational change. To distinguish between these possibilities, we used a modified form of the order-of-incubation experiment described in the legend for Fig. 4 (see Fig. 5A for a schematic diagram). T1L ISVPs were allowed to undergo the μ1 conformational change in the absence of RBCs by incubation with Cs+ ions at 32°C and then removed to ice. The chilled sample was divided into four aliquots. Aliquots 1 and 4 were left untreated. Aliquot 2 was incubated with trypsin on ice, after which the protease was inactivated with soybean trypsin inhibitor. Aliquot 3 was treated with inactivated trypsin generated by preincubation of protease with soybean trypsin inhibitor. RBCs were then added to all four aliquots, and hemolysis reactions were allowed to proceed on ice (Fig. 5B). Parallel reaction mixtures were subjected to SDS-PAGE and Coomassie staining to visualize the viral proteins (Fig. 5C). As expected, aliquot 1 was wholly competent for hemolysis. However, aliquot 2 was wholly deficient for hemolysis. Comparison of the protein compositions of viral particles within aliquots 1 and 2 revealed that loss of hemolytic capacity correlated with a substantial reduction in the complement of μ1 in aliquot 2. The mere presence of extra proteins (i.e., trypsin and soybean trypsin inhibitor) did not explain the inhibition of hemolysis, since inactivated trypsin in aliquot 3 had little or no effect on either the extent of hemolysis or μ1 content (aliquot 3). Also ruled out was the possibility that trypsin inhibits hemolysis by acting on RBCs rather than viral particles; RBCs pretreated with trypsin were lysed efficiently (aliquot 4). These findings strongly suggest that a protease-sensitive conformer of μ1 is necessary for hemolysis.
FIG. 5.

Requirement of a protease-sensitive μ1 conformer for hemolysis. (A) Schematic diagram for the experiment. Nonpurified T1L ISVPs (4 × 1012 particles/ml) were incubated in reaction buffer containing CsCl (300 mM) for 8 min at 32°C and then removed to ice. The sample was divided into four aliquots, and each was treated as indicated. ISVP∗, particle type derived from ISVPs that have undergone μ1 conformational change (see text for more information); try, trypsin; sbti, soybean trypsin inhibitor; try†, trypsin (1 mg/ml) pretreated with soybean trypsin inhibitor (3 mg/ml) for 20 min on ice; rbc†, RBCs pretreated first with trypsin for 20 min on ice and then with soybean trypsin inhibitor for 10 min. (B) The amount of hemolysis in each sample was determined. Averages ± standard deviations from three trials are shown. (C) Same as panel B except that Laemmli sample buffer was added instead of RBCs. Samples were boiled and subjected to SDS-PAGE, and the viral proteins were visualized by Coomassie staining.
Hydrophobicity of viral particles increases in concert with μ1 conformational change.
Hemolysis induced by ISVPs almost certainly involves the interaction of viral protein regions with the RBC membrane bilayer. Since change in μ1 conformation is required for hemolysis, we suspected that this change may expose hydrophobic sequences previously buried within ISVPs. In the following experiments, we used two different assays to detect changes in the hydrophobicity of viral particles: fluorescence of the polarity-sensitive dye bis-ANS and partitioning of particles into detergent Triton X-114.
bis-ANS fluorescence.
bis-ANS is essentially nonfluorescent in aqueous solution but becomes strongly fluorescent when dissolved in nonpolar solvents or when bound to surface-accessible hydrophobic sites within proteins (46). T1L ISVPs were incubated with bis-ANS and either Na+ or Cs+ ions for different times at 32°C and then removed to ice. Fluorescence in each sample was measured in a microplate reader. Parallel samples were exposed to trypsin on ice to assess the conformational status of μ1 (data not shown). Little or no change in bis-ANS fluorescence was seen when T1L ISVPs were incubated with Na+ ions (i.e., at conditions that did not induce μ1 conformational change over the entire time course) (Fig. 6A). In contrast, when T1L ISVPs were incubated with Cs+ ions, a large increase in bis-ANS fluorescence was observed. The onset time of this increase corresponded to that of μ1 conformational change (Fig. 6A). Experiments with T3D ISVPs in the presence of Na+ ions confirmed that bis-ANS fluorescence increased in concert with the acquisition of a protease-sensitive conformation by protein μ1 (data not shown). These findings suggest that viral particles expose hydrophobic sequences to the aqueous milieu in association and temporal correlation with μ1 conformational change.
FIG. 6.

Effect of μ1 conformational change on bis-ANS fluorescence. (A) Nonpurified T1L ISVPs (4 × 1012 particles/ml) were incubated in reaction buffer containing bis-ANS (25 μm) and NaCl (squares) or CsCl (circles) (300 mM) for different times at 32°C. The amount of bis-ANS fluorescence was then measured. Results from two trials are shown. The conformational state of μ1 in each sample was assessed by trypsin treatment and is indicated below the graph. + and −, loss and retention of δ after trypsin treatment, respectively. μ1∗, protease-sensitive conformer of μ1. (B) Schematic diagram for the order-of-incubation experiment. The experiment was performed as for Fig. 5 except that bis-ANS (25 μM) was added to samples instead of RBCs. (C) The amount of bis-ANS fluorescence in each sample from panel B was determined. Results from two trials are shown as superimposed open and filled bars.
Triton X-114 partitioning.
Nonionic detergent Triton X-114 forms a homogeneous phase in aqueous solution at 4°C but separates into discrete detergent-rich and detergent-poor phases as the temperature is elevated past 20°C (8). Proteins that form mixed micelles with detergent by virtue of their exposed hydrophobic regions (e.g., integral membrane proteins) partition into the detergent-rich phase, whereas proteins lacking extensive hydrophobic surfaces (e.g., many cytosolic proteins) partition into the detergent-poor phase. T1L ISVPs were incubated with Na+ or Cs+ ions at 32°C and removed to ice. Triton X-114 was then added to the samples. After further incubation on ice, detergent-rich and -poor phases were separated by incubation at 37°C, and the detergent-rich phase was floated by centrifugation. Both phases were harvested, and the distribution of viral proteins in each fraction was examined by SDS-PAGE and Coomassie staining. When T1L ISVPs were incubated with Na+ ions at 32°C prior to Triton X-114 extraction, viral proteins were predominantly located in the detergent-poor fraction, indicating that most particles had partitioned into the detergent-poor phase (Fig. 7A). In contrast, when T1L ISVPs were incubated with Cs+ ions at 32°C before Triton X-114 extraction, viral proteins were predominantly located in the detergent-rich fraction, indicating that most particles had shifted into the detergent-rich phase (Fig. 7A). Trypsin treatment of aliquots removed before Triton X-114 extraction indicated that, as expected, μ1 had acquired a protease-sensitive conformation in particles incubated at 32°C with Cs+ ions but not Na+ ions (data not shown). These findings, together with those with bis-ANS (Fig. 6A), strongly suggest that μ1 conformational change causes hydrophobic protein regions to be exposed at the surfaces of viral particles.
FIG. 7.
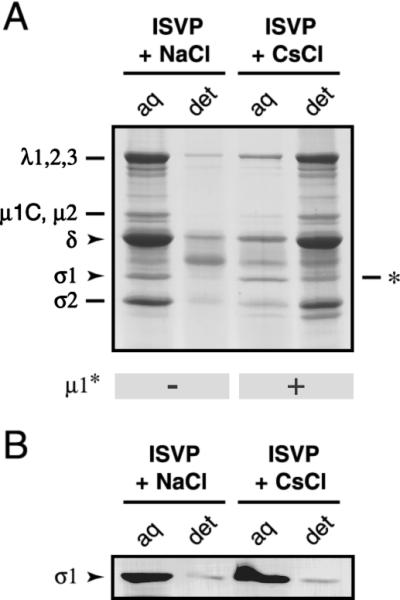
Effect of μ1 conformational change on Triton X-114 partitioning of viral proteins. Nonpurified T1L ISVPs (4 × 1011 particles/ml) were incubated in reaction buffer containing NaCl or CsCl (200 mM) for 40 min at 37°C and then removed to ice. Samples were extracted with Triton X-114 as described in Materials and Methods. The detergent-poor (aq) and detergent-rich (det) fractions were subjected to SDS-PAGE, followed by Coomassie staining to visualize the viral proteins (A) or immunoblot analysis with a T1L σ1-specific polyclonal antiserum (B). ∗, position of an unidentified proteolytic fragment that comigrated with σ1. The conformational state of μ1 in each fraction was assessed by trypsin treatment and is indicated below the gel.
μ1 exposes hydrophobic regions as a result of its conformational change.
The preceding data make μ1 a prime candidate to be the viral protein that exposes hydrophobic regions preceding hemolysis. To test that hypothesis, we performed an order-of-incubation experiment exactly as described for Fig. 5, with the exception that bis-ANS was added to the reaction mixtures instead of RBCs (see Fig. 6B for a schematic diagram). T1L ISVPs were incubated with Cs+ ions at 32°C to allow the μ1 conformational change to occur and then removed to ice. Reaction mixtures were split into three aliquots. Aliquot 1 was left untreated, aliquot 2 was treated with trypsin, and aliquot 3 was treated with inactivated trypsin. bis-ANS was then added to the reaction mixtures, and the fluorescence in each was measured after a further incubation on ice (Fig. 6C). bis-ANS fluorescence in aliquot 2 was substantially reduced (by ∼80%) relative to that in aliquots 1 and 3. Comparison of the protein compositions of the three samples revealed that loss of fluorescence correlated with a substantial reduction in the complement of μ1 in aliquot 2 (data not shown). The findings suggest that μ1 exposes previously buried hydrophobic regions as a result of its conformational change.
Receptor-binding protein σ1 is lost from particles in concert with μ1 conformational change.
Closer examination of Coomassie-stained gels from the Triton X-114 experiment revealed that, although most viral proteins from the particles preincubated with Cs+ ions were predominantly located in the detergent-rich fraction, a 50,000-Mr band migrating at the expected position of receptor-binding protein σ1 appeared to be enriched in the detergent-poor fraction (Fig. 7A). Immunoblot analysis of detergent-rich and -poor fractions with a σ1-specific antibody confirmed that most σ1 proteins did not partition into the detergent-rich phase along with other viral proteins (Fig. 7B). This observation suggests that μ1 conformational change is associated with the loss of σ1 from viral particles and that the hydrophobicity of σ1 does not increase substantially in conjunction with its elution from particles.
To test further whether viral particles that had undergone μ1 conformational change had also lost σ1, we attempted to purify those particles. T1L ISVPs were incubated with Na+ or Cs+ ions at 32°C and then transferred to ice. Trypsin treatment of aliquots removed prior to purification confirmed that μ1 had acquired a protease-sensitive conformation in the viral particles incubated at 32°C with Cs+ ions but not Na+ ions (data not shown). Remaining samples were loaded atop preformed CsCl gradients, and viral particles (density [ρ] ≈ 1.38 g/cm3) were separated from particle-free proteins (ρ ≈ 1.30 g/cm3) according to density by ultracentrifugation. Visual inspection of the gradients revealed that viral particles incubated with Na+ ions before centrifugation had formed a homogeneous band with an opalescent appearance, as typically seen when ISVPs are concentrated on CsCl density gradients (Fig. 8A). In contrast, particles incubated with Cs+ ions before centrifugation produced an inhomogeneous band composed of white flocculated material (Fig. 8A). A flocculent band was also obtained with T3D ISVPs that had been incubated with Na+ ions at 32°C (data not shown). The opalescent and flocculent bands migrated at similar positions in CsCl gradients, indicating that the particles in them had similar densities (data not shown). The change in morphology of the particle band from opalescent to flocculent suggested that viral particles that had undergone μ1 conformational change were prone to aggregation at high concentrations. This agrees with our finding that μ1 conformational change enhances the hydrophobicity of particles (Fig. 6 and 7). To determine the protein compositions of viral particles in the opalescent and flocculent bands, the samples were subjected to SDS-PAGE and monitored by Coomassie staining or immunoblot analysis with a σ1-specific antibody. As expected, particles in the opalescent band closely resembled ISVPs; the resemblance extended to the presence of μ1 fragment δ and protein σ1 (Fig. 8B). Particles comprising the flocculent band also resembled ISVPs in containing an approximately full complement of the δ fragment of μ1; however, they contained little or no σ1 (Fig. 8B). These findings confirmed that viral particles that had undergone μ1 conformational change had also lost σ1. They also indicated that the central δ fragment of μ1 remained associated with particles even after μ1 acquired a protease-sensitive conformation.
FIG. 8.
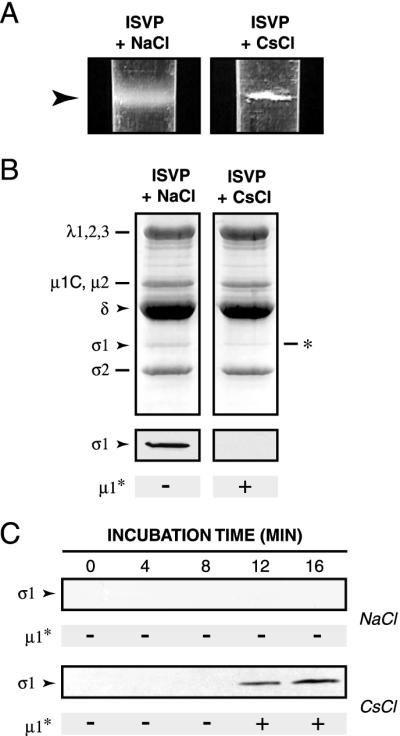
Effect of μ1 conformational change on particle association of protein σ1. (A) Nonpurified T1L ISVPs (4 × 1012 particles/ml) were incubated in reaction buffer containing NaCl or CsCl (300 mM) for 20 min at 32°C and then removed to ice. Each sample was overlaid onto a preformed CsCl gradient (ρ = 1.30 to 1.45 g/cm3, 3.2 ml) and subjected to centrifugation in a Beckman SW60 rotor (30,000 rpm for 2 h at 5°C). Gradients were photographed with a Nikon Coolpix digital camera. Images were cropped and resized in Photoshop, version 5.5 (Adobe Systems, San Jose, Calif.). Arrowhead, position of the band in each gradient. (B) The banded material in panel A was concentrated by TCA precipitation and subjected to SDS-PAGE, followed by Coomassie staining to visualize the viral proteins (top) or immunoblot analysis to visualize σ1. ∗, position of an unidentified proteolytic fragment that comigrated with σ1. The conformational state of μ1 in each sample was assessed by trypsin treatment and is indicated below the gels and immunoblots. (C) Nonpurified T1L ISVPs (4 × 1012 particles/ml) were incubated in reaction buffer containing NaCl or CsCl (300 mM) for different times at 32°C and then removed to ice. Each sample was overlaid onto a sucrose cushion (20% [wt/vol], 500 μl) and subjected to centrifugation in a Beckman TLA 100.2 rotor (90,000 rpm for 1 h at 5°C). A 200-μl fraction was removed from the top of each gradient, and the proteins in this fraction were concentrated by TCA precipitation and subjected to SDS-PAGE. Protein σ1 was visualized by immunoblot analysis. The conformational state of μ1 in each sample was assessed by trypsin treatment and is indicated below each immunoblot.
We next sought to define the temporal relationship between μ1 conformational change and loss of σ1 from particles. T1L ISVPs were incubated with Na+ or Cs+ ions for different times at 32°C and then removed to ice. Each sample was loaded onto a sucrose cushion, and viral particles were pelleted by ultracentrifugation. A portion of supernatant from the top of each tube (Fig. 8C) and the particle pellet at the bottom of each tube (data not shown) were subjected to SDS-PAGE and immunoblot analysis with a σ1-specific antibody. Particles incubated with Na+ ions did not undergo μ1 conformational change and released little or no σ1 over the time course. In contrast, particles incubated with Cs+ ions underwent μ1 conformational change and released most or all of their σ1 fibers, with similar onset times for both structural changes. These findings indicate that σ1 is lost from viral particles in concert with acquisition of the protease-sensitive conformation of μ1.
Viral particles that contain a protease-sensitive μ1 conformer and lack σ1 are activated for mRNA synthesis.
Both virions and ISVPs are inactive with regard to genome-dependent synthesis of viral mRNAs. In contrast, cores, which lack outer-capsid proteins μ1, σ3, and σ1 are activated for mRNA synthesis (3, 21, 37, 60). Derepression of the core-associated transcriptases correlates with protease digestion of μ1, and a difference among strains in the capacity of particles to undergo transcriptase activation is genetically determined by M2 (11, 21, 37). Conversely, addition of μ1 and σ3 to cores represses mRNA synthesis (24). These observations strongly suggest that the outer capsid in general, and protein μ1 in particular, play a role in regulating the activity of the core-associated transcriptases. Other experiments showed that incubation of ISVPs with K+ or Cs+ ions at 37°C is sufficient for derepression of transcriptase activity and that proteolytic removal of μ1 from viral particles is not required (9, 12, 23, 57). In the present study, we found that incubation of ISVPs with Cs+ ions accelerates, in concert, both conformational change in μ1 (Fig. 3) and loss of σ1 from particles (Fig. 7 and 8). Putting previous and present findings together, we speculated that one or more structural changes in ISVPs identified in this study may also play a role in transcriptase activation. To test that hypothesis, T1L virions or ISVPs were incubated with Na+ or Cs+ ions at 32°C and then removed to ice. Samples were incubated at 37°C in a transcription reaction mixture containing all four ribonucleoside triphosphates, [α-32P]GTP, Mg2+ ions, and an ATP-regenerating system. Long transcription products were separated from oligonucleotides and free nucleotides by precipitation with TCA, and 32P incorporation into the precipitated RNA was quantitated by scintillation counting. Viral particles containing a protease-resistant conformer of μ1 (i.e., virions incubated with Na+ or Cs+ ions and ISVPs incubated with Na+ ions) remained inactive at mRNA synthesis (Fig. 9). In contrast, particles containing a protease-sensitive conformer of μ1 (i.e., ISVPs incubated with Cs+ ions) were active for mRNA synthesis (Fig. 9). These results suggest that μ1 conformational change or loss of σ1 from particles or both are necessary for derepression of the core-associated viral transcriptases.
FIG. 9.
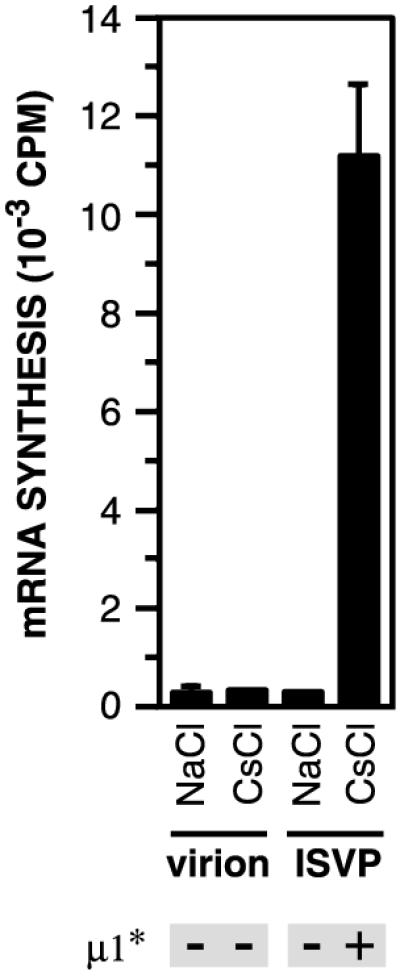
Effect on μ1 conformational change on activity of the particle-associated viral transcriptases. Purified T1L virions and nonpurified T1L ISVPs (4 × 1012 particles/ml) were incubated in reaction buffer containing NaCl or CsCl (300 mM) at 32°C for 20 min and then removed to ice. Samples were then incubated with a transcription reaction mixture including ribonucleoside triphosphates and [α-32P]GTP for 1 h at 37°C as described previously (44). The amount of 32P incorporated into reovirus mRNA was measured by TCA precipitation followed by liquid scintillation counting (44). Averages ± standard deviations of three trials are shown. The conformational state of μ1 in each sample was assessed by trypsin treatment and is indicated below the graph.
DISCUSSION
In this report we present evidence that reovirus ISVPs can undergo a structural transition to a distinct particle type, the ISVP*, when incubated at physiological temperature. We show that ISVP*s contain an altered conformer of the penetration protein μ1 (i.e., μ1*), in which previously buried hydrophobic regions are now exposed (Fig. 2 to 8 and 10). We also show that ISVP*s have lost receptor-binding protein σ1 (Fig. 7, 8, and 10). Induction of the ISVP-to-ISVP* transition is required for permeabilization of RBC membranes (Fig. 4) and activation of the core-associated viral transcriptases (Fig. 9). Furthermore, the μ1* conformer is itself required for hemolytic activity (Fig. 5). Our findings support an entry model in which the ISVP per se does not mediate membrane penetration but, instead, is first converted to the ISVP* form, which then plays the central role in membrane penetration. We propose that the μ1 conformational change occurring as part of the ISVP-to-ISVP* transition is analogous to the entry-related conformational transitions in enveloped-virus fusion machines, such as the influenza virus HA and TBEV E proteins, and nonenveloped virus penetration machines, such as the poliovirus VP1 and VP4 proteins (Fig. 10).
FIG. 10.

Parallels between the membrane penetration machines of enveloped and nonenveloped viruses. It is proposed that the membrane penetration protein of the nonenveloped reoviruses (E) resembles the membrane fusion proteins of enveloped viruses such as influenza virus (B) and TBEV (C) as well as the membrane penetration proteins of other nonenveloped viruses such as poliovirus (D). All of these proteins appear to undergo primed and triggered rearrangements that yield a hydrophobic protein conformer capable of interacting with membranes. Protein-membrane interactions have different consequences for the enveloped and nonenveloped viruses: membrane fusion for the former and either membrane pore formation or membrane perforation for the latter. Some essential structural and functional properties of viral particles containing each type of protein conformer are summarized in panel A. (E) Schematic of radial sections of reovirus particle forms containing the different conformers of membrane penetration protein μ1. Also indicated are other changes that accompany the structural transitions in reovirus particles: σ3 degradation (virion to ISVP) and σ1 elution and λ2 conformational change (ISVP to ISVP*). Proteins are colored as in Fig. 1A.
Primed and triggered conformational changes in metastable viral structures: parallels between enveloped viruses and the nonenveloped reoviruses.
Influenza virus HA homotrimers are synthesized and assembled as HA0 precursors, which are inactive at fusion. HA0 undergoes an endolytic cleavage by a furin-like protease to yield HA1 and HA2. This cleavage primes HA for fusion by liberating sequences in HA2 for fusion-related conformational changes (39, 63). Monomers of E protein in fusion-inactive precursor TBEV particles exist in heterodimeric complex with protein prM. Proteolytic degradation of a region of prM by a furin-like protease allows homodimerization of the E protein, thereby priming E for its conformational change (32, 33). Priming by proteolytic cleavage at a single site within the fusion protein itself or more extensive processing of an interacting protein provides a mechanism to prevent inappropriate induction of conformational changes in the fusion machine that may inactivate viral particles or kill host cells.
The proteolytic degradation of σ3 and/or more-limited cleavage(s) of μ1 by lysosomal proteases (1, 2, 40) may represent analogous priming mechanisms for reovirus particles (Fig. 10). Virions, which contain σ3 in complex with uncleaved μ1 homotrimers, do not undergo the μ1-to-μ1* change (Fig. 9) and cannot mediate hemolysis (Fig. 1). In contrast, ISVPs, which lack σ3 and which contain μ1 mostly as cleaved fragments, are competent to do both (Fig. 2). The endolytic cleavage of μ1 at the δ-φ junction during conversion of virions to ISVPs appears to play no part in priming μ1 (15, 16), however, suggesting a primary role for σ3 degradation in this process (36). How might σ3 degradation prime μ1 for its conformational change? Examination of the recently determined crystal structure of the (μ1)3/(σ3)3 heterohexamer (42) reveals that each σ3 monomer interacts extensively with two adjacent subunits in the μ1 trimer. σ3 may thus function as a molecular clamp, inhibiting the conformational freedom of individual μ1 subunits within the trimer.
Primed HA1-HA2 trimers and E dimers change conformation in response to a low-pH trigger. HA can also be induced to change conformation by nonphysiological stimuli such as elevated temperature and pressure (14, 28). For both HA and E, the conformationally rearranged form of the fusion protein is more thermostable than its precursor (18, 65). Such observations led to the hypothesis that the primed fusion proteins are kinetically trapped in a metastable state (14, 65). We show here that reovirus ISVPs, but not virions, are accelerated to undergo the μ1-to-μ1* change by Cs+ ions (Fig. 3). The rate of this change is also increased by other monovalent cations, such as K+, and by elevated particle concentration (57; M. L. Nibert and K. Chandran, unpublished data). Moreover, we recently showed that thermal inactivation of ISVPs is associated with a conformational change in μ1 that renders it protease sensitive and that ISVPs are less thermostable than virions (36, 47). Although the mechanistic bases for the accelerating effects of monovalent cations, particle concentration, and temperature on μ1 conformational change are poorly understood, the above findings lead us to speculate that removal of σ3 renders the μ1 trimers in ISVPs metastable, that is, energetically poised to undergo a conformational change in response to a triggering stimulus (Fig. 10). The identity of the physiological trigger for the μ1-to-μ1* change is not yet known (but see below).
Changes in μ1 structure.
μ1 folds into four domains in ISVPs. Domains I to III are mainly α-helical in nature and form the intertwined body of the μ1 trimer. Domain IV is a jelly roll β-barrel that constitutes the top domain of each subunit within the trimer (42). The δ region makes up portions of domains I to III and domain IV in its entirety. Our observation that δ is extensively protease sensitive in μ1* (Fig. 2 and 3) indicates that sequences within this region have undergone refolding during the μ1-to-μ1* transition. The results of partial-proteolysis experiments suggest that most or all of these refolding events occur in the lower domains (I to III) and not in the top domain (IV) (35; M. L. Nibert and K. Chandran, unpublished data). Domains I to III contain several hydrophobic sequences and long amphipathic α-helices that may become reorganized during μ1 conformational change and that may account for the hydrophobicity of μ1* as well as the putative μ1*-membrane interactions that lead to hemolysis. Most of these sequences are buried within the μ1 trimer, and partial or total disruption of intersubunit interactions may be necessary to permit their exposure.
Although the present study focused on changes in the central δ region of μ1, the N- and/or C-terminal regions of μ1 (μ1N and φ, respectively) may also rearrange as part of the μ1 conformational change. The highly apolar μ1N region (N-myristoyl group and 41 amino acids), a prime candidate for mediating μ1*-membrane interactions, is buried within the base of the μ1 trimer in ISVPs (42). Its exposure during the μ1-to-μ1* change and the subsequent insertion into a membrane seem to require not only the reorganization of sequences from the δ region that sandwich μ1N in the trimer but also autolytic cleavage at the μ1N-δ junction (42, 53; A. L. Odegard and M. L. Nibert, unpublished data). The nature of changes in μ1 tertiary and quaternary structure during the μ1-to-μ1* transition, as well as identities of the μ1* sequences that insert into membrane, are currently unknown but are topics of ongoing investigation in our laboratory.
Common features of nonenveloped-virus penetration proteins.
It has been proposed that externalization of a buried N-myristoylated peptide plays a role in membrane penetration by the nonenveloped picornaviruses and polyomaviruses (19, 34). With poliovirus, associated structural changes include externalization of the hydrophobic N terminus of capsid protein VP1 (26) and extensive “tectonic” movements throughout the capsid (5). These conformational changes in poliovirus are triggered by receptor binding to the capsid surface (30). In addition, cleavages of the poliovirus capsid polyprotein are required for these events (34), and the mature capsid is metastable (68). The rotavirus penetration protein (VP4) is not N-myristoylated but contains an internal hydrophobic sequence, similar to a fusion peptide, near which cleavage must occur before the protein is active (29, 49). The Ad penton base protein is neither N-myristoylated nor cleaved; moreover, conformational changes in this protein appear to occur but are not well characterized (58). Thus, although few if any features have been shown to be consistently shared by the penetration proteins of all nonenveloped viruses, some common features include N-myristoylation, priming by proteolytic cleavage(s), metastability of the primed proteins, and triggered conformational changes with exposure of the myristoyl group and/or other hydrophobic regions in association with membrane interaction (Fig. 10). The lack of uniformly consistent features may reflect the fact that several variations to the membrane penetration mechanism are employed by the different nonenveloped viruses.
Loss of σ1 from particles and putative conformational change in λ2.
Our finding that σ1 loss and μ1 conformational change are temporally correlated (Fig. 8) suggests that the two events are mechanistically linked. Since these proteins are thought not to contact each other in particles, σ1 loss may be coupled to μ1 conformational change via a third protein that interacts with both of them. Several observations implicate λ2 as this protein. First, σ1 binds at the center of the pentameric “shutter” atop the λ2 turret and μ1 subunits make extensive contacts with the walls of the λ2 turret around each fivefold axis (22). Second, when ISVPs are digested to cores in vitro, λ2 becomes capable of opening the pentameric shutter (22). Third, σ1 cannot stably bind to this more open conformer of the turret (K. Chandran and M. L. Nibert, unpublished data). We thus conclude that the λ2 turret opens either constitutively or transiently during the ISVP-to-ISVP* transition and that σ1 is lost from viral particles as a consequence of this change in the turret.
The recently determined structure of a portion of σ1 revealed a striking resemblance to the Ad fiber protein (17, 69). Both proteins form trimers comprising a fibrous tail and a globular head. Both tails are composed in part of a β-spiral motif, and both heads contain eight-stranded β-barrel domains. Ad fiber and σ1 are both bound to a pentameric structure at the fivefold axes of a viral particle, the penton base for the Ad fiber (64) and the λ2 turret for σ1 (22). Both proteins mediate attachment by binding to cell surface receptors (4, 6). Interestingly, in addition to their structural and functional similarities, the Ad fiber and σ1 may have similar fates during the entry-related disassembly of their respective particles. The Ad fiber dissociates from particles after viral attachment but before penetration (31, 48), and σ1 loss occurs in concert with activation of particles for membrane permeabilization (Fig. 8). We propose that σ1, like the Ad fiber, dissociates from particles after attachment to cells but before or during the membrane penetration step.
Triggering the ISVP-to-ISVP* transition.
Loss of the fiber protein from Ad virions is thought to require a conformational change in the penton base, triggered by its binding to the integrin coreceptor (31, 48). Our observation that T1L ISVPs undergo σ1 loss and the μ1-to-μ1* change only slowly at physiological temperature (Fig. 2 and 6 to 8) suggests that a triggering stimulus is also required to induce the ISVP-to-ISVP* transition within cells. One possibility is that the binding of σ1 to the receptor induces the opening of the λ2 turret and σ1 loss. The λ2 conformational change may then propagate to μ1 and trigger it to change conformation. In support of this idea, it has been suggested that reovirus attachment involves conformational changes in σ1 that propagate to other outer-capsid proteins (17, 25). A σ1-receptor interaction is unlikely to be the sole trigger for the ISVP-to-ISVP* transition, however, since viral particles can undergo this transition in the absence of the receptor (Fig. 3) or σ1 (16; K. Chandran and M. L. Nibert, unpublished data). We favor, instead, a model in which the μ1-to-μ1* change initiates the ISVP-to-ISVP* transition. The μ1 conformational change then induces constitutive or transient opening of the λ2 turret, which in turn results in σ1 release. Consistent with this alternative model is our previous observation that addition of μ1, but not σ1, to cores induces the reverse conformational change, namely, closure of the λ2 turret (16). The μ1-to-μ1* transition may be triggered during entry by K+ ions (57) (data not shown), which, like Cs+ ions, accelerate the transition in vitro (Fig. 3). It is also conceivable that direct interaction of μ1 with a host cell factor such as a lipid or protein is the inducing signal.
Contributions of μ1, σ1, and λ2 to membrane penetration and derepression of the viral transcriptases.
Our present findings indicate that μ1 plays the major role in membrane penetration but do not exclude participation of the other outer-capsid proteins. The following observations, however, suggest that involvement of σ1 and/or λ2 is unlikely to take the form of protein interactions with the membrane bilayer. (i) The hydrophobicity of σ1 does not increase in conjunction with its dissociation from particles (Fig. 7); (ii) σ1 affects neither the extent nor the rate of hemolysis (16; K. Chandran and M. L. Nibert, unpublished data); and (iii) cores, which lack μ1 and contain a more open conformer of the λ2 turret, do not lyse RBCs (Fig. 1) (16). An alternative possibility is that σ1 and λ2 participate not by interacting with the target membrane but by facilitating μ1*-membrane interactions. For example, the opening of the λ2 turret followed by release of σ1 during the ISVP-to-ISVP* transition may be necessary to allow μ1* to approach the membrane. In addition, loss of σ1 may be necessary to disengage infecting particles from receptors and permit their release into the cytoplasm after penetration. We speculate that release of Ad fibers from particles may fulfill similar functions in membrane penetration by that virus.
Derepression of the reovirus core-associated transcriptases coincides with the ISVP-to-ISVP* transition (Fig. 8). A role for the μ1-to-μ1* change in this process is certain, given previous evidence implicating M2 and μ1 as determinants of both transcriptase activation and repression (21, 23, 24). How does μ1* allow derepression of the viral transcription machines located within the inner capsid? In ISVPs, large loops that extend from each μ1 trimer contact the raised nodules composed of inner-capsid protein σ2 (22, 42, 56). σ2 may therefore relay a signal from μ1* to the transcription machines. Alternatively or in addition, μ1* may signal the transcriptases by inducing the λ2 turret to open. The λ2 conformational change may effect activation by propagating to the capsid interior via inner-capsid protein λ1 or σ2 or both. Furthermore, because viral mRNAs are thought to exit the particle via the central channel of the λ2 turret, both the opening of the turret and the loss of σ1 may be necessary for unimpeded mRNA egress and continued transcription (24; M. Yeager, S. Weiner, and K. M. Coombs, Abstr. 40th Meet. Biophys. Soc., abstr. 116, 1996). Our hypothesis that ISVP*s are, or closely resemble, primary transcriptase particles is consistent with previous observations that the latter resemble ISVPs, and not cores, in protein composition (Fig. 8) (38, 62). Unlike activation of the virus-membrane interaction, however, activation of the core-associated viral transcriptases requires only the μ1-to-μ1* change and not the continued presence of μ1* (37) (data not shown). Thus, μ1* may be removed from particles during or after membrane penetration.
Acknowledgments
We thank L. A. Breun and E. Freimont for excellent technical support. Many thanks also go to the other members of our laboratory, S. Liemann, and S. C. Harrison for helpful discussions and to M. Agosto, S. C. Harrison, K. S. Myers, A. L. Odegard, and J. S. L. Parker for critical reviews of the manuscript.
This work was supported by NIH grants R29 AI39533 and R01 AI46440 (to M.L.N.). K.C. was additionally supported by a predoctoral fellowship from the Howard Hughes Medical Institute and a Fields postdoctoral fellowship made available to the Department of Microbiology and Molecular Genetics through the generosity of Ruth Peedin Fields.
Footnotes
This paper is dedicated to the memory of Lakshmi Chandran.
REFERENCES
- 1.Baer, G. S., and T. S. Dermody. 1997. Mutations in reovirus outer-capsid protein σ3 selected during persistent infections of L cells confer resistance to protease inhibitor E64. J. Virol. 71:4921-4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baer, G. S., D. H. Ebert, C. J. Chung, A. H. Erickson, and T. S. Dermody. 1999. Mutant cells selected during persistent reovirus infection do not express mature cathepsin L and do not support reovirus disassembly. J. Virol. 73:9532-9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerjee, A. K., and A. J. Shatkin. 1970. Transcription in vitro by reovirus-associated ribonucleic acid-dependent polymerase. J. Virol. 6:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barton, E. S., J. C. Forrest, J. L. Connolly, J. D. Chappell, Y. Liu, F. J. Schnell, A. Nusrat, C. A. Parkos, and T. S. Dermody. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441-451. [DOI] [PubMed] [Google Scholar]
- 5.Belnap, D. M., D. J. Filman, B. L. Trus, N. Cheng, F. P. Booy, J. F. Conway, S. Curry, C. N. Hiremath, S. K. Tsang, A. C. Steven, and J. M. Hogle. 2000. Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J. Virol. 74:1342-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bewley, M. C., K. Springer, Y. B. Zhang, P. Freimuth, and J. M. Flanagan. 1999. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 286:1579-1583. [DOI] [PubMed] [Google Scholar]
- 7.Blumenthal, R., P. Seth, M. C. Willingham, and I. Pastan. 1986. pH dependent lysis of liposomes by adenovirus. Biochemistry 25:2231-2237. [DOI] [PubMed] [Google Scholar]
- 8.Bordier, C. 1981. Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem. 256:1604-1607. [PubMed] [Google Scholar]
- 9.Borsa, J., D. G. Long, T. P. Copps, M. D. Sargent, and J. D. Chapman. 1974. Reovirus transcriptase activation in vitro: further studies on the facilitation phenomenon. Intervirology 3:15-35. [DOI] [PubMed] [Google Scholar]
- 10.Borsa, J., B. D. Morash, M. D. Sargent, T. P. Copps, P. A. Lievaart, and J. G. Szekely. 1979. Two modes of entry of reovirus particles into L cells. J. Gen. Virol. 45:161-170. [DOI] [PubMed] [Google Scholar]
- 11.Borsa, J., M. D. Sargent, T. P. Copps, D. G. Long, and J. D. Chapman. 1973. Specific monovalent cation effects on modification of reovirus infectivity by chymotrypsin digestion in vitro. J. Virol. 11:1017-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borsa, J., M. D. Sargent, D. D. Ewing, and M. Einspenner. 1982. Perturbation of the switch-on of transcriptase activity in intermediate subviral particles from reovirus. J. Cell. Physiol. 112:10-18. [DOI] [PubMed] [Google Scholar]
- 13.Borsa, J., M. D. Sargent, P. A. Lievaart, and T. P. Copps. 1981. Reovirus: evidence for a second step in the intracellular uncoating and transcriptase activation process. Virology 111:191-200. [DOI] [PubMed] [Google Scholar]
- 14.Carr, C. M., C. Chaudhry, and P. S. Kim. 1997. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. USA 94:14306-14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandran, K., and M. L. Nibert. 1998. Protease cleavage of reovirus capsid protein μ1/μ1C is blocked by alkyl sulfate detergents, yielding a new type of infectious subvirion particle. J. Virol. 72:467-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandran, K., S. B. Walker, Y. Chen, C. M. Contreras, L. A. Schiff, T. S. Baker, and M. L. Nibert. 1999. In vitro recoating of reovirus cores with baculovirus-expressed outer-capsid proteins μ1 and σ3. J. Virol. 73:3941-3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chappell, J. D., A. E. Prota, T. S. Dermody, and T. Stehle. 2002. Crystal structure of reovirus attachment protein σ1 reveals evolutionary relationship to adenovirus fiber. EMBO J. 21:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen, J., S. A. Wharton, W. Weissenhorn, L. J. Calder, F. M. Hughson, J. J. Skehel, and D. C. Wiley. 1995. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in E. coli into the low-pH-induced conformation. Proc. Natl. Acad. Sci. USA 92:12205-12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen, X. S., T. Stehle, and S. C. Harrison. 1998. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J. 17:3233-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coombs, K. M. 1998. Stoichiometry of reovirus structural proteins in virus, ISVP, and core particles. Virology 243:218-228. [DOI] [PubMed] [Google Scholar]
- 21.Drayna, D., and B. N. Fields. 1982. Activation and characterization of the reovirus transcriptase: genetic analysis. J. Virol. 41:110-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dryden, K. A., G. Wang, M. Yeager, M. L. Nibert, K. M. Coombs, D. B. Furlong, B. N. Fields, and T. S. Baker. 1993. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J. Cell Biol. 122:1023-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ewing, D. D., M. D. Sargent, and J. Borsa. 1985. Switch-on of transcriptase function in reovirus: analysis of polypeptide changes using 2-D gels. Virology 144:448-456. [DOI] [PubMed] [Google Scholar]
- 24.Farsetta, D. L., K. Chandran, and M. L. Nibert. 2000. Transcriptional activities of reovirus RNA polymerase in recoated cores. Initiation and elongation are regulated by separate mechanisms. J. Biol. Chem. 275:39693-39701. [DOI] [PubMed] [Google Scholar]
- 25.Fernandes, J., D. Tang, G. Leone, and P. W. Lee. 1994. Binding of reovirus to receptor leads to conformational changes in viral capsid proteins that are reversible upon virus detachment. J. Biol. Chem. 269:17043-17047. [PubMed] [Google Scholar]
- 26.Fricks, C. E., and J. M. Hogle. 1990. Cell-induced conformational change in poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 64:1934-1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furlong, D. B., M. L. Nibert, and B. N. Fields. 1988. σ1 protein of mammalian reoviruses extends from the surfaces of viral particles. J. Virol. 62:246-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaspar, L. P., A. C. Silva, A. M. Gomes, M. S. Freitas, A. P. Ano Bom, W. D. Schwarcz, J. Mestecky, M. J. Novak, D. Foguel, and J. L. Silva. 2001. Hydrostatic pressure induces the fusion-active state of enveloped viruses. J. Biol. Chem. 277:8433-8439. [DOI] [PubMed] [Google Scholar]
- 29.Gilbert, J. M., and H. B. Greenberg. 1997. Virus-like particle-induced fusion from without in tissue culture cells: role of outer-layer proteins VP4 and VP7. J. Virol 71:4555-4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomez Yafal, A., G. Kaplan, V. R. Racaniello, and J. M. Hogle. 1993. Characterization of poliovirus conformational alteration mediated by soluble cell receptors. Virology 197:501-505. [DOI] [PubMed] [Google Scholar]
- 31.Greber, U. F. 1998. Virus assembly and disassembly: the adenovirus cysteine protease as a trigger factor. Rev. Med. Virol. 8:213-222. [DOI] [PubMed] [Google Scholar]
- 32.Guirakhoo, F., F. X. Heinz, C. W. Mandl, H. Holzmann, and C. Kunz. 1991. Fusion activity of flaviviruses: comparison of mature and immature (prM-containing) tick-borne encephalitis virions. J. Gen. Virol. 72:1323-1329. [DOI] [PubMed] [Google Scholar]
- 33.Heinz, F. X., and S. L. Allison. 2001. The machinery for flavivirus fusion with host cell membranes. Curr. Opin. Microbiol. 4:450-455. [DOI] [PubMed] [Google Scholar]
- 34.Hindiyeh, M., Q. H. Li, R. Basavappa, J. M. Hogle, and M. Chow. 1999. Poliovirus mutants at histidine 195 of VP2 do not cleave VP0 into VP2 and VP4. J. Virol. 73:9072-9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hooper, J. W., and B. N. Fields. 1996. Role of the μ1 protein in reovirus stability and capacity to cause chromium release from host cells. J. Virol. 70:459-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jané-Valbuena, J., M. L. Nibert, S. M. Spencer, S. B. Walker, T. S. Baker, Y. Chen, V. E. Centonze, and L. A. Schiff. 1999. Reovirus virion-like particles obtained by recoating infectious subvirion particles with baculovirus-expressed σ3 protein: an approach for analyzing σ3 functions during virus entry. J. Virol. 73:2963-2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joklik, W. K. 1972. Studies on the effect of chymotrypsin on reovirions. Virology 49:700-715. [DOI] [PubMed] [Google Scholar]
- 38.Joklik, W. K., and M. R. Roner. 1995. What reassorts when reovirus genome segments reassort? J. Biol. Chem. 270:4181-4184. [DOI] [PubMed] [Google Scholar]
- 39.Klenk, H. D., R. Rott, M. Orlich, and J. Blodorn. 1975. Activation of influenza A viruses by trypsin treatment. Virology 68:426-439. [DOI] [PubMed] [Google Scholar]
- 40.Kothandaraman, S., M. C. Hebert, R. T. Raines, and M. L. Nibert. 1998. No role for pepstatin-A-sensitive acidic proteinases in reovirus infections of L or MDCK cells. Virology 251:264-272. [DOI] [PubMed] [Google Scholar]
- 41.Lee, P. W. K., E. C. Hayes, and W. K. Joklik. 1981. Protein σ1 is the reovirus cell attachment protein. Virology 108:156-163. [DOI] [PubMed] [Google Scholar]
- 42.Liemann, S., K. Chandran, T. S. Baker, M. L. Nibert, and S. C. Harrison. 2002. Structure of the reovirus membrane-penetration protein, μ1, in a complex with is protector protein, σ3. Cell 108:283-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lucia-Jandris, P., J. W. Hooper, and B. N. Fields. 1993. Reovirus M2 gene is associated with chromium release from mouse L cells. J. Virol. 67:5339-5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luongo, C. L., K. A. Dryden, D. L. Farsetta, R. L. Margraf, T. F. Severson, N. H. Olson, B. N. Fields, T. S. Baker, and M. L. Nibert. 1997. Localization of a C-terminal region of λ2 protein in reovirus cores. J. Virol. 71:8035-8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luongo, C. L., K. M. Reinisch, S. C. Harrison, and M. L. Nibert. 2000. Identification of the guanylyltransferase region and active site in reovirus mRNA capping protein λ2. J. Biol. Chem. 275:2804-2810. [DOI] [PubMed] [Google Scholar]
- 46.McClure, W. O., and G. M. Edelman. 1966. Fluorescent probes for conformational states of proteins. I. Mechanism of fluorescence of 2-p-toluidinylnaphthalene-6-sulfonate, a hydrophobic probe. Biochemistry 5:1908-1919. [DOI] [PubMed] [Google Scholar]
- 47.Middleton, J. K., T. F. Severson, K. Chandran, A. L. Gillian, J. Yin, and M. L. Nibert. 2002. Thermostability of reovirus disassembly intermediates (ISVPs) correlates with genetic, biochemical, and thermodynamic properties of major surface protein μ1. J. Virol. 76:1051-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakano, M. Y. B., K. M. Suomalainen, R. P. Stidwill, and U. F. Greber. 2000. The first step of adenovirus type 2 disassembly occurs at the cell surface, independently of endocytosis and escape to the cytosol. J. Virol. 74:7085-7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nandi, P., A. Charpilienne, and J. Cohen. 1992. Interaction of rotavirus particles with liposomes. J. Virol. 66:3363-3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nibert, M. L. 1998. Structure of mammalian orthoreovirus particles. Curr. Top. Microbiol. Immunol. 238(Reovirus i):1-30. [DOI] [PubMed] [Google Scholar]
- 51.Nibert, M. L., and B. N. Fields. 1992. A carboxy-terminal fragment of protein μ1/μ1C is present in infectious subvirion particles of mammalian reoviruses and is proposed to have a role in penetration. J. Virol. 66:6408-6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nibert, M. L., and L. A. Schiff. 2001. Reoviruses and their replication, p. 1679-1728. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 53.Nibert, M. L., L. A. Schiff, and B. N. Fields. 1991. Mammalian reoviruses contain a myristoylated structural protein. J. Virol. 65:1960-1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Noble, S., and M. L. Nibert. 1997. Characterization of an ATPase activity in reovirus cores and its genetic association with core-shell protein λ1. J. Virol. 71:2182-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parker, M. W., A. D. Tucker, D. Tsernoglou, and F. Pattus. 1990. Insights into membrane insertion based on studies of colicins. Trends Biochem. Sci. 15:126-129. [DOI] [PubMed] [Google Scholar]
- 56.Reinisch, K. M., M. L. Nibert, and S. C. Harrison. 2000. Structure of the reovirus core at 3.6 Å resolution. Nature 404:960-967. [DOI] [PubMed] [Google Scholar]
- 57.Sargent, M. D., D. G. Long, and J. Borsa. 1977. Functional analysis of the interactions between reovirus particles and various proteases in vitro. Virology 78:354-358. [DOI] [PubMed] [Google Scholar]
- 58.Seth, P., M. C. Willingham, and I. Pastan. 1985. Binding of adenovirus and its external proteins to Triton X-114. J. Biol. Chem. 260:14431-14434. [PubMed] [Google Scholar]
- 59.Shai, Y. 1999. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1462:55-70. [DOI] [PubMed] [Google Scholar]
- 60.Shatkin, A. J., and J. D. Sipe. 1968. RNA polymerase activity in purified reoviruses. Proc. Natl. Acad. Sci. USA 61:1462-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shepard, D. A., J. G. Ehnstrom, and L. A. Schiff. 1995. Association of reovirus outer capsid proteins σ3 and μ1 causes a conformational change that renders σ3 protease sensitive. J. Virol. 69:8180-8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Silverstein, S. C., C. Astell, D. H. Levin, M. Schonberg, and G. Acs. 1972. The mechanisms of reovirus uncoating and gene activation in vivo. Virology 47:797-806. [DOI] [PubMed] [Google Scholar]
- 63.Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531-569. [DOI] [PubMed] [Google Scholar]
- 64.Stewart, P. L., R. M. Burnett, M. Cyrklaff, and S. D. Fuller. 1991. Image reconstruction reveals the complex molecular organization of adenovirus. Cell 67:145-154. [DOI] [PubMed] [Google Scholar]
- 65.Stiasny, K., S. L. Allison, C. W. Mandl, and F. X. Heinz. 2001. Role of metastability and acidic pH in membrane fusion by tick-borne encephalitis virus. J. Virol. 75:7392-7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sturzenbecker, L. J., M. Nibert, D. Furlong, and B. N. Fields. 1987. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J. Virol. 61:2351-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tosteson, M. T., M. L. Nibert, and B. N. Fields. 1993. Ion channels induced in lipid bilayers by subvirion particles of the nonenveloped mammalian reoviruses. Proc. Natl. Acad. Sci. USA 90:10549-10552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsang, S. K., P. Danthi, M. Chow, and J. M. Hogle. 2000. Stabilization of poliovirus by capsid-binding antiviral drugs is due to entropic effects. J. Mol. Biol. 296:335-340. [DOI] [PubMed] [Google Scholar]
- 69.van Raaij, M. J., A. Mitraki, G. Lavigne, and S. Cusack. 1999. A triple β-spiral in the adenovirus fiber shaft reveals a new structural motif for a fibrous protein. Nature 401:935-938. [DOI] [PubMed] [Google Scholar]
- 70.Virgin, H. W., IV, M. A. Mann, B. N. Fields, and K. L. Tyler. 1991. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J. Virol. 65:6772-6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Werck-Reichhart, D., I. Benveniste, H. Teutsch, F. Durst, and B. Gabriac. 1991. Glycerol allows low-temperature phase separation of membrane proteins solubilized in Triton X-114: application to the purification of plant cytochromes P-450 and b5. Anal. Biochem. 197:125-131. [DOI] [PubMed] [Google Scholar]