

Abstract
The H subunit of the yeast V-ATPase is an extended structure with two relatively independent domains, an N-terminal domain consisting of amino acids 1–348 and a C-terminal domain consisting of amino acids 352–478 (Sagermann, M., Stevens, T. H., and Matthews, B. W. (2001) Proc. Natl. Acad. Sci. U. S. A. 98, 7134–7139). We have expressed these two domains independently and together in a yeast strain lacking the H subunit (vma13Δmutant). The N-terminal domain partially complements the growth defects of the mutant and supports ~25% of the wild-type Mg2+-dependent ATPase activity in isolated vacuolar vesicles, but surprisingly, this activity is both largely concanamycin-insensitive and uncoupled from proton transport. The C-terminal domain does not complement the growth defects, and supports no ATP hydrolysis or proton transport, even though it is recruited to the vacuolar membrane. Expression of both domains in a vma13Δstrain gives better complementation than either fragment alone and results in higher concanamycin-sensitive ATPase activity and ATP-driven proton pumping than the N-terminal domain alone. Thus, the two domains make complementary contributions to structural and functional coupling of the peripheral V1 and membrane Vo sectors of the V-ATPase, but this coupling does not require that they be joined covalently. The N-terminal domain alone is sufficient for activation of ATP hydrolysis in V1, but the C-terminal domain is essential for proper communication between the V1 and Vo sectors.
V-ATPases2 are ATP-driven proton pumps responsible for acidification of intracellular organelles in all eukaryotic cells and for proton transport across the plasma membrane in certain cells (1, 2). V-ATPases are comprised of a peripheral complex containing the sites for ATP hydrolysis, the V1 sector, attached to a membrane complex containing the proton pore, the Vo sector (2). The yeast V-ATPase has proved to be an excellent model system for eukaryotic V-ATPases. In yeast, eight subunits, designated A, B, C, D, E, F, G, and H, make up the V1 sector, and at least six subunits designated a, c, c′, c″, d, and e subunits make up the Vo sector (3, 4). All of these subunits have homologues in higher eukaryotes, and in many cases, these homologues have been found to functionally substitute for each other.
V-ATPases share a common evolutionary ancestor with F1Fo-ATP synthases (5). The core of the catalytic machinery, specifically the ATP hydrolyzing A subunit, the B subunit, and proteolipid subunits (c, c′, and c″), show significant homology with ATP synthase subunits. However, V-ATPases are dedicated proton pumps, while ATP synthases operate primarily in the direction of ATP synthesis in vivo. The catalytic mechanism is fundamentally similar between these two types of enzymes and involves subunit rotation (6, 7). In ATP synthases, two stalk structures connect the peripheral headgroup containing the ATP binding subunits and the integral membrane complex containing the proton pore. These two structures act as a “rotor,” which is driven to move sequentially between the catalytic sites by proton movement through the pore, and as a “stator,” which fixes part of the pore to the catalytic headgroup, allowing for relative rotation of the rotor with respect to the catalytic sites (8). Although there must be a stator and rotor for V-ATPases to accomplish rotational catalysis, there is surprisingly little homology between the stalk subunits of the F- and V-type ATPases. This has made assignment of structural and functional roles to the 6–8 V-ATPase stalk subunits difficult. Some consensus is emerging for placement of the d, D, and F subunits in the central, rotor stalk (9), and the E, G, C, and H subunits in the peripheral, stator stalks (10–14), but some aspects of these assignments are still disputed (15, 16). Portions of the V1 A and Vo a subunits have also been implicated as part of the stator stalk (17, 18).
Regulation of V-ATPases and the F1Fo-ATP synthase also appears to be very different, consistent with their distinct cellular localizations and functions. One unique feature of V-ATPases is reversible disassembly in response to changes in growth conditions (19, 20). Intact V-ATPases exist in dynamic equilibrium with cytosolic V1 sectors and membrane-bound Vo sectors in the cell. In yeast, this equilibrium can be perturbed by changes in extracellular carbon source; assembled V-ATPase complexes are rapidly and reversibly disassembled into free V1 and Vo sectors in response to a brief glucose deprivation (21). Disassembly both inactivates the physiologically relevant MgATPase activity in V1 sectors and prevents proton transport through Vo. This mode of regulation, which has been observed only in eukaryotes, imposes additional constraints on the stator of V-ATPases, which must balance the stability needed for productive rotational catalysis with the regulated instability implied by reversible disassembly.
V-ATPase subunit H, which is found in all eukaryotic V-ATPases but not in the archaebacterial V-ATPases, has a number of distinctive features. It is the only subunit that is not necessary for assembly of the full V-ATPase (22, 23). In a yeast vma13Δstrain, which lacks the H subunit, V1-Vo complexes are assembled and transported to the vacuole (22). These complexes are not active, however, leading to the conclusion that the H subunit is an activator of the fully assembled V-ATPase complex. The H subunit remains with the V1 sector upon disassembly from Vo, and in contrast to its role in the intact complex, acts as an inhibitor of ATP hydrolysis in the free V1 sectors (24). Taken together, these results suggest the H subunit has a critical function both in sensing attachment between the V1 and Vo sectors and allowing ATP-driven proton transport and in silencing ATP hydrolysis in V1 that has detached from Vo. These contrasting functions indicate that the H subunit is central to establishment of structural and functional coupling of V1 with Vo.
The yeast H subunit was the first V-ATPase subunit to be crystallized (25). The crystal structure revealed that the H subunit was a rather elongated protein that consists of two structurally independent domains joined by a four amino acid linker. In this work, we have expressed those two domains independently and together to assess their roles in establishing a functional connection between the V1 and Vo sectors and in communicating conformational information between the two sectors. Surprisingly, we find that one of the two domains, consisting of the N-terminal 348 amino acids of the H subunit, is partially functional in vivo. Closer characterization reveals a number of enzymatic defects, however, and the implications of these characteristics for H subunit function are discussed.
EXPERIMENTAL PROCEDURES
Materials and Media
Concanamycin A was purchased from Wako Biochemicals, and Zymolyase-100T was purchased from MP Biomedicals, Inc. G418 was obtained from Sigma. Yeast genomic DNA was obtained using the Masterpure yeast DNA preparation kit (Epicenter). Restriction enzymes were from New England Biolabs, and LA-Taq (Panvera) was used for PCR unless otherwise indicated. Yeast medium were prepared as described previously (26). Monoclonal antibody 9E10, against the Myc epitope, was obtained from Roche Applied Science.
Strains and Plasmids
Wild-type yeast strain SF838–5A and SF838–5A vma13Δ::LEU2 have been described previously (24). To construct a congenic strain containing a vma13Δ::kanMX deletion, the vma13Δ::kanMX allele was PCR-amplified from strain BY4741 vma13Δ::kanMX from the yeast deletion mutant array purchased from Research Genetics. This strain contains a precise deletion of the VMA13 open reading frame with the kanMX marker flanked by a set of marker sequences (27). Genomic DNA was prepared from this strain, and the vma13Δmutant allele was amplified with oligonucleotides VMA13 Δ600 (5′-GGTTACAGGTATCATGTGTGTTTCGTTTG and VMA13–200 (5′-GCATTACCAATCACGCACGCACGCAGTC-GG) to obtain a product containing ~600 bp of VMA13 upstream sequence and 200 bp of VMA13 downstream sequence. The PCR product was used directly to transform wild-type strain SF838–5A, and transformants were selected by growth on YEPD (yeast extract/peptone/dextrose medium) containing 200 μg/ml G418. Replacement of the wild-type VMA13 with the mutant allele was confirmed by PCR from genomic DNA isolated from transformants. The resulting strain, SF838–5A vma13Δ::kanMX, showed the full range of VmaΔgrowth phenotypes, as did the BY4741 vma13Δ::kanMX strain. The BY4741 vma13Δ::kanMX strain was also transformed with the wild-type and mutant plasmids and gave comparable results.
Construction and characterization of the wild-type VMA13 plasmid containing an N-terminal Myc tag was described previously (24). This plasmid was used as template for construction of the VMA13-NT and VMA13-CT mutant plasmids using the QuikChange XL site-directed mutagenesis kit (Stratagene). The VMA13-NT plasmid (which contains a deletion of amino acids 349–478 of Vma13p) was constructed using the following primers: GGAAATCCTAGAAAACTAAAGATATAGAAGACCG (Δ349–478aa) and CGGTCTTCTATATCTTTAGTTTTCTAGGATTTCC (Δ349–478aa rc). The VMA13-CT plasmid (which contains a deletion of amino acids 2–352, but includes the N-terminal Myc-tag) was constructed using the following primers: CTGAAGAAGACTTGTTGACCTCCTTCGATG (Δ2–352aa) and CATCGAAGGAGGTCAACAAGTCTTCTTCAG (Δ2–352aa rc). Following mutagenesis, the remaining VMA13 open reading frame was sequenced to confirm incorporation of the deletion mutations and absence of any additional mutations. To allow co-transformation of the NT- and CT-containing plasmids, the VMA13-NT insert was sub-cloned into pRS315 by removing the insert with SacI, and cloning into pRS316 cut with the same enzyme. All plasmids were introduced into the SF838–5A vma13Δ::kanMX strain using an overnight lithium acetate transformation protocol (28), and transformants were selected on supplemented minimal medium lacking uracil (SD-uracil) for pRS316-based plasmids, lacking leucine (SD-leucine) for pRS315-based plasmids, or lacking both uracil and leucine (for co-transformants containing both NT and CT plasmids).
Biochemical Methods
Vacuoles were obtained as described previously, except that strains containing plasmids were grown in supplemented minimal medium to maintain the plasmid prior to lysis, and the cells were grown to an A600 of ~1.0 before harvesting. Yeast whole cell lysates and vacuolar vesicles were prepared for SDS-polyacrylamide gel electrophoresis and immunoblotting as described (29).
ATP hydrolysis rates were assessed using a coupled enzyme assay at 37 °C, and concanamycin inhibition was measured by adding concanamycin A directly to the assay mixture to a final concentration of 100 nm. (Concanamycin A was prepared as a 100 μM stock in dimethyl sulfoxide. Addition of a comparable concentration of dimethyl sulfoxide to the assay mix did not affect activity.) Inhibition by N-ethyl maleimide (NEM) was determined by mixing ~5 μg of vacuolar vesicles with 50 μM N-ethyl maleimide and incubating for 10 min on ice. This mixture was then added to the ATPase assay mixture and hydrolysis rates were determined. Proton pumping was measured on a SPEX Fluorolog-3–21 fluorometer by the ACMA quenching assay as described by Shao and Forgac (17). Vacuolar vesicles (10 μg) were added to 3 ml of transport buffer containing 1 μM ACMA and stirred. Pumping was initiated by addition of a solution containing ATP and MgSO4 to give a final concentration of 0.5 mm ATP and 1.0 mm MgSO4 in the assay cuvette. Fluorescence intensity was monitored continuously at an excitation wavelength of 410 nm and an emission wavelength of 490 nm. Where indicated, concanamycin A was added to a final concentration of 100 nm. Coupling efficiencies are measured by dividing the ATP hydrolysis rate (in the absence of concanamycin A, expressed at μmol/min/mg protein), by the change in fluorescence intensity during the first 20 s following addition of MgATP. The fluorescence intensity was corrected for changes in fluorescence arising solely from MgATP addition by subtracting the values obtained when concanamycin A was present throughout the assay.
Quinacrine Staining of Whole Cells
Quinacrine labeling was performed as described (30), and labeled cells were observed within 10 min of labeling. Concanamycin A was included to a 500 nm final concentration after the quinacrine labeling step where indicated. Quinacrine fluorescence was observed on a Zeiss Axioskop II fluorescence microscope equipped with a Hamamatsu ORCA CCD camera.
RESULTS
The x-ray crystal structure of isolated yeast subunit H (25) indicates that this subunit has an elongated structure consisting of two major domains (Fig. 1). The N-terminal domain is composed of amino acids 1–348, and consists of a series of five HEAT repeats. The C-terminal domain consists of amino acids 353–478. In the crystal structure, these two domains are joined by a four-amino acid linker (amino acids 349–352) but appear to be structurally rather independent. We engineered constructs to express the N-terminal domain (VMA13-NT) and C-terminal domain (VMA13-CT) independently by deleting amino acids 349–478 or amino acids 2–352, respectively. The deletions were introduced into a low copy plasmid that places a Myc epitope tag at the N terminus of the H subunit (24), so that both deletion constructs are also N-terminally Myc-tagged.
Figure 1. Ribbon diagram of the yeast H subunit (Vma13p) domain structure.
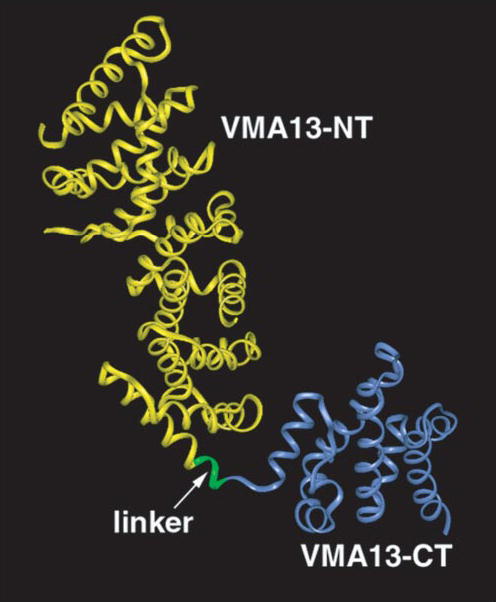
Coordinates deposited as PDB 1HO8 (25) were visualized using Insight II (Accelrys). Backbone structure for amino acids 1–348 (corresponding to the VMA13-NT domain) is shown in yellow, and the backbone structure of amino acids 352– 478 (VMA13-CT domain) are shown in blue. These two amino acids are joined by a four-amino acid linker in the intact subunit and this linker is shown in green.
To determine whether the two domains would be stably expressed in yeast, the VMA13-NT and VMA13-CT constructs were transformed individually into a vma13Δ strain, which lacks any other copy of subunit H, and then whole cell lysates were prepared from the transformants. The Myc-tagged wild-type H subunit has a relative molecular mass of ~54 kDa, and the NT and CT domains have the expected relative molecular masses of ~40 and 14 kDa (data not shown). We also inserted the deletion constructs into low copy plasmids with different nutritional markers and co-transformed the vma13Δ strain with both plasmids. The VMA13-NT + CT-containing strain contains Myc-tagged proteins of both 40 and 14 kDa. The relative level of the two proteins in the VMA13-NT + CT strain was somewhat variable because they are coexpressed from different plasmids and there may be some variation in the relative copy numbers of the plasmids.
We next asked whether the individual domains were able to support function of the V-ATPase. Loss of V-ATPase activity gives an easily measurable growth phenotype. Mutants lacking all V-ATPase activity, like the vma13Δ mutant, are able to grow at pH 5, but show little or no growth at an extracellular pH > 7 or high extracellular calcium concentrations (31). As shown in Fig. 2, the Myc-tagged wild-type VMA13 plasmid restores growth to the vma13Δstrain at both pH 7.5 and in pH 7.5 medium containing 60 mm CaCl2, as demonstrated by growth as a patch at the highest density of cells (left) on all three plates and as well defined colonies at the lowest density (right). The vma13Δstrain containing the VMA13-NT plasmid showed some growth at pH 7.5, but very little growth on pH 7.5 + CaCl2 plates. (Higher levels of V-ATPase activity are required for growth at pH 7.5 when extra calcium is added (32).) The strain containing VMA13-CT did not grow under either of these conditions, suggesting there was little or no complementation of the Vma−growth phenotype. Interestingly, the strain containing both VMA13-NT and VMA13-CT grew better at pH 7.5 than either strain containing the individual fragments. (Note that the two spots of lower cell density for the VMA13-NT + CT strain have much better growth than the VMA13-NT strain at pH 7.5. Neither the NT nor the NT + CT strain formed well-defined colonies at even the highest density plating on the pH 7.5 + CaCl2 plate.) This suggests that the NT and CT fragments of the H subunit are able to functionally complement each other, at least to some extent, under conditions where they are not joined covalently.
Figure 2. Expression of VMA13 NT and CT domains in yeast and complementation of growth phenotypes in a vma13Δ mutant.
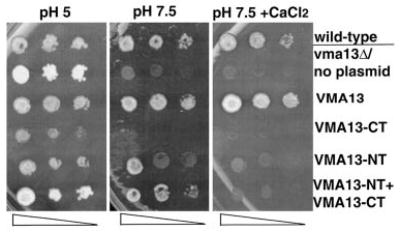
Wild-type yeast cells (top row) and vma13Δ yeast cells carrying no plasmid or the indicated VMA13 fragments on plasmids were grown to log phase, subjected to serial 10-fold dilutions, and then transferred to a YEPD plate buffered pH 5 (pH 5), YEPD buffered to pH 7.5 (pH 7.5), or YEPD buffered to pH 7.5 containing 60 mm CaCl2. The growth of the strains was compared after 2– 4 days at 30 °C.
To examine the properties of V-ATPases containing the various fragments of subunit H in more detail, we isolated vacuolar vesicles from all of the strains. As shown by an immunoblot probed with the anti-Myc antibody in Fig. 3A, all of the Myc-tagged H subunit fragments were isolated with the vacuolar vesicles, suggesting that they retained enough binding to other V-ATPase subunits to keep them with the membranes during isolation. The levels of several other V-ATPase subunits in the isolated vacuoles were also determined by immunoblot. Both the membrane-bound Vo subunit a and the peripheral V1 subunits A, B, C, and E were present in vacuoles isolated from the all of the strains (Fig. 3B). There was a considerable loss of all of the V1 subunits from the VMA13-CT. There is somewhat less of the V1 subunits, particularly the C subunit, in the VMA13-NT-containing strain. There was better retention of all of the V1 subunits on the vacuolar membrane when both the VMA13-NT and VMA13-CT fragments were present.
Figure 3. Levels of V-ATPase subunits in vacuoles containing the VMA13 fragments.
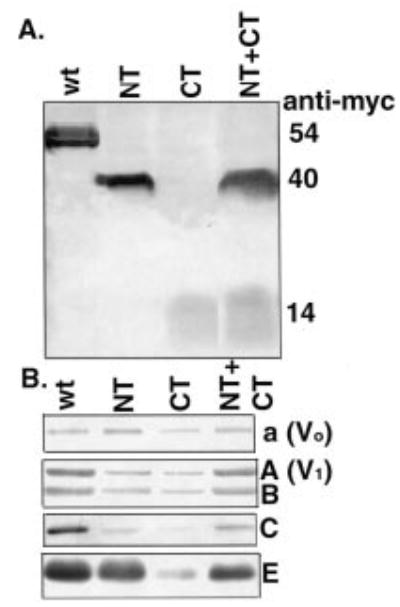
Vacuolar vesicles were isolated from vma13δ mutants expressing plasmid-borne copies of the intact H subunit gene (wt) or the indicated H subunit fragments (NT, CT, and NT+CT). Vacuolar proteins was solubilized, separated by SDS-PAGE, and transferred to nitrocellulose before blotting with antibodies specific for the Myc epitope on the wild-type and mutant H subunits and for different V-ATPase subunits. A, immunoblot of vacuolar vesicle protein (15 μg) probed with anti-Myc antibody. B, immunoblot of vacuolar vesicle protein probed with antibodies to Vo subunit a, V1 subunit A, V1 subunit B, V1 subunit C, and V1 subunit E. 5 μg of protein was loaded for visualization of subunits A, B, and E, and 15 μg was loaded for visualization of subunits a and C.
TABLE ONE shows the total ATPase activity measured in each of the vacuolar vesicle preparations. The VMA13-CT construct supports very little ATPase activity, even though there was some catalytic subunit A present, but the VMA13-NT vacuolar vesicles contained 25.4% of the total ATPase activity found in vesicles with the full-length VMA13. The VMA13-NT + CT vesicles had almost twice as much ATPase activity as the VMA13-NT vesicles. The ATPase activity in the VMA13-NT vesicles was also notably less stable than that of wild-type vesicles. Vesicles from cells containing the wild-type VMA13 retain most of the ATPase activity after an overnight incubation on ice or through extended freezing. The VMA13-NT vesicles rapidly lost activity under these conditions, and the VMA13-NT+ CT vesicles showed an intermediate level of stability. We generally assess V-ATPase specific activity by measuring the proportion of the total ATPase activity that is sensitive to 100 nm concanamycin A, and as shown in TABLE ONE, 86.4% of the ATPase activity in the wild-type vacuolar vesicles was inhibited by concanamycin A. However, we found that vacuolar vesicles from both the VMA13-NT and VMA13-NT + CT cells had a much higher level of concanamycin A-insensitive ATPase activity than the wild-type vesicles, with only 34% inhibition in VMA13-NT and 49% inhibition in VMA13-NT + CT (TABLE ONE).
TABLE ONE.
ATPase activity in vacuolar vesicles
| Strain | Total ATPase specific activity | Wild-type activity | Average inhibition by concanamycin A | Average inhibition by NEM |
|---|---|---|---|---|
| μmol/min/mg | % | % | % | |
| vma13Δ/VMA13 | 1.14 ± 0.13 (n = 8) | 100 | 86.4 ± 4.3 (n = 8) | 80 ± 4.8 (n = 3) |
| vma13Δ/VMA13-NT | 0.29 ± 0.04 (n = 9) | 25.4 | 33.5 ± 4.6 (n = 9) | 66.8 ± 4.5 (n = 4) |
| vma13Δ/VMA13-CT | 0.02 ± 0.01 (n = 3) | 1.5 | 9.8 ± 3.5 (n = 3) | N/Aa |
| vma13Δ/VMA13-NT 3 VMA13-CT | 0.54 ± 0.09 (n = 6) | 47.3 | 48.7 ± 11.3 (n = 6) | 72.3 ± 2.1 (n = 3) |
Not applicable.
The concanamycin A-insensitive ATPase activity in wild-type vacuolar vesicles is generally attributed to enzymes other than V-ATPases, in part because it is also insensitive to other specific inhibitors of V-ATPase activity. (The percentage of the ATPase activity in wild-type vesicles that is insensitive to concanamycin A ranges from 80–95%, and its source is unknown.) Contamination of the vacuolar vesicles from the VMA13-NT mutant with other membranes might result in a lower V-ATPase specific activity and account for concanamycin insensitivity, but loss of concanamycin sensitivity in the V-ATPase itself could also account for these results. Therefore, we compared the inhibition by 50 μM NEM with that by 100 nm concanamycin A. At this concentration, NEM is also a specific inhibitor of V-ATPase, but it acts by a completely different mechanism from concanamycin A. Whereas concanamycin A inhibits by binding to the proteolipid subunits in the Vo sector of the V-ATPase (33–35), NEM inhibits by binding to a conserved cysteine that is located at the catalytic sites in the V1 sector (36). As shown in TABLE ONE, the ATPase activity in vacuoles from cells containing wild-type VMA13 is inhibited to nearly same extent by concanamycin A and NEM, with slightly less inhibition by NEM on average. In contrast, ATPase activity in the VMA13-NT and VMA13-NT + CT vesicles was much more sensitive to NEM than to concanamycin A. These results indicate that the V-ATPase has ATPase activity in the VMA13-NT containing vesicles, but has lost much of its sensitivity to concanamycin A.
We also assayed the proton-pumping capability of the V-ATPase in the wild-type and mutant vacuoles by ACMA fluorescence quenching. As shown in Fig. 4A, vesicles containing the full-length H subunit show quenching of ACMA fluorescence upon addition of MgATP. This quenching is reversed upon addition of 100 nm concanamycin A, and addition of 100 nm concanamycin A before MgATP prevented almost all of the quenching, other than the drop that occurs immediately upon MgATP addition. There is a small but detectable quenching of ACMA fluorescence in the VMA13-NT + CT vesicles as well, and this quenching is both reversible by addition of concanamycin A and prevented by addition of concanamycin A before MgATP (Fig. 4B). The VMA13-NT vacuoles (Fig. 4C) show very little MgATP-dependent quenching, however, and there is only a small difference in the extent of quenching in the presence or absence of concanamycin A. The VMA13-CT vacuoles showed no pumping at all. By calculating the ratio between the initial rate of proton pumping (in the first 20 s following the drop because of MgATP addition) and the ATPase specific activity, we can estimate a relative coupling ratio for the strains that exhibited both V-ATPase and proton pumping activity. Based on this calculation, the coupling ratio for the full-length Vma13p vacuoles is 325, the coupling ratio of the VMA13-NT + CT vacuoles is 197, and the initial rate of pumping in the VMA13-NT and VMA13-CT vacuoles was too low to allow measurement of a coupling ratio. These results indicate that the V-ATPase activity seen in the VMA13-NT vacuolar vesicles is very poorly coupled to proton pumping, but that some coupling efficiency can be recovered by addition of the CT domain.
Figure 4. Proton pumping in vacuolar vesicle preparations containing intact VMA13 and VMA13 fragments.
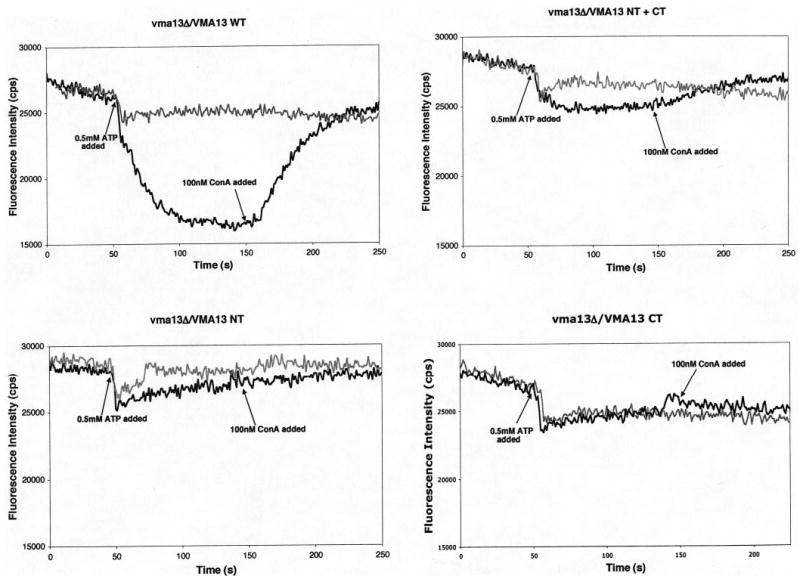
Vacuolar vesicles (10 μg of protein per assay) isolated from the indicated strains were mixed with 1 μM ACMA in transport buffer (50 mm NaCl, 30 mm KCl, 20 mm HEPES, pH 7) in a fluorometer cuvette. Fluorescence emission intensity (black line for each plot) was monitored continuously as the mixture was stirred at 25 °C, and at the indicated point a mixture of MgSO4 and ATP was added to give final concentrations of 1 mm MgSO4 and 0.5 mm ATP (indicated as 0.5 mm ATP on each plot). After the fluorescence decrease had stabilized, 100 nm concanamycin A was added to the cuvette at the indicated time. The gray plots represent control experiments conducted for each strain in which 100 nm concanamycin A was present throughout the assay to inhibit any V-ATPase-dependent changes in fluorescence.
The VMA13-NT strain was able to partially complement the growth defects of a vma13Δ mutant strain (Fig. 2). This suggests that the VMA13-NT mutant must support some level of proton pumping in vivo. To address this, we examined quinacrine staining of the vacuoles in the VMA13-NT strain. Vacuoles from the vma13Δstrain are completely unable to take up the lysosomotropic amino quinacrine and therefore show no staining (data not shown), and the vma13Δstrain containing only the VMA13-CT plasmid also showed no staining (Fig. 5E). As shown in Fig. 5, in vma13Δcells containing the full-length VMA13 plasmid, there is staining of the vacuole by quinacrine, and this staining is missing when 500 nm concanamycin A is added after the initial labeling with quinacrine because proton pumping must continue for quinacrine localization to be maintained. Vacuoles of cells containing VMA13-NT were stained with quinacrine to almost the same level as cells with wild-type VMA13, suggesting that there must be some coupling of ATP hydrolysis and proton pumping in vivo, even though little or no pumping was detected in vitro. Interestingly, when we included 500 nm concanamycin A, quinacrine straining of the vacuole was partially, but not completely inhibited, in the VMA13-NT strain. This result is surprising because it suggests that in the presence of this mutation, the V-ATPase is able to maintain a proton gradient across the vacuolar membrane even in the presence of concanamycin A. We also examined quinacrine staining in cells containing the VMA13-NT + CT plasmids (data not shown). Most of the vacuoles in this strain also stained with quinacrine, but the extent of concanamycin inhibition was variable. We attribute both the variability in quinacrine staining and concanamycin inhibition to differences in the balance of the two plasmids and relative expression of the NT and CT fragments between different cells. Therefore, cells containing approximately equal amounts of NT and CT may show better concanamycin inhibition of quinacrine staining, whereas those that express higher relative amounts of NT or CT show a greater resemblance to the cells containing NT or CT fragment alone.
Figure 5. Quinacrine uptake into vacuoles of wild-type and mutant cells.
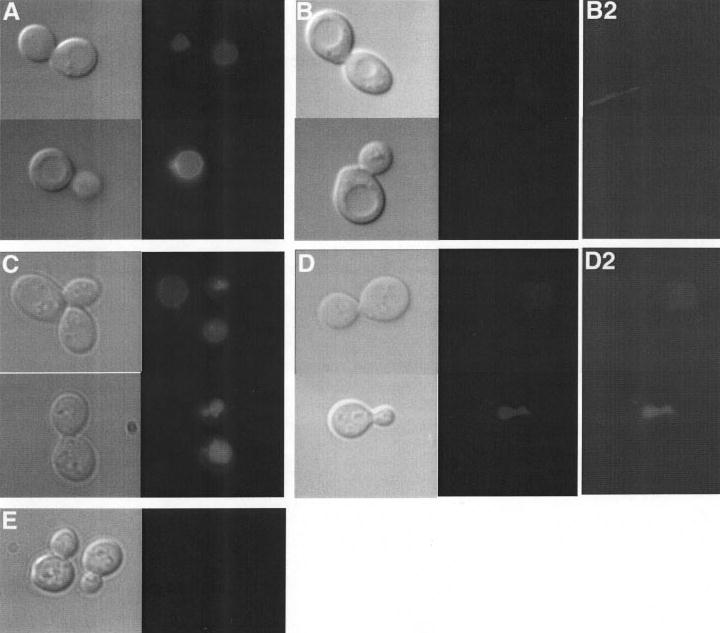
vma13Δ cells carrying plasmids containing the wild-type VMA13 (A and B), VMA13-NT (C and D), or VMA13-CT (E) were stained with the lysosomotropic amine quinacrine and visualized under Nomarski optics (left panel of each set) and fluorescein fluorescence optics (right panel of each set). In B and D, concanamycin A was added to the cells after quinacrine staining but before visualization. The vacuole appears as a clear indentation under Nomarski optics in cells containing wild-type VMA13 that corresponds to the site of quinacrine accumulation in A. The vacuolar morphology in cells containing the VMA13 fragments (C, D, and E) appears to be altered, but quinacrine accumulation is still readily visible in the fluorescence images in C, and even present in D, whereas it is completely missing from E. All fluorescence images were exposed for identical times, but the contrast is changed (to the same extent) in B2 and D2 to more clearly show the staining present in D2 but not in B2.
The level of V-ATPase activity appears to be regulated in response to energy supply in vivo by regulated disassembly of the V1 and Vo sectors (21, 37). Thus, the yeast V-ATPase is predominantly in the assembled state in cells growing in glucose, the preferred carbon source, but rapidly and reversibly disassembles into free V1 and Vo sectors when cells are deprived of glucose for as little as 2–5 min. Recently, Shao and Forgac (17) demonstrated that disassembly in response to glucose deprivation does not occur in the absence of proton gradient across the vacuolar membrane, suggesting that the V-ATPase is “sensing” the absence of a proton gradient and preventing disassembly of V1 from Vo. This result suggests that long range communication between the membrane-bound subunits and the V1 sector is important for disassembly in the absence of glucose. We hypothesized that disassembly might also be affected in the VMA13-NT mutant because it shows altered coupling behavior. We measured V-ATPase assembly and disassembly by comparing the level of Vo subunits co-precipitated with V1 via a monoclonal antibody against subunit A to the level of Vo subunits precipitated by a monoclonal antibody that recognizes the 100 kDa subunit (subunit a) only when Vo is not bound to V1 (21, 29). In cells containing the intact Myc-VMA13 plasmid, 62% of the Vo sectors are assembled with V1 in the presence of glucose, 25% are assembled after a 15-min glucose deprivation, and 55% are assembled after restoration to glucose to glucose-deprived cells. In cells containing the VMA13-NT, 47% are assembled in the presence of glucose, 22% are assembled after glucose deprivation, and 55% are assembled after glucose restoration. Thus, the VMA13-NT plasmid supports both disassembly and reassembly of the V-ATPase in response to changes in glucose concentration.
DISCUSSION
The discovery that the V-ATPase could assemble into an inactive V1Vo complex when subunit H was missing led to the suggestion that the H subunit might serve to functionally bridge the V1 and Vo sectors in order to establish the long distance communication between the catalytic sites and proton pore that is critical for rotational catalysis (22, 23). Our mutagenesis results are consistent with this model, but provide two surprising additions that would not have been predicted from the crystal structure or the biochemical characterization of complexes lacking subunit H. First, we find that the two domains suggested by the H subunit crystal structure can operate together, at least to some degree, even when they are not covalently attached. Even more surprising are the results with the VMA13-NT domain alone. This domain provides partial complementation of the growth defects of a vma13Δ mutant, suggesting that it supports V-ATPase function in vivo to some extent, but our results indicate that this domain has lost important features of V1-Vo communication.
Electron microscopy and image analysis of both the yeast and bovine clathrin-coated vesicle V-ATPases have placed the H subunit in the peripheral stalk, near the bottom of the V1 sector and potentially at the intersection of V1 and Vo (38). At this location, the H subunit might be expected to participate in the stator function in the V-ATPase catalytic mechanism, stably connecting the catalytic headgroup to the Vo a sub-unit in order to allow productive rotational catalysis. In the Escherichiacoli F1Fo-ATPase, the stator is comprised of the membrane-bound b2 dimer linked to the soluble δ-subunit, and these subunits form a number of connections to the F1 headgroup (8). The stator stalk has a very different subunit composition in V-ATPases, and involves many more, predominantly soluble, V1 subunits. This may reflect the added demands on stator function in the V-ATPases, particularly reversible disassembly. The stator subunits of F1Fo-ATPase can be quite tolerant of mutations, possibly because there are multiple interactions that provide stability and disruption of one or a few of these interactions can therefore be tolerated (39–41). A similar explanation may account for the ability of the VMA13-NT and VMA13-CT to provide partial complementation when expressed together. Under this explanation, these two domains may each have important interactions with V1 and Vo subunits that they need to achieve, but a tight covalent interaction between the two halves is not essential because there are multiple weak interactions among the other stator subunits that allow for the overall stator function to be accomplished with reduced efficiency. Alternatively, the crystal structure may have captured the H subunit in its most extended form, and the VMA13-NT and VMA13-CT domains may in fact be closer in vivo and able to interact non-covalently. It is notable that the presence of both halves of subunit H seems to stabilize the other peripheral stalk subunits in isolated vacuolar membranes. As shown in Fig. 3, there is some loss of all of the V1 subunits, but particularly the C and E subunits of the peripheral stalk, in the VMA13-CT vesicles, but the E subunit is much better retained in the VMA13-NT vesicles. When both halves of the H subunit are present, both the E and C subunits are at near wild-type levels. Recent cross-linking experiments place the C subunit near the V1Vo-interface (51), so partial loss of this subunit from the VMA13-NT vesicles is consistent with defects in structural communication between V1 and Vo in this mutant (see below).
The VMA13-CT domain supported very little ATPase activity (TABLE ONE), and it was completely incapable of complementing the Vma−phenotype, supporting proton pumping, or supporting quinacrine uptake into vacuoles in vivo. We believe that the VMA13-CT is contributing to V-ATPase function when co-expressed with the VMA13-NT, however. First, the strain carrying both plasmids shows somewhat better growth at pH 7.5 than the strain containing the VMA13-NT alone (Fig. 2). Second, vacuoles from cells containing both plasmids not only contained both domains, but also a significantly higher level of concanamycin A-sensitive ATPase activity and much better proton pumping than the vacuoles containing VMA13-NT alone. There was more variability between different vacuole preparations from the strain co-transformed with two separate plasmids, but we also observed some variation in the levels of expression of the two domains between different co-transformants, as assessed by Western blotting of whole cell lysates (data not shown). Although both domains are expressed from low copy plasmids, these plasmids are not strictly single copy (42) and differences in relative plasmid copy number may account for the variation. Nevertheless, as long as both domains were present in whole cell lysates, we found that there was an improvement in growth and in levels of concanamycin A-sensitive ATP hydrolysis over the strain containing the VMA13-NT construct alone.
The phenotypes of the VMA13-NT mutant subunit expressed alone indicate a novel defect in communication between the V1 and Vo sectors. This defect is apparent at several levels. First, the presence of concanamycin A-insensitive ATPase activity in isolated yeast vacuolar vesicles is very unusual, but the sensitivity of much of this activity to NEM strongly suggests that the ATPase activity is coming from V1 sectors. Both concanamycin A and NEM are highly specific inhibitors of V-ATPase activity at these concentrations, but concanamycin A inhibits by binding to the Vo c subunits (33, 35), while NEM inhibits by binding at the catalytic sites in V1 (36, 43). Because of the tight coupling of ATP hydrolysis to proton pumping, these two inhibitors usually give similar results. In bovine clathrin-coated vesicles, significant concanamycin A-insensitive ATP hydrolysis been observed in the intact V-ATPase, but this activity was Ca2+-dependent and attributed to functionally uncoupled, although structurally attached, V1 sectors (44). (Virtually all Mg2+-dependent activity was concanamycin inhibited.) In contrast, the ATPase activity in the VMA13-NT-containing vacuoles is both Mg2+-dependent and uncoupled, based on the loss of ATP-driven proton transport. Other yeast mutants have exhibited decreases in MgATP-dependent coupling efficiency, but in these mutants, the Mg2+-dependent ATPase activity could still be fully inhibited by concanamycin A (17, 45), and overexpression of VMA13 has a similar effect (46). The most logical explanation for our results is that binding of concanamycin A to the Vo sector is no longer able to inhibit ATP hydrolysis in the V1 sector because of the defect in functional coupling. Interestingly,certain mutations in the b- and δ-subunits of the E. coli F1Fo-ATPase stator have also been shown to uncouple ATP hydrolysis and proton transport (47, 48).
Second, partial complementation of the growth phenotype by the VMA13-NT strain suggests that some coupling ATP hydrolysis and proton transport must occur in vivo, despite the very poor coupling in vitro, and consistent with this, quinacrine uptake into vacuoles of the VMA13-NT strain in vivo indicates that the vacuoles are acidified (Fig. 5). However, the vacuolar acidification is not completely inhibited by concanamycin A, even at the relatively high concentrations (500 nm) that completely inhibit quinacrine uptake in wild-type cells. One explanation for this would be vacuolar acidification by a second mechanism, independent of the V-ATPase, but this is not consistent with the complete inhibition of quinacrine uptake by concanamycin A in wild-type cells (Fig. 5) or the complete lack of quinacrine uptake into vma13Δ cells (data not shown). (We cannot test for presence of a V-ATPase-independent activity in vivo with NEM because there are too many potential targets for this inhibitor.) Under current models proposed for inhibition by concanamycin A and related inhibitors, it is hard to envision how concanamycin A-independent proton pumping might be induced by an H subunit mutation. It is highly unlikely that an H subunit mutation could affect concanamycin A binding to the Vo sector. However, it has been proposed that bafilomycins and concanamycins inhibit by binding to subunit c and preventing the ring of c subunits from rotating past subunit a, which is held in a fixed position by the stator subunits (35). Defects in stator structure arising from subunit H mutations might allow more movement of the a subunit, thus permitting some rotation of the c ring even in the presence of the inhibitors. Not all of the c subunits are likely to have inhibitor bound, so increased flexibility at the region of a-c contact might allow a subunit c with concanamycin bound to pass, and therefore, also allow some proton pumping to occur via c subunits not bound to inhibitor. Regardless of the mechanism, this result is again consistent with a defect in communication between the Vo and the V1 sectors.
The presence of an uncoupled ATPase activity arising from mutant V-ATPases might be expected to have an adverse effect on growth of the cell. This may be the case, although it is somewhat difficult to distinguish growth defects arising from a partial loss of proton pumping and those arising from uncoupled ATP hydrolysis at this time. The VMA13-NT mutant plasmid allowed only partial complementation of the pH-dependent growth phenotype of the vma13Δ mutant (Fig. 2), and did not complement the growth defect at high pH and calcium. In contrast, the extent of vacuolar acidification in this mutant, at least when judged by quinacrine accumulation, would appear to be sufficient for complementation. For example, rav1Δ mutants have a much more severe loss of quinacrine accumulation but exhibit little or no Vma− growth phenotype at 30 °C (49, 50). This type of assessment is highly qualitative, since the full physiological basis of the Vma−growth phenotype is not understood, but it is possible that the VMA13-NT mutant is relieving some of the problems that lead to poor growth in the vma13Δ mutant strain by permitting some vacuolar acidification, but creating others by generating some uncoupled ATP hydrolysis. Further work will be necessary to assess the full physiological consequences of this mutation.
Acknowledgments
We thank Richard Cross for the use of the fluorometer.
Footnotes
This work was supported by National Institutes of Health Grant 1R01GM50322 (to P. M. K.).
The abbreviations used are: V-ATPase, vacuolar proton-translocating ATPase; SD-uracil, fully supplemented minimal medium lacking uracil; ACMA, 9-amino-6-chloro-2-me-thoxyacridine; VMA13-NT, N-terminal fragment of the yeast H subunit, consisting of amino acids 1–348; VMA13-CT, C-terminal fragment of the yeast H subunit, consisting of amino acids 352– 478; NEM, N-ethylmaleimide; aa, amino acid.
References
- 1.Wieczorek H, Brown D, Grinstein S, Ehrenfeld J, Harvey WR. Bioes-says. 1999;21:637–648. doi: 10.1002/(SICI)1521-1878(199908)21:8<637::AID-BIES3>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 2.Nishi T, Forgac M. Nat Rev Mol Cell Biol. 2002;3:94–103. doi: 10.1038/nrm729. [DOI] [PubMed] [Google Scholar]
- 3.Kane PM, Smardon AM. J Bioenerg Biomembr. 2003;35:313–321. doi: 10.1023/a:1025724814656. [DOI] [PubMed] [Google Scholar]
- 4.Sambade M, Kane PM. J Biol Chem. 2004;279:17361–17365. doi: 10.1074/jbc.M314104200. [DOI] [PubMed] [Google Scholar]
- 5.Nelson N, Taiz L. Trends Biochem Sci. 1989;14:113–116. doi: 10.1016/0968-0004(89)90134-5. [DOI] [PubMed] [Google Scholar]
- 6.Imamura H, Nakano M, Noji H, Muneyuki E, Ohkuma S, Yoshida M, Yokoyama K. Proc Natl Acad Sci U S A. 2003;100:2312–2315. doi: 10.1073/pnas.0436796100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirata T, Iwamoto-Kihara A, Sun-Wada GH, Okajima T, Wada Y, Futai M. J Biol Chem. 2003;278:23714–23719. doi: 10.1074/jbc.M302756200. [DOI] [PubMed] [Google Scholar]
- 8.Dunn SD, McLachlin DT, Revington M. Biochim Biophys Acta. 2000;1458:356–363. doi: 10.1016/s0005-2728(00)00086-4. [DOI] [PubMed] [Google Scholar]
- 9.Iwata M, Imamura H, Stambouli E, Ikeda C, Tamakoshi M, Nagata K, Makyio H, Hankamer B, Barber J, Yoshida M, Yokoyama K, Iwata S. Proc Natl Acad Sci U S A. 2004;101:59–64. doi: 10.1073/pnas.0305165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fethiere J, Venzke D, Diepholz M, Seybert A, Geerlof A, Gentzel M, Wilm M, Bottcher B. J Biol Chem. 2004;279:40670–40676. doi: 10.1074/jbc.M407086200. [DOI] [PubMed] [Google Scholar]
- 11.Yokoyama K, Nagata K, Imamura H, Ohkuma S, Yoshida M, Tamakoshi M. J Biol Chem. 2003;278:42686–42691. doi: 10.1074/jbc.M305853200. [DOI] [PubMed] [Google Scholar]
- 12.Arata Y, Baleja JD, Forgac M. J Biol Chem. 2002;277:3357–3363. doi: 10.1074/jbc.M109967200. [DOI] [PubMed] [Google Scholar]
- 13.Arata Y, Baleja JD, Forgac M. Biochemistry. 2002;41:11301–11307. doi: 10.1021/bi0262449. [DOI] [PubMed] [Google Scholar]
- 14.Jones RP, Durose LJ, Findlay JB, Harrison MA, Moore LL, Fulton AM, Harrison ML, Geahlen RL, Pali T, Whyteside G, Dixon N, Kee TP, Ball S, Finbow ME, Marsh D, Harrison M, Durose L, Song CF, Barratt E, Trinick J, Jones R, Hunt IE, Jaeger J, Ward A, O’Reilly J, Barratt EA, Powell B, Cobine PA, George GN, Winzor DJ, Harrison MD, Mogahaddas S, Dameron CT, Murray J, Kim YI, Cobine P, Meier S, Jones PC, Hughes G, Griffiths DE, Jones P. Biochemistry. 2005;44:3933–3941. doi: 10.1021/bi048402x. [DOI] [PubMed] [Google Scholar]
- 15.Gruber G, Radermacher M, Ruiz T, Godovac-Zimmermann J, Canas B, Kleine-Kohlbrecher D, Huss M, Harvey WR, Wieczorek H. Biochemistry. 2000;39:8609–8616. doi: 10.1021/bi000103u. [DOI] [PubMed] [Google Scholar]
- 16.Chaban YL, Coskun U, Keegstra W, Oostergetel GT, Boekema EJ, Gruber G. J Biol Chem. 2004;279:47866–47870. doi: 10.1074/jbc.M408460200. [DOI] [PubMed] [Google Scholar]
- 17.Shao E, Forgac M. J Biol Chem. 2004;279:48663–48670. doi: 10.1074/jbc.M408278200. [DOI] [PubMed] [Google Scholar]
- 18.Landolt-Marticorena C, Williams KM, Correa J, Chen W, Manolson MF. J Biol Chem. 2000;275:15449–15457. doi: 10.1074/jbc.M000207200. [DOI] [PubMed] [Google Scholar]
- 19.Wieczorek H, Gruber G, Harvey WR, Huss M, Merzendorfer H, Zeiske W. J Exp Biol. 2000;203:127–135. doi: 10.1242/jeb.203.1.127. [DOI] [PubMed] [Google Scholar]
- 20.Kane PM, Parra KJ. J Exp Biol. 2000;203:81–87. doi: 10.1242/jeb.203.1.81. [DOI] [PubMed] [Google Scholar]
- 21.Kane PM. J Biol Chem. 1995;270:17025–17032. [PubMed] [Google Scholar]
- 22.Ho MN, Hirata R, Umemoto N, Ohya Y, Takatsuki A, Stevens TH, Anraku Y. J Biol Chem. 1993;268:18286–18292. [PubMed] [Google Scholar]
- 23.Xie XS, Crider BP, Ma YM, Stone DK. J Biol Chem. 1994;269:25809–25815. [PubMed] [Google Scholar]
- 24.Parra KJ, Keenan KL, Kane PM. J Biol Chem. 2000;275:21761–21767. doi: 10.1074/jbc.M002305200. [DOI] [PubMed] [Google Scholar]
- 25.Sagermann M, Stevens TH, Matthews BW. Proc Natl Acad Sci U S A. 2001;98:7134–7139. doi: 10.1073/pnas.131192798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherman, F., Fink, G.R., and Hicks, J.B. (1982) Methods in Yeast Genetics, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
- 27.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volck-aert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 28.Elble R. BioTechniques. 1992;13:18–20. [PubMed] [Google Scholar]
- 29.Kane PM, Kuehn MC, Howald-Stevenson I, Stevens TH. J Biol Chem. 1992;267:447–454. [PubMed] [Google Scholar]
- 30.Roberts CJ, Raymond CK, Yamashiro CT, Stevens TH. Methods Enzymol. 1991;194:644–661. doi: 10.1016/0076-6879(91)94047-g. [DOI] [PubMed] [Google Scholar]
- 31.Nelson H, Nelson N. Proc Natl Acad Sci U S A. 1990;87:3503–3507. doi: 10.1073/pnas.87.9.3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Curtis KK, Francis SA, Oluwatosin Y, Kane PM. J Biol Chem. 2002;277:8979–8988. doi: 10.1074/jbc.M111708200. [DOI] [PubMed] [Google Scholar]
- 33.Huss M, Ingenhorst G, Konig S, Gassel M, Drose S, Zeeck A, Altendorf K, Wieczorek H. J Biol Chem. 2002;277:40544–40548. doi: 10.1074/jbc.M207345200. [DOI] [PubMed] [Google Scholar]
- 34.Bowman BJ, Bowman EJ. J Biol Chem. 2002;277:3965–3972. doi: 10.1074/jbc.M109756200. [DOI] [PubMed] [Google Scholar]
- 35.Bowman EJ, Graham LA, Stevens TH, Bowman BJ. J Biol Chem. 2004;279:33131–33138. doi: 10.1074/jbc.M404638200. [DOI] [PubMed] [Google Scholar]
- 36.Feng Y, Forgac M. J Biol Chem. 1992;267:5817–5822. [PubMed] [Google Scholar]
- 37.Parra KJ, Kane PM. Mol Cell Biol. 1998;18:7064–7074. doi: 10.1128/mcb.18.12.7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilkens S, Inoue T, Forgac M. J Biol Chem. 2004;279:41942–41949. doi: 10.1074/jbc.M407821200. [DOI] [PubMed] [Google Scholar]
- 39.McCormick KA, Deckers-Hebestreit G, Altendorf K, Cain BD. J Biol Chem. 1993;268:24683–24691. [PubMed] [Google Scholar]
- 40.Sorgen PL, Caviston TL, Perry RC, Cain BD. J Biol Chem. 1998;273:27873–27878. doi: 10.1074/jbc.273.43.27873. [DOI] [PubMed] [Google Scholar]
- 41.Hazard AL, Senior AE. J Biol Chem. 1994;269:418–426. [PubMed] [Google Scholar]
- 42.Ohya Y, Botstein D. Genetics. 1994;138:1041–1054. doi: 10.1093/genetics/138.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng Y, Forgac M. J Biol Chem. 1994;269:13224–13230. [PubMed] [Google Scholar]
- 44.Crider BP, Xie XS. J Biol Chem. 2003;278:44281–44288. doi: 10.1074/jbc.M307372200. [DOI] [PubMed] [Google Scholar]
- 45.Xu T, Forgac M. J Biol Chem. 2000;275:22075–22081. doi: 10.1074/jbc.M002983200. [DOI] [PubMed] [Google Scholar]
- 46.Curtis KK, Kane PM. J Biol Chem. 2001;20:20. [Google Scholar]
- 47.Caviston TL, Ketchum CJ, Sorgen PL, Nakamoto RK, Cain BD. FEBS Lett. 1998;429:201–206. doi: 10.1016/s0014-5793(98)00597-3. [DOI] [PubMed] [Google Scholar]
- 48.Hazard AL, Senior AE. J Biol Chem. 1994;269:427–432. [PubMed] [Google Scholar]
- 49.Seol JH, Shevchenko A, Deshaies RJ. Nat Cell Biol. 2001;3:384–391. doi: 10.1038/35070067. [DOI] [PubMed] [Google Scholar]
- 50.Smardon AM, Tarsio M, Kane PM. J Biol Chem. 2002;277:13831–31839. doi: 10.1074/jbc.M200682200. [DOI] [PubMed] [Google Scholar]
- 51.Inoue T, Forgac M. J Biol Chem. 2005;280:27896–27903. doi: 10.1074/jbc.M504890200. [DOI] [PubMed] [Google Scholar]