

Abstract
The DNA polymerase holoenzyme of herpes simplex virus type 1 (HSV-1) is a stable heterodimer consisting of a catalytic subunit (Pol) and a processivity factor (UL42). HSV-1 UL42 differs from most DNA polymerase processivity factors in possessing an inherent ability to bind to double-stranded DNA. It has been proposed that UL42 increases the processivity of Pol by directly tethering it to the primer and template (P/T). To test this hypothesis, we took advantage of the different sensitivities of Pol and Pol/UL42 activities to ionic strength. Although the activity of Pol is inhibited by salt concentrations in excess of 50 mM KCl, the activity of the holoenzyme is relatively refractory to changes in ionic strength from 50 to 125 mM KCl. We used nitrocellulose filter-binding assays and real-time biosensor technology to measure binding affinities and dissociation rate constants of the individual subunits and holoenzyme for a short model P/T as a function of the ionic strength of the buffer. We found that as observed for activity, the binding affinity and dissociation rate constant of the Pol/UL42 holoenzyme for P/T were not altered substantially in high- versus low-ionic-strength buffer. In 50 mM KCl, the apparent affinity with which UL42 bound the P/T did not differ by more than twofold compared to that observed for Pol or Pol/UL42 in the same low-ionic-strength buffer. However, increasing the ionic strength dramatically decreased the affinity of UL42 for P/T, such that it was reduced more than 3 orders of magnitude from that of Pol/UL42 in 125 mM KCl. Real-time binding kinetics revealed that much of the reduced affinity could be attributable to an extremely rapid dissociation of UL42 from the P/T in high-ionic-strength buffer. The resistance of the activity, binding affinity, and stability of the holoenzyme for the model P/T to increases in ionic strength, despite the low apparent affinity and poor stability with which UL42 binds the model P/T in high concentrations of salt, suggests that UL42 does not simply tether the Pol to DNA. Instead, it is likely that conformational alterations induced by interaction of UL42 with Pol allow for high-affinity and high-stability binding of the holoenzyme to the P/T even under high-ionic-strength conditions.
The DNA polymerase of herpes simplex virus type 1 (HSV-1) is a heterodimer composed of a catalytic subunit (Pol), encoded by the UL30 gene, and a double-stranded (ds) DNA binding protein, encoded by the UL42 gene (6, 13, 15, 31). The UL42 protein stimulates the activity of Pol on nicked and/or gapped DNA templates in high-salt buffer and increases the processivity of Pol on singly primed single-stranded (ss) DNA templates (12, 15, 17). The results of biochemical and genetic analyses have revealed that both Pol and UL42 protein are required for extensive leading and lagging strand DNA synthesis in cell-free (in vitro) assays (10, 25, 35, 36) and for viral DNA replication in infected cells (in vivo) (20, 22, 27, 33). The carboxyl-terminal (C-terminal) 227-amino-acid domain of Pol is sufficient for physical interaction with UL42 protein in vitro (8). Although deletion of as few as 35 residues from the Pol C terminus destroys viral DNA replication capability in vivo and processive DNA replication in vitro, removal of the entire C-terminal interaction domain has little effect on the basal, unstimulated activity of Pol on nicked and gapped templates (7, 38). These results indicate that stable interaction of the Pol and UL42 protein is required for fully processive DNA synthesis in vitro and in vivo, though not for inherent Pol catalytic activity.
The mechanism(s) by which UL42 protein increases the processivity of HSV-1 Pol is not known. However, it may be different from that of the more extensively studied ring-like processivity factors such as the β subunit of Escherichia coli Pol III and proliferating cell nuclear antigen (PCNA) (reviewed in reference 24). These processivity factors require ATP and clamp loading proteins to assemble the ring structures and their respective polymerases on DNA templates, and they appear to work by sliding along the DNA, thus tethering the polymerase to the template. Furthermore, these processivity factors possess no strong inherent capacity to bind to the DNA (4). The processivity factor of the bacteriophage T7 DNA polymerase, the E. coli thioredoxin, differs in that it requires no ATP or clamp loading proteins and appears to increase the processivity of the polymerase by increasing its affinity for DNA template (19, 37). Nevertheless, thioredoxin alone does not bind to DNA (18). Thus, the UL42 protein is distinguished from each of these processivity factors by its ability to bind to DNA in the absence of Pol (13, 14). Although it has been proposed that the DNA binding properties of UL42 may serve to tether HSV-1 Pol to DNA template (15), it is not clear how such binding would allow unimpeded elongation without also acting as a brake (5, 40).
We tested the tethering hypothesis using several means to quantify binding of the subunits and holoenzyme to a model DNA primer and template (P/T). We took advantage of the fact that Pol and Pol/UL42 possess different optimum conditions for catalytic activity to test predictions of the tethering model for binding in the low- and high-ionic-strength buffer conditions. In this report, we have analyzed affinities and dissociation rate constants of the subunits and holoenzyme for the model P/T using nitrocellulose filter-binding assays and real-time biosensor technology. The use of this model P/T to define the kinetic parameters of the enzymatic activities of Pol and Pol/UL42 in low-and high-ionic strength will be described elsewhere (M. Chaudhuri and D. Parris, unpublished data). Our results indicate that the affinity and stability of UL42 for this P/T are greatly reduced by increases in ionic strength. By contrast, the Pol/UL42 holoenzyme activity displays lack of salt sensitivity as does the complex's binding affinity and half-life for association with P/T. These results are inconsistent with a model for processivity which relies on simple tethering of the Pol by the UL42 subunit.
MATERIALS AND METHODS
Cells and viruses.
Recombinant baculoviruses which express the HSV-1 Pol or UL42 were the gifts of Robert Lehman (Stanford University) and Mark Challberg (National Institutes of Health), respectively. Sf9 insect cells were used to propagate recombinant baculoviruses as previously described (29). Cells were infected singly with either the Pol or UL42 recombinant at a multiplicity of infection of 10 PFU per cell for the purification of the individual subunits of the DNA polymerase holoenzyme or were infected with 10 PFU of each per cell for the purification of the Pol/UL42 complex.
Protein purification.
Buffers used were buffer B-2 (20 mM Tris-Cl [pH 7.5 or 8.2], 50 mM NaCl, 5 mM 2-mercaptoethanol, 1 mM EDTA, 1% glycerol) for protein purifications and buffer C (50 mM Tris-Cl [pH 7.5], 5 mM 2-mercaptoethanol, 1 mM EDTA, 1% glycerol) containing 50 or 125 mM KCl, as indicated, for enzyme, nitrocellulose filter-binding, and DNA mobility shift assays. Nuclear extracts were prepared from infected cells 40 h postinfection essentially as described previously (12) and dialyzed extensively against B-2 buffer, pH 8.2. Insoluble protein was removed by low-speed centrifugation following dialysis. Protein fractionation was conducted at room temperature with the assistance of a Pharmacia fast protein liquid chromatography system, and all fractions were collected at 0°C. The presence of the relevant protein(s) in column fractions was determined by immunoblotting with Pol- and/or UL42-specific antiserum and by DNA polymerase assay (for Pol and Pol/UL42) as previously described (28), except that activity was measured in the presence of 50 and 125 mM KCl for Pol and Pol/UL42, respectively. The soluble protein fraction (containing Pol, UL42, or Pol/UL42 complex) was applied to a 9-ml DEAE-Sepharose column equilibrated with B-2 buffer, pH 8.2. The protein was eluted with a 60-ml linear salt gradient from 0 to 0.5 M KCl in B-2 buffer, pH 8.2. Fractions containing the relevant HSV-1 protein(s) were pooled and diluted in B-2 buffer (pH 7.5) to contain a final concentration of 0.2 M KCl and applied to a 5-ml Blue-Sepharose column. A 60-ml linear gradient from 0 to 1 M KCl in B-2 buffer (pH 7.5) was used for elution of proteins. For the third step in the purification of Pol, fractions positive for Pol activity were pooled and applied to a 9-ml heparin-Sepharose column. Fractions were collected over a linear salt gradient (60 ml) from 0 to 1 M KCl in B-2 buffer, pH 7.5. For the third step in the purification of UL42 or Pol/UL42 complex, fractions from the Blue-Sepharose column containing the relevant proteins were applied to a 9-ml Q-Sepharose column, and the proteins were eluted with an 80-ml linear gradient from 0 to 1 M KCl in B-2 buffer, pH 7.5. This final column for the purification of Pol/UL42 holoenzyme ensured that no free Pol was present inasmuch as the latter protein fails to bind Q-Sepharose under the buffer conditions used (results not shown). Following the third column step, the Pol, UL42, and Pol/UL42 preparations were each concentrated by passage through Centricon-30 ultrafiltration units (Amicon, Bedford, Mass.) and adjusted to buffer C containing 200 mM KCl for storage. Prior to use, the salt concentration was adjusted with KCl as indicated. The protein concentration of each preparation was determined initially using the Coomassie dye-based method of Bio-Rad (Hercules, Calif.), according to the instructions of the manufacturer, and preparations were maintained at 4°C.
Preparation of DNA P/T.
All synthetic oligonucleotides were purchased from Integrated DNA Technologies, Inc. (Coralville, Iowa) as gel-purified products. The DNA template strand was a 67-mer (5′ATTTGCTGACCTTTGTTCTGGATGAGTTGGTTGGACGGCTGCGAGGCGATCAAGGTGTCGTAGTGGC 3′), and the primer strand was a 45-mer (5′GCCACTACGACACCTTGATCGCCTCGCAGCCGTCCAACCAACTCA 3′). For BIAcore binding experiments, the primer strand was purchased with a biotin tag at the 5′ end. In some experiments, the template strand contained a covalently linked digoxigenin (DIG) tag at the 5′ end. For DNA mobility shift and nitrocellulose filter-binding experiments, the primer strand was labeled at the 5′ end with [γ-32P]ATP by standard procedures. For annealing, equimolar amounts of primer and template strands were mixed, heated to 50°C for 10 min, and slowly cooled to room temperature. The annealed product was stored at −20°C for later use.
DNA mobility shift.
The number of molecules of Pol, UL42, or Pol/UL42 capable of binding to a P/T was determined by incubation of 32P-radiolabeled P/T with increasing concentrations of purified Pol, UL42, or Pol/UL42 complex for 10 min at room temperature in binding buffer C containing 50 mM KCl. The ability of the proteins to impede the migration of P/T and the number of complexes formed were assessed after electrophoresis through a nondenaturing 6% polyacrylamide gel as previously described (13).
Immobilization of DNA P/T to biosensor chips.
The BIAcore 2000 system and biosensor-SA chips were obtained from Pharmacia Biosensor AB or Biacore, Inc. (Piscataway, N.J.). Buffer A, used for all BIAcore binding experiments, contained 50 mM Tris-Cl (pH 7.5), 1 mM EDTA, and 1% glycerol and was adjusted to contain KCl as indicated. The annealed biotinylated P/T was adjusted to a concentration of 100 nM in buffer A containing 0.3 M KCl and injected at a rate of 5 μl/min over one surface of a biosensor-SA chip, which contained streptavidin covalently linked to the surface. Injection was halted when the desired number of response units (RU) was bound, as indicated for each experiment. For experiments to measure binding to blocked P/T, monoclonal antibody 21H8 to DIG (Abcam Ltd., Cambridge, Mass.) was passed over the appropriate DNA-bound surfaces until steady-state binding was achieved. The surfaces were washed extensively in buffer A containing 1 M KCl without detectable loss of bound antibody (results not shown), and new baselines were established for each surface prior to the addition of test protein. Following each round of protein binding, chip surfaces containing bound Pol or UL42 were regenerated with buffer A containing 1 M KCl, while those containing bound Pol/UL42 were regenerated with buffer A containing 2 M KCl without detectable loss of bound DNA (results not shown).
Biosensor assays.
Increasing concentrations (25 to 600 nM) of purified test protein were injected at room temperature at a flow rate of 5 μl/min for 8 min over the biosensor chip surface to which the P/T had been immobilized and simultaneously injected over a second control surface lacking bound DNA. Binding of proteins to the chip surfaces resulted in an increase in RU. The change in RU as a function of time was determined after subtraction of the values for interaction of proteins with the control surface to produce a sensorgram. After the binding period, buffer A was passed over the DNA-bound and control chip surfaces at a flow rate of 5 μl/min, and the dissociation of the protein was recorded by a decrease in RU.
The apparent association rate constants (kon) were calculated from the sensorgrams by nonlinear global curve fitting with Biaevaluation version 3.0 software, using the simple homogeneous binding model, A + B = AB, which assumes pseudo-first-order binding kinetics and uses a single apparent dissociation rate constant (koff) for calculation of kon. The dissociation of the protein from the chip surface was assumed to follow first-order kinetics and is described by the following equation:
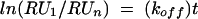 |
(1) |
where RU1 is the number of RU recorded at time t1, RUn is the number of RU recorded at time tn and t is the time interval tn − t1. The equilibrium dissociation constant (Kd) was calculated from the apparent association and dissociation constants obtained for BIAcore experiments using the following equation:
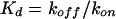 |
(2) |
Nitrocellulose filter-binding assays.
Filter-binding experiments were performed by the double-filter method devised by Wong and Lohman (43) in which the DNA-protein complexes are trapped on a nitrocellulose filter and the remaining, unbound DNA is trapped on a DEAE-cellulose membrane placed directly under the nitrocellulose filter. Both filters were purchased from Schleicher and Schuell (Keene, N.H.). A 5′-end-labeled P/T, prepared as described above, was used as the DNA probe. For determination of Kd, purified Pol or Pol/UL42 in binding buffer C containing 50 or 125 mM KCl, respectively, was incubated with increasing concentrations of DNA probe for at least 20 min at room temperature. The samples were applied in duplicate to the nitrocellulose/DEAE filter stack using a vacuum manifold and immediately washed briefly with binding buffer. The radioactivity bound to each filter was quantified using a Molecular Dynamics PhosphorImager and ImageQuant software (Sunnyvale, Calif.). The radioactivity in the bound and free fractions was normalized as a percentage of total DNA recovered to account for loading errors as previously described (43). Bound DNA [E · D] as a proportion of active protein concentration [E] was plotted against total DNA concentration [D], and the data were fitted (21, 32) to the following quadratic equation:
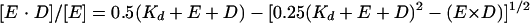 |
(3) |
To determine the stability of preformed protein-P/T complexes, Pol and Pol/UL42 were allowed to bind to the labeled P/T in low- or high-salt buffer for 20 min at room temperature. Unlabeled activated calf thymus DNA (final concentration, 500 μg/ml) was then added to trap dissociated protein, and samples were removed in duplicate at various times thereafter and applied to a double-filter stack. The bound and free DNA were quantified by phosphorimager analysis as described above. The dissociation rate constant was calculated according to equation 1, except that the amount remaining bound to nitrocellulose was substituted for RU.
RESULTS
Stoichiometry of proteins bound to model P/T.
In an effort to describe the kinetic parameters which define the polymerization reaction of the HSV-1 Pol and the mechanism by which UL42 increases its processivity, we have designed a model synthetic P/T for use in single turnover experiments. The DNA P/T, which was blunt on one end, contained a 67-mer template strand which produced a 22-nucleotide 5′ overhang when annealed to a shorter complementary primer strand (45-mer). Although the central portions of the P/T correspond to a shorter P/T used in single turnover experiments with the T7 DNA polymerase (32), the ss overhang and ds regions were extended to fully accommodate the amount of a synthetic DNA hairpin which was protected from DNase I digestion by a bound Pol/UL42 heterodimer (14). This design was expected to maximize DNA-protein contacts while minimizing the possibility of binding of multiple subunits or complexes to the DNA.
The purity of the preparations used for binding studies was assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis followed by silver staining of the gel (Fig. 1). The high levels of purity of UL42, Pol, and Pol/UL42 are demonstrated by the virtual absence of other stained polypeptide species. Moreover, the Pol and Pol/UL42 protein preparations were determined to be approximately 70% active, based on kinetic, active-site titration experiments using the model P/T (Chaudhuri and Parris, unpublished).
FIG. 1.

Purity of UL42, Pol, and Pol/UL42 preparations. Proteins were expressed in insect cells infected with recombinant baculoviruses and purified by column chromatography as described in Materials and Methods. Concentrated preparations (2 μg) of UL42 (lane 1), Pol (lane 2), or Pol/UL42 (lane 3) were subjected to electrophoresis through a denaturing gradient polyacrylamide gel (5 to 15%) followed by silver staining. Preparations were estimated to be at least 95% pure.
To assess the actual stoichiometry with which the HSV-1 DNA polymerase subunits and holoenzyme could bind the P/T, we performed DNA mobility shift assays with radioactively labeled P/T in buffer containing 50 mM KCl. Figure 2 demonstrates that increasing concentrations of Pol, UL42, or Pol/UL42 yielded specific DNA-protein complexes which were not present when the probe was incubated alone or in the presence of 400 nM bovine serum albumin (BSA). Pol formed a single major complex with the DNA at protein concentrations at or below 200 nM (Fig. 2, lane 4). In this particular experiment, a second complex of lower mobility was formed at protein concentrations of 300 and 400 nM and was of lower relative abundance than the higher-mobility complex (lanes 5 and 6). This second complex was not observed in all experiments and may represent unresolved DNA-protein aggregates. Alternatively, though less likely due to high excess of DNA probe, it may represent low-affinity binding of Pol to the 3′ blunt end on molecules also containing Pol bound at the primer terminus, as previously reported with a different DNA probe (41). As expected from the design of the P/T, Pol/UL42 formed only a single major complex with the DNA even at the highest concentration tested, 400 nM (Fig. 2, lane 20). Because Pol has been reported to be a monomer in solution and Pol/UL42 is a stable heterodimer (6, 13, 15), it is likely that the major shifted DNA species represents a 1:1 stoichiometry of Pol/UL42 and DNA. By contrast, UL42 produced predominantly a faster-migrating complex with P/T at the lower protein concentrations tested (Fig. 2, lanes 9 and 10), but at a 400 nM concentration of UL42, the predominant shifted species was the slower-migrating complex (Fig. 2, lane 12). The latter result suggests that up to two molecules or multimers of UL42 can bind to each molecule of the model P/T at the highest concentration of protein tested. When complexes of DNA with each of the proteins were run on the same gel, the slower-migrating UL42 complex migrated to a position just below that of the Pol/UL42 complex (results not shown), while the faster-migrating UL42 complex had an even higher mobility than the major Pol complex, as noted previously (13, 41). On the basis of these relative mobilities and the fact that UL42 exists as a monomer in solution (13, 15), it is likely that the faster- and slower-migrating complexes represent 1:1 and 2:1 stoichiometries, respectively, of UL42 to P/T.
FIG. 2.

Electrophoretic mobility shift analysis of protein binding to model P/T. Purified Pol (lanes 3 to 6), UL42 (lanes 9 to 12), or Pol/UL42 heterodimer (lanes 15 to 20) preparations were incubated with 32P-end-labeled P/T (100 nM), and the formation of complexes was analyzed by electrophoresis through nondenaturing 6% polyacrylamide gels. The final concentrations of Pol and UL42 tested were 100, 200, 300, and 400 nM, while those tested for the Pol/UL42 heterodimer were 25, 50, 100, 200, 300, and 400 nM (concentration shown by the thickness of the triangle over the lanes). The mobility of the P/T probe (P) alone (lanes 1, 7, and 13) or in the presence of 400 nM BSA (B) (lanes 2, 8, and 14) also is shown. In some preparations of probe, nonspecific (ns) higher-mobility bands were noted. Specifically shifted complexes are indicated by arrows.
Detection of binding to P/T by nitrocellulose filter assays.
We and others have reported that the optimum salt concentration for Pol/UL42 activity on nicked and gapped DNA substrates is substantially higher than that for Pol alone (1, 16). Figure 3 shows the effects of KCl concentration on the activities of the purified Pol and Pol/UL42 preparations using maximally activated calf thymus DNA as the substrate. The results confirm the salt sensitivity of the catalytic subunit alone, such that activity decreased with increasing KCl concentrations above 50 mM. By contrast, the Pol/UL42 holoenzyme activity was relatively resistant to changes in ionic strength over KCl concentrations ranging from 50 to 125 mM. Similar salt resistance was observed for Pol/UL42 holoenzyme reconstituted from the individual subunits (39).
FIG. 3.

Effect of ionic strength on polymerase activities of purified Pol and Pol/UL42. Polymerase activities were measured by the incorporation of [3H]dTTP into acid-insoluble form using activated calf thymus DNA as the template essentially as described previously (12) in buffer containing the indicated concentrations of KCl. The relative activities of 1.2 nM Pol (•) or 2.5 nM Pol/UL42 (▪) were calculated as a percentage of the activities in 50 mM KCl, corresponding to 1,200 and 1,050 units, respectively. A unit of activity was defined as the number of femtomoles of [3H]dTTP incorporated in 20 min at 37°C.
In order to correlate the effect of ionic strength on the binding of Pol and Pol/UL42 to a defined P/T with the effect on enzymatic activity, we selected buffer containing 50 mM KCl as representative of low-ionic-strength conditions and buffer containing 125 mM KCl as representative of high-ionic strength conditions for all subsequent experiments. The use of KCl concentrations below 50 mM was impractical due to the poor stability of the enzyme preparations in extremely low-ionic-strength buffer (results not shown). Because UL42 bound the model P/T with a stoichiometry of at least 2:1 (Fig. 2), nitrocellulose filter-binding assays were not employed to assess UL42 binding.
We employed a fixed concentration of Pol or Pol/UL42 as indicated and tested increasing concentrations of radioactively labeled P/T for binding to protein after a 20-min preincubation period. These protein concentrations were selected to be at or above the estimated Kd for the respective protein preparations based on active-site titrations (Table 1; Chaudhuri and Parris, unpublished). Figure 4A shows the binding isotherm of the P/T for Pol (52 nM) in 50 mM KCl. Pol binding approached but did not reach saturation over the concentrations of DNA tested. However, on the basis of nonlinear curve fitting, we estimated the maximum ratio of bound P/T to protein molecules to be 1.2, which closely approximated a 1:1 stoichiometry of P/T to active Pol molecules. The concentration of DNA required for half-maximal binding of Pol to P/T was calculated as the Kd (83.0 ± 1.2 nM). With 125 mM KCl, we were unable to detect significant retention of DNA by Pol on nitrocellulose filters, even at the highest concentration (100 nM) of P/T tested (results not shown).
TABLE 1.
Summary of apparent kinetic constants for protein binding to model DNA P/Ta
| Protein | KCl concn (mM) | Nitrocellulose filter-binding assayb
|
Biosensor assayc
|
Enzyme kinetics assaydKd (nM) | ||||
|---|---|---|---|---|---|---|---|---|
| kon (M−1 s−1) (koff/Kd) | koff (s−1) | t1/2 (min) | Kd (nM) | Kd (nM) [koff/kon] | Rel. Kd | |||
| UL42 | 50 | NDe | ND | ND | ND | 35.3 | 2.1 | ND |
| 125 | ND | ND | ND | ND | 150,000 | 8,770 | ND | |
| Pol | 50 | 1.87 × 104 | 1.55 × 10−3 | 7.5 | 83.0 | 17.1 | 1.0 | 44.2 |
| 125 | If | I | I (very fast) | I | 47.7 | 2.8 | ND | |
| Pol/UL42 | 50 | 1.27 × 105 | 8.37 × 10−4 | 13.8 | 6.60 | 9.02 | 0.5 | ND |
| 125 | 1.16 × 105 | 8.76 × 10−4 | 13.2 | 7.51 | 30.7 | 1.8 | 7.43 | |
All kinetic constants are apparent except for those determined by pre-steady-state kinetic analysis.
Values for Kd and koff were determined as described in the legends to Fig. 4 and 5, respectively. The values of t1/2 were derived from the equation ln 0.5 = −kt1/2, and the value of kon was determined according to equation 2.
All kinetic constants were derived by global fitting of the binding and dissociation curves from five different protein concentrations using the Biaevaluation version 3.0 software. All binding assumed the langmuir model A + B = AB. Rel. Kd, relative or normalized Kd.
Derived by active-site titration of enzyme activity on the model P/T under pre-steady-state conditions (Chaudhuri and Parris, unpublished).
ND, not done.
I, indeterminate.
FIG. 4.

Nitrocellulose filter-binding assays. (A to C) Binding isotherms. Purified Pol with an active concentration of 52 nM (A) or purified Pol/UL42 complexes with active concentrations of 10 nM (B) and 11 nM (C) were incubated with increasing concentrations of 32P-labeled P/T in buffer C containing either 50 mM (A and B) or 125 mM (C) KCl and applied in duplicate to a filter stack containing nitrocellulose and DEAE to trap DNA-protein complexes and free DNA, respectively. The data were fitted to equation 3 to predict the maximum stoichiometry of DNA-protein complexes and the DNA concentration at half-maximum binding (Kd). (D) Scatchard plots of the same data fitted by linear regression analysis. The negative values of the reciprocals of the slopes give estimates of P/T binding Kds of 11.5 nM for Pol (•) and 1.7 nM for Pol/UL42 (▪) in 50 mM KCl and 1.0 nM for Pol/UL42 in 125 mM KCl (▴).
Binding isotherms for purified Pol/UL42 with the P/T under low- and high-salt buffer conditions (active concentrations of 11 and 10 nM, respectively) are shown in Fig. 4B and C, respectively. Interestingly, the binding isotherms of Pol/UL42 for P/T under high- and low-salt buffer conditions were nearly identical, consistent with the similarities in holoenzyme activity in low- and high-ionic-strength reaction conditions. Plots of both data sets estimated a maximum binding stoichiometry of 1:1 for P/T to the Pol/UL42 complex, as predicted from electrophoretic mobility shift assays (Fig. 2). With low-salt buffer conditions, the concentration of P/T at half-maximal binding for Pol/UL42 complex (Kd) was estimated to be 6.60 ± 0.47 nM, demonstrating a 12-fold-higher affinity for P/T than that observed with Pol under the same buffer conditions. The Kd of Pol/UL42 for P/T in high-ionic-strength buffer was estimated to be 7.51 ± 0.92 nM, virtually the same as that under low-salt buffer conditions. These values are summarized in Table 1.
Nonlinear regression analysis to fit the binding data to a quadratic function is the most appropriate for measuring Kd when binding is relatively strong (21). However, we also quantified the data by Scatchard analysis (Fig. 4D) for comparison with similarly derived values previously reported for binding of Pol and Pol/UL42 to other DNA substrates. The negative values of the reciprocals of the slopes of the lines provide estimates of Kd, which were 11.5 nM for Pol in 50 mM KCl and 1.7 and 1.0 nM for Pol/UL42 in 50 and 125 mM KCl, respectively. The Kd values for Pol and Pol/UL42 complex we obtained in 50 mM KCl based on Scatchard plots of the data are similar to those obtained by Scatchard analysis of binding in 50 mM NaCl (7.1 and 0.78 nM, respectively) using a synthetic hairpin substrate (14).
To provide an indication of the stability of the protein-P/T complexes once formed, we incubated the labeled P/T with purified Pol or Pol/UL42 (52 and 11 nM, respectively) for 20 min to allow binding to reach equilibrium. The apparent dissociation rate constants were determined by measuring the amount of DNA-protein complex remaining after the addition of a high concentration of cold activated DNA in order to trap unbound protein and that which dissociated from the P/T. Because dissociation follows first-order kinetics, the data were transformed to produce a linear plot of the natural logarithm of the proportion of bound DNA remaining at each time point versus time (Fig. 5). The slope of the plot provides an estimate of −koff, and the half-life of each of the DNA-protein complexes was calculated according to equation 1. The results, summarized in Table 1, demonstrate that in low-ionic-strength buffer, the stability of the complex of HSV-1 Pol catalytic subunit with P/T was approximately one-half that of the holoenzyme-DNA complex. However, in high-ionic-strength buffer, the Pol-DNA complex was so unstable that we were unable to detect significant amounts bound to the nitrocellulose after as little as 2 min following the addition of cold DNA trap (results not shown). By contrast, the dissociation rate constant of Pol/UL42 holoenzyme from P/T was approximately the same in high- and low-ionic-strength buffer. The instability of the Pol-DNA complex was not due to instability of the Pol in 125 mM KCl, since all of our Pol preparations are routinely stored in buffer containing 200 mM KCl and are adjusted to lower salt concentrations immediately prior to experiments. Thus, a high concentration of salt significantly reduces the ability of the Pol catalytic subunit to form and maintain a stable complex with the model P/T in the absence of UL42, while having little effect on the binding and dissociation properties of the holoenzyme for P/T.
FIG. 5.

Stability of protein-DNA complexes. Purified Pol (52 nM) (A) or Pol/UL42 (11 nM) (B and C) was equilibrated with 6 nM labeled P/T in buffer C containing either 50 (A and B) or 125 mM KCl (C). At time zero, excess unlabeled activated calf thymus DNA trap was added and the amount of labeled DNA-protein complexes remaining as a function of time was determined by nitrocellulose filter-binding activity as described in the legend to Fig. 4. The data were transformed to produce linear plots of the natural logarithm of the proportion remaining versus time.
Theory and experimental rationale of BIAcore binding assays.
It was possible that the high-affinity binding of the holoenzyme to P/T was imparted by high-affinity binding of the UL42 accessory subunit. Alternatively, it was possible that the conformation of Pol within the holoenzyme was altered by its interaction with UL42. To distinguish between these possibilities, it was necessary to measure the effect of ionic strength on binding of the accessory protein to the model P/T. However, as indicated in Fig. 2, the stoichiometry of UL42 binding to the P/T was at least 2:1, such that nitrocellulose filter retention assays would not accurately measure affinity or stability of UL42-P/T complexes. Therefore, we employed biosensor technology to assess the binding of UL42 compared to that of Pol and Pol/UL42 to P/T in low- and high-ionic-strength buffer.
Using the BIAcore biosensor, it is possible to independently measure association and dissociation of proteins with DNA molecules in real time. This analytical method is useful for comparisons of binding and dissociation events under similar conditions. We predicted that if UL42 served to simply tether Pol to the P/T, it should have a higher affinity than that of Pol for the P/T. Moreover, like the holoenzyme, binding of UL42 to the P/T should be relatively resistant to ionic strength within the 50 to 125 mM KCl range.
Real-time binding experiments were performed using biotinylated DNA P/T. In order to orient the P/T to the chip surface to allow uniform access to the recessed 3′ OH end of the primer, we employed commercial gold chips to which streptavidin was covalently linked (biosensor chip SA). Although the gel mobility experiments suggested that at high protein concentrations, two molecules of Pol might be able to associate with a small proportion of the P/T in solution, the attachment of the blunt end to the chip surface would likely render this low-affinity site (41) unavailable to Pol. In initial experiments, we tested binding of Pol to chips containing either 120 or 500 RU of immobilized DNA but found little difference in the observed binding and dissociation kinetics. Therefore, we elected to compare binding parameters in all subsequent experiments with the lower amount (120 RU) of bound DNA substrate to reduce bulk mass transport, to minimize the possibility for rebinding, and to minimize the amount of purified protein required for experiments (2).
Detection of binding of polymerase subunits and holoenzyme to P/T using the BIAcore 2000 biosensor.
Binding experiments were performed by passage of a protein solution of known concentration over two surfaces of the biosensor chip, one of which contained no bound DNA and the other which contained 120 RU of biotinylated P/T. The amount of protein bound to the control surface was subtracted from that bound to the surface containing the DNA to produce a sensorgram (examples of which are shown in Fig. 6). Control experiments demonstrated that BSA at concentrations as high as 600 nM failed to bind to surfaces containing the model P/T at a level significantly higher than to non-DNA-containing surfaces (39).
FIG. 6.

Sensorgrams of binding of purified proteins to model P/T. Binding of Pol (A and D), UL42 (B and E), and Pol/UL42 heterodimer (C and F) to biosensor SA-gold chips to which 120 RU of biotinylated P/T was bound was measured using the BIAcore 2000 system as described in Materials and Methods. Curves of increasing thickness denote binding of 25, 50, 150, 300, and 600 nM protein. Binding and dissociation were performed in buffer A containing either 50 mM KCl (A to C) or 125 mM KCl (D to F). Sensorgrams shown are representative of at least two independent experiments for each protein and the concentrations indicated.
Binding of increasing concentrations (25 to 600 nM) of Pol, UL42, or Pol/UL42 complex to the P/T in buffer containing either 50 or 125 mM KCl is displayed in Fig. 6 as overlay plots of sensorgrams. Following an 8-min association phase, dissociation was monitored by passage of buffer without protein over the chip surface. Kinetic constants were derived by global fitting and are summarized in Table 1. All constants derived in this way should be viewed as apparent and are most useful for comparisons of the constants.
To facilitate comparisons, the same scale has been used to display all binding data. In each case, the 8-min binding period was sufficient to attain or approach steady-state (association events equivalent to dissociation events) at the highest concentration of protein tested (600 nM). In 125 mM KCl, binding of Pol was dramatically reduced from that observed in 50 mM KCl (compare Fig. 6D with A) at all protein concentrations tested. These results explain why binding of Pol to P/T in 125 mM KCl could not be measured by nitrocellulose filter retention assays and are consistent with the effect of salt on Pol activity. The apparently slower dissociation of Pol in high-salt conditions observed in the biosensor assay compared to that in the nitrocellulose filter retention assay most likely reflects rebinding of dissociated Pol (2), since faster flow rates during the dissociation phase reduced the amount of Pol remaining on the chip (results not shown). Consistent with the results of nitrocellulose filter-binding assays (Fig. 4B and C), the binding curves for Pol/UL42 holoenzyme were similar in low- and high-ionic-strength buffer (Fig. 6C and F, respectively). These results support the utility of real-time biosensor assays for assessing and comparing binding behavior.
The sensorgrams readily indicate the sensitivity of UL42 for binding the P/T in high-ionic-strength buffer (compare Fig. 6E with B). Moreover, the observed dissociation rate of UL42 from the P/T was demonstrably greater than that of Pol or Pol/UL42 at either salt concentration, even by visual inspection of sensorgrams. The rate was so high that significant binding of UL42 in high-ionic-strength buffer (Fig. 6E) was detected only at the highest concentration of protein tested (600 nM). These results are consistent with our inability to detect significant binding of UL42 to the P/T in the high-ionic-strength buffer by nitrocellulose filter-binding assays (results not shown).
Table 1 displays the relative apparent Kd value for each protein compared to that observed for Pol in 50 mM KCl (normalized to 1.0). The apparent Kds of UL42 and Pol for P/T in 50 mM KCl differ by only a factor of 2, indicative of similar affinities. However, the apparent Kd observed for UL42 binding to P/T in 125 mM KCl was higher than that for Pol by more than 3 orders of magnitude. Comparison of the kinetics of dissociation indicates that UL42 dissociates much more rapidly from the P/T under high-salt conditions than does Pol (Fig. 6). By contrast, Pol/UL42 binding to P/T is relatively refractory to ionic strength by all measures. Using biosensor technology, the apparent Kd for P/T binding by Pol/UL42 does not differ from the reference (Pol at 50 mM KCl) by more than twofold, even in high-salt buffer (Table 1). The Kd for Pol/UL42 with P/T calculated from nitrocellulose filter retention assays also showed little change (6.6 and 7.5 nM) in low- versus high-salt buffer, respectively, and the stability of the DNA-protein complex once formed was similar in low- and high-salt buffer (Table 1). It is also worth noting that the apparent Kd for Pol/UL42 complex with our model P/T observed by nitrocellulose filter-binding assays is nearly identical to that calculated by pre-steady-state kinetic analysis (7.43 nM [Table 1; also Chaudhuri and Parris, unpublished]).
Because of the small amount of UL42 which was capable of associating with the P/T in buffer containing 125 mM KCl, we performed similar BIAcore binding experiments in buffers containing 75 and 100 mM KCl. The apparent Kd of UL42 for the P/T was calculated by global curve fitting and plotted as a function of salt concentration (Fig. 7). The results confirm that even at moderate ionic strength, the affinity of UL42 for P/T is reduced from that at low-ionic strength. We observed similar relative reductions in binding of mutant UL42 proteins i206 and T142A to the P/T as a function of increasing ionic strength (39; also results not shown).
FIG. 7.

Effect of salt concentration on binding affinity of UL42 for P/T. The apparent Kd for binding of UL42 to P/T was determined by BIAcore analysis and global curve fitting as described in the legend to Fig. 6 except that binding and dissociation buffers contained the KCl concentrations indicated on the x axis.
To assess whether the dissociation kinetics of UL42 we observed in the biosensor assays was due to sliding off the free end of the P/T, as suggested by the model presented by Randell and Coen (34), we tested UL42 binding to blocked P/T. P/T containing a 5′ biotinylated primer was annealed to a 5′ DIG-tagged template strand, and the free 5′ ss overhang was blocked by the attachment of DIG-specific antibody in situ to the P/T bound to the chip surface. The antibody to DIG was passed over surfaces containing DIG-labeled P/T, standard P/T, or no DNA until maximum (steady-state) binding of antibody was obtained. For an additional control, a fourth surface contained DIG-labeled P/T which was not exposed to antibody was used. With high concentrations of salt, we observed too little binding of UL42 to estimate association or dissociation binding constants with confidence, although there was no difference in binding or dissociation kinetics among all three P/Ts (not shown). Therefore, we report only the results of binding experiments in low-salt buffer.
In 50 mM KCl, the association rate constant for UL42 binding was 2 orders of magnitude lower for the blocked P/T than for either standard P/T or nonblocked DIG-labeled P/T (Table 2). Despite slower association kinetics with the blocked P/T, similar amounts of UL42 were bound to all P/Ts by the end of the 8-min association phase (not shown). Interestingly, we observed no difference between the apparent dissociation rate constants of UL42 from the antibody-blocked and nonblocked DIG-labeled P/T. On the basis of results of other control experiments comparing the binding behavior of UL42 to standard and DIG-labeled P/T, we do not consider the slight differences in rate constants observed for standard and nonblocked DIG-labeled P/T in this experiment to be significant. Therefore, the results demonstrate that the dissociation kinetics of the UL42 from P/T measured by the biosensor assay are either not due to sliding off of the ss end, or the sliding rate is slower than the rate of dissociation from the internal portion of P/T. It is of interest to note that the half-life for association of the UL42 with the blocked or unblocked P/Ts linked to the chip (10.5 to 11.5 min) is similar to the 13.3-min half-life observed in solution from streptavidin-blocked DNA or the12-min half-life observed with long DNA molecules (34), indicating that our biosensor results reflect inherent binding affinity of UL42 to DNA rather than sliding behavior. Because access to the ds end was effectively blocked by tethering the P/T to the chip surface, these experiments do not address the possibility that in solution, UL42 can slide off a free ds end faster or more efficiently than it can dissociate from a blocked or free end with a 5′ ss overhang.
TABLE 2.
Comparison of apparent kinetic constants for binding of UL42 to blocked and unblocked P/T in 50 mM KCla
| Binding surface | kon (M−1 s−1) | koff (s−1) | Kd (M) |
|---|---|---|---|
| Std. P/T + Abb | 1.5 × 104 | 3.7 × 10−3 | 7.4 × 10−8 |
| DIG P/T, no Abc | 6.2 × 103 | 1.1 × 10−3 | 1.8 × 10−7 |
| DIG P/T + Abd | 7.7 × 101 | 1.0 × 10−3 | 4.8 × 10−5 |
Binding to the indicated P/T was measured using the biosensor assay. All kinetic constants were derived by global fitting as indicated in footnote c of Table 1.
The binding surface contained the standard (Std.) biotinylated P/T over which DIGoxigenin-specific antibody (Ab) had been passed.
The binding surface contained DIG-tagged, biotinylated P/T.
The binding surface contained DIG-tagged, biotinylated P/T over which DIG-specific Ab had been passed.
Taken together, these results demonstrate that the Pol accessory protein UL42 does not have an inherently higher affinity, compared to the catalytic subunit, for our model P/T, at any of the buffer conditions we tested. In fact, under the high-ionic strength conditions in which Pol/UL42 holoenzyme retains full activity, UL42 when tested alone, binds with lower affinity and less stably than does Pol. These results indicate that the stable association of Pol/UL42 for the P/T in high salt cannot be explained by simple tethering of the Pol by UL42. However, they do not exclude the possibility that conformational changes conferred to either or both of the subunits by their physical interaction may alter the nature and stability of the contacts of the respective subunits with P/T when bound by the holoenzyme.
DISCUSSION
DNA polymerase processivity factors allow the catalytic polymerase to remain associated with the DNA template through many successive additions of deoxynucleoside triphosphates for the replication of complete genomes within a limited time frame. Although the mechanism by which toroid-shaped processivity factors, such as PCNA and E. coli DNA polymerase III β subunit, allow their cognate polymerases to assemble onto and remain attached to the DNA template has been well established (24), it is less clear how nontoroid processivity factors control the ability of their polymerases to bind and remain attached to DNA template for long-chain DNA synthesis (23). A common feature of the latter class of processivity factors, which includes thioredoxin (for T7 DNA polymerase) and the HSV-1 UL42 protein, is the lack of a requirement for assembly factors and ATP hydrolysis (30). The crystal structure of the essential N-terminal two-thirds of the UL42 protein (ΔC320) was recently reported, and it was found to have striking similarity to the structure of PCNA, despite its lack of sequence homology with PCNA or other sliding clamps (44). However, the relationship between the two topologically similar domains of UL42 protein differs from those of PCNA, leading to a flatter profile and inability to be assembled as subunits of a ring capable of closely encircling DNA without conformational change. Thus, the mechanism by which UL42 increases the processivity of its cognate polymerase remains unclear.
Intrinsic binding properties of each subunit to model P/T differs from that of the holoenzyme.
UL42 protein is unusual in its ability to bind DNA, and it has been proposed that it may tether the Pol to its template via these interactions. In low-salt buffer, the apparent affinity by which monomeric UL42 binds to P/T does not differ substantially from that of the Pol catalytic subunit. However, the intrinsic binding capability of UL42 is dramatically reduced with increased ionic strength. Likewise, two independent measures, nitrocellulose filter-binding assays and biosensor assays, demonstrated that the apparent affinity of the Pol catalytic subunit to P/T is reduced substantially by increased ionic strength. The activity of the Pol catalytic subunit is also highly sensitive to increases in ionic strength. Thus, the resistance of the activity of the holoenzyme to a wide range of salt concentrations (50 to 125 mM KCl) reflects the intrinsic binding behavior of neither subunit alone but is consistent with the binding behavior of the complex as a function of ionic strength.
We interpret these results to indicate that UL42 does not increase Pol processivity by virtue of a high affinity for DNA, at least under high-ionic-strength conditions. Studies which have previously reported a strong binding affinity of UL42 for DNA have utilized relatively low-ionic-strength buffers. For example, Weisshart et al. (41) scored single UL42-DNA complexes under extremely low-salt conditions (10 mM KCl) using gel shift analysis to estimate a Kd of 1.4 nM. Although we did not test for binding under such low-salt conditions, our results (Fig. 7) indicate that interactions between UL42 and DNA are extremely sensitive to KCl concentrations above 50 mM. In another study, Gottlieb and Challberg (14) estimated a low Kd (1.1 nM) for UL42-DNA interactions in 50 mM NaCl using nitrocellulose filter-binding and mobility shift experiments with a synthetic hairpin DNA substrate. However, the ds portion of the substrate contained 31 bp and would be capable of binding more than one monomer of UL42 over most of the range of concentrations of protein which they utilized. Thus, it is possible that the Kd reported by these researchers was underestimated.
In contrast to the results reported herein, Franz et al. (11) reported that UL42 binding to a 159-bp DNA fragment was resistant to 150 mM ammonium sulfate, although they did not quantify the amount of binding observed. It is not clear why their results are different from those we have shown. However, differences may be due to the higher concentration of UL42 used by Franz and coworkers (greater than fivefold) compared to those we have used, the possibility that their purified fractions contained a subpopulation of UL42 molecules not found in ours (e.g., different levels of phosphorylation), and/or the larger DNA fragment used for their binding studies. In support of the latter possibility, Randell and Coen have noted that dissociation of UL42 in solution is more rapid from smaller DNA fragments than from larger DNA fragments (34).
Comparison of HSV-1 UL42 with other nontoroidal polymerase processivity factors.
Nontoroidal processivity factors, such as thioredoxin, share many common features, but it is not yet clear whether they utilize similar mechanisms to increase the processivity of their respective Pols (23). Like thioredoxin, UL42 protein when bound to its cognate polymerase catalytic subunit increases the processivity of the Pol catalytic subunit. Thioredoxin also reduces the Kd of the holoenzyme for P/T from that of the catalytic subunit, although it does not possess intrinsic DNA binding capability. In this report, we reveal that strong DNA binding ability of UL42 is restricted to low-salt conditions and does not correlate with the activity of the holoenzyme. Interestingly, the Kd of the T7 gene 5 protein/thioredoxin holoenzyme was shown to be dependent on salt (19), in contrast to what we observed for the HSV-1 DNA polymerase holoenzyme. The salt dependence of the Kd for the gene 5 protein/thioredoxin complex indicated that the holoenzyme possessed more charge-charge interaction with the DNA than did the catalytic subunit alone. Thus, our results suggest that the enhanced affinity of the HSV-1 Pol/UL42 for DNA occurs by a mechanism less dependent upon increased electrostatic interactions than that observed for the T7 holoenzyme.
Instead, the properties we have ascribed to the HSV-1 DNA polymerase holoenzyme and its subunits may more closely resemble those of the human mitochondrial DNA Pol γ. The p55 accessory subunit of Pol γ, like UL42 protein, possesses intrinsic ability to bind to DNA and binds preferentially to ds DNA (26). Moreover, p55 binds Pol γ with tight affinity in the absence of DNA to form a heterodimer and increases its processivity on complex activated DNA templates and on a poly(rA)-oligo(dT) template. As observed for the HSV-1 Pol catalytic subunit, Pol γ is highly sensitive to salt, and like UL42, the p55 accessory subunit stimulates the activity of its cognate Pol on activated salmon sperm DNA templates in a salt-dependent manner (26). Moreover, under low-salt conditions, both accessory proteins inhibit their respective polymerases (1, 26). Taken together, these features suggest that the p55 subunit of Pol γ and UL42 protein may share a common mechanism for increasing the processivity of their cognate Pols. Nevertheless, we were unable to observe any significant homology between UL42 and murine Pol γ accessory protein at either the amino acid or structural level (results not shown).
Implications for the method by which UL42 increases Pol processivity.
Given the salt sensitivity for binding of each HSV-1 DNA polymerase subunit alone to P/T, how, then, is the binding of the holoenzyme to the P/T maintained to permit the processive DNA synthesis required for a replicative DNA polymerase? It has been shown by circular dichroism that interaction between the Pol catalytic subunit and DNA alters the conformation of Pol (42). Nevertheless, our results and those of others (41) suggest a relatively rapid dissociation rate of Pol from the DNA, leading to lower processivity compared to that of the holoenzyme. We propose a model in which the interaction between HSV-1 Pol and UL42 also confers a substantial conformational change on one or both protein subunits (Fig. 8). Interaction between Pol and UL42 is mediated by the C terminus of Pol (8). The latter domain may not be well-ordered in the absence of UL42, but upon binding to UL42, its conformation could change. Zuccola et al. (44) have resolved the crystal structure of a short C-terminal Pol peptide bound to a large N-terminal domain of UL42. Their results, compared to the results of a previous nuclear magnetic resonance analysis of the Pol peptide, demonstrate that the Pol peptide is better ordered when bound to UL42 than it is in solution (3, 44). Thus, the C terminus of the HSV-1 Pol may form a flexible domain, similar to the extended loop in the thumb domain of T7 Pol which binds to thioredoxin (9). As suggested for T7 DNA polymerase (9, 23), binding of the UL42 protein to a flexible or disordered C-terminal Pol domain could lead to a more closed conformation around the DNA, resulting in a greatly reduced dissociation rate and increased processivity, particularly under high-ionic-strength conditions. Proof of such a model must await revelation of the structural details of HSV-1 Pol with and without UL42.
FIG. 8.

Conformational model for mechanism by which UL42 increases Pol processivity. Pol and Pol/UL42 are represented schematically before (left) and after (right) binding to P/T. (Top) Although Pol alone displays some change in conformation upon DNA binding, it dissociates rapidly from the P/T, leading to low processivity. (Bottom) Binding of UL42 to the Pol C terminus alters the conformation of Pol and in the presence of DNA, the two proteins together serve as a clamp, decreasing the dissociation rate and increasing processivity.
Acknowledgments
We thank members of the Parris laboratory for many helpful discussions during the course of this work and Tim Vojt for computer-aided design of the model in Fig. 8.
This work was supported in part by grant GM34930 from the National Institutes of Health. Core services were provided by the Department of Molecular Virology, Immunology, and Medical Genetics of Ohio State University and by a Comprehensive Cancer Center core grant from the National Cancer Institute (P30 CA16058).
REFERENCES
- 1.Berthommé, H., S. J. Monahan, D. S. Parris, B. Jacquemont, and A. L. Epstein. 1995. Cloning, sequencing, and functional characterization of the two subunits of the pseudorabies virus DNA polymerase holoenzyme: evidence for specificity of interaction. J. Virol. 69:2811-2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bondeson, K., A. Frostell-Karlsson, L. Fagerstam, and G. Magnusson. 1993. Lactose repressor-operator DNA interactions: kinetic analysis by a surface plasmon resonance biosensor. Anal. Biochem. 214:245-251. [DOI] [PubMed] [Google Scholar]
- 3.Bridges, K. G., Q.-X. Hua, M. R. Brigham-Burke, J. D. Martin, P. Hensley, C. E. Dahl, P. Digard, M. A. Weiss, and D. M. Coen. 2000. Secondary structure and structure-activity relationships of peptides corresponding to the subunit interface of herpes simplex virus DNA polymerase. J. Biol. Chem. 275:472-478. [DOI] [PubMed] [Google Scholar]
- 4.Burgers, P. M. J., and B. L. Yoder. 1993. ATP-independent loading of the proliferating cell nuclear antigen requires DNA ends. J. Biol. Chem. 268:19923-19926. [PubMed] [Google Scholar]
- 5.Chow, C. S., and D. M. Coen. 1995. Mutations that specifically impair the DNA binding activity of the herpes simplex virus protein UL42. J. Virol. 69:6965-6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crute, J. J., and I. R. Lehman. 1989. Herpes simplex-1 DNA polymerase. Identification of an intrinsic 5′-3′ exonuclease with ribonuclease H activity. J. Biol. Chem. 264:19266-19270. [PubMed] [Google Scholar]
- 7.Digard, P., C. S. Chow, L. Pirrit, and D. M. Coen. 1993. Functional analysis of the herpes simplex virus UL42 protein. J. Virol. 67:1159-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Digard, P., and D. M. Coen. 1990. A novel functional domain of an α-like DNA polymerase. The binding site on the herpes simplex virus polymerase for the viral UL42 protein. J. Biol. Chem. 26:17393-17396. [PubMed] [Google Scholar]
- 9.Doublié, S., S. Tabor, A. M. Long, C. C. Richardson, and T. Ellenberger. 1998. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature (London) 391:251-258. [DOI] [PubMed] [Google Scholar]
- 10.Falkenberg, M., I. R. Lehman, and P. Elias. 2000. Leading and lagging strand DNA synthesis in vitro by a reconstituted herpes simplex virus type 1 replisome. Proc. Natl. Acad. Sci. USA 97:3896-3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franz, C., F. J. P. Kühn, and C. W. Knopf. 1999. DNA and protein interactions of the small subunit of herpes simplex virus type 1 DNA polymerase. Virology 253:55-64. [DOI] [PubMed] [Google Scholar]
- 12.Gallo, M. L., D. I. Dorsky, C. S. Crumpacker, and D. S. Parris. 1989. The essential 65-kilodalton DNA-binding protein of herpes simplex virus stimulates the virus-encoded DNA polymerase. J. Virol. 63:5023-5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallo, M. L., D. H. Jackwood, M. Murphy, H. S. Marsden, and D. S. Parris. 1988. Purification of the herpes simplex virus type 1 65-kilodalton DNA-binding protein: properties of the protein and evidence of its association with the virus-encoded DNA polymerase. J. Virol. 62:2874-2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gottlieb, J., and M. D. Challberg. 1994. Interaction of herpes simplex virus type 1 DNA polymerase and the UL42 accessory protein with a model primer template. J. Virol. 68:4937-4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gottlieb, J., A. I. Marcy, D. M. Coen, and M. D. Challberg. 1990. The herpes simplex virus type 1 UL42 gene product: a subunit of DNA polymerase that functions to increase processivity. J. Virol. 64:5976-5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hart, G. J., and R. E. Boehme. 1992. The effect of the UL42 protein on the DNA polymerase activity of the catalytic subunit of the DNA polymerase encoded by herpes simplex virus type 1. FEBS Lett. 305:97-100. [DOI] [PubMed] [Google Scholar]
- 17.Hernandez, T. R., and I. R. Lehman. 1990. Functional interaction between the herpes simplex-1 DNA polymerase and UL42 protein. J. Biol. Chem. 265:11227-11232. [PubMed] [Google Scholar]
- 18.Huber, H. E., M. Russel, P. Model, and C. C. Richardson. 1986. Interaction of mutant thioredoxins of Escherichia coli with the gene 5 protein of phage T7. The redox capacity of thioredoxin is not required for stimulation of DNA polymerase activity. J. Biol. Chem. 261:15006-15012. [PubMed] [Google Scholar]
- 19.Huber, H. E., S. Tabor, and C. C. Richardson. 1987. Escherichia coli thioredoxin stabilizes complexes of bacteriophage T7 DNA polymerase and primed templates. J. Biol. Chem. 262:16224-16232. [PubMed] [Google Scholar]
- 20.Jofre, J. T., P. A. Schaffer, and D. S. Parris. 1977. Genetics of resistance to phosphonoacetic acid in strain KOS of herpes simplex virus type 1. J. Virol. 23:833-836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson, K. A. 1995. Rapid quench analysis of polymerases, adenosine triphosphatases, and enzyme intermediates. Methods Enzymol. 249:38-61. [DOI] [PubMed] [Google Scholar]
- 22.Johnson, P. A., M. G. Best, T. Friedmann, and D. S. Parris. 1991. Isolation of a herpes simplex virus type 1 mutant deleted for the essential UL42 gene and characterization of its null phenotype. J. Virol. 65:700-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelman, Z., J. Hurwitz, and M. O'Donnell. 1998. Processivity of DNA polymerases: two mechanisms, one goal. Structure 6:121-125. [DOI] [PubMed] [Google Scholar]
- 24.Kelman, Z., and M. O'Donnell. 1995. Structural and functional similarities of prokaryotic and eukaryotic DNA polymerase sliding clamps. Nucleic Acids Res. 23:3613-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klinedinst, D., and M. D. Challberg. 1994. Helicase-primase complex of herpes simplex virus type 1: a mutation in the UL52 subunit abolishes primase activity. J. Virol. 68:3693-3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim, S. E., M. J. Longley, and W. C. Copeland. 1999. The mitochondrial p55 accessory subunit of human DNA polymerase γ enhances DNA binding, promotes processive DNA synthesis, and confers N-ethylmaleimide resistance. J. Biol. Chem. 274:38197-38202. [DOI] [PubMed] [Google Scholar]
- 27.Marcy, A. I., P. D. Olivo, M. D. Challberg, and D. M. Coen. 1990. Enzymatic activities of overexpressed herpes simplex virus DNA polymerase purified from recombinant baculovirus-infected cells. Nucleic Acids Res. 18:1207-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monahan, S. J., T. F. Barlam, C. S. Crumpacker, and D. S. Parris. 1993. Two regions of the herpes simplex virus type 1 UL42 protein are required for its functional interaction with the viral DNA polymerase. J. Virol. 67:5922-5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monahan, S. J., L. A. Grinstead, W. Olivieri, and D. S. Parris. 1998. Interaction between the herpes simplex virus type 1 origin-binding and DNA polymerase accessory proteins. Virology 241:122-130. [DOI] [PubMed] [Google Scholar]
- 30.O'Donnell, M. E., P. Elias, and I. R. Lehman. 1987. Processive replication of single-stranded DNA templates by the herpes simplex virus-induced DNA polymerase. J. Biol. Chem. 262:4252-4259. [PubMed] [Google Scholar]
- 31.Parris, D. S., A. Cross, L. Haarr, A. Orr, M. C. Frame, M. Murphy, D. J. McGeoch, and H. S. Marsden. 1988. Identification of the gene encoding the 65-kilodalton DNA-binding protein of herpes simplex virus type 1. J. Virol. 62:818-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel, S. S., I. Wong, and K. A. Johnson. 1991. Presteady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry 30:511-525. [DOI] [PubMed] [Google Scholar]
- 33.Purifoy, D. J. M., R. B. Lewis, and K. L. Powell. 1977. Identification of the herpes simplex virus DNA polymerase gene. Nature (London) 269:621-623. [DOI] [PubMed] [Google Scholar]
- 34.Randell, J. C., and D. M. Coen. 2001. Linear diffusion on DNA despite high-affinity binding by a DNA polymerase processivity factor. Mol. Cell 8:911-920. [DOI] [PubMed] [Google Scholar]
- 35.Sherman, G., J. Gottlieb, and M. D. Challberg. 1992. The UL8 subunit of the herpes simplex virus helicase-primase complex is required for efficient primer utilization. J. Virol. 66:4884-4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skaliter, R., and I. R. Lehman. 1994. Rolling circle DNA replication in vitro by a complex of herpes simplex virus type 1-encoded enzymes. Proc. Natl. Acad. Sci. USA 91:10665-10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tabor, S., H. E. Huber, and C. C. Richardson. 1987. Escherichia coli thioredoxin confers processivity on the DNA polymerase activity of the gene 5 protein of bacteriophage T7. J. Biol. Chem. 262:16212-16223. [PubMed] [Google Scholar]
- 38.Tenney, D. J., P. A. Micheletti, J. T. Stevens, R. K. Hamatake, J. T. Matthews, A. R. Sanchez, W. W. Hurlburt, M. Bifano, and M. G. Cordingley. 1993. Mutations in the C terminus of herpes simplex virus type 1 DNA polymerase can affect binding and stimulation by its accessory protein UL42 without affecting basal polymerase activity. J. Virol. 67:543-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thornton, K. E., M. Chaudhuri, S. J. Monahan, L. A. Grinstead, and D. S. Parris. 2000. Analysis of in vitro activities of herpes simplex virus type 1 UL42 mutant proteins: correlation with in vivo function. Virology 275:373-390. [DOI] [PubMed] [Google Scholar]
- 40.Tsurumi, T., T. Daikoku, and Y. Nishiyama. 1994. Further characterization of the interaction between Epstein-Barr virus DNA polymerase catalytic subunit and its accessory subunit with regard to the 3′-to-5′ exonucleolytic activity and stability of initiation complex at primer terminus. J. Virol. 68:3354-3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weisshart, K., C. S. Chow, and D. M. Coen. 1999. Herpes simplex virus processivity factor UL42 imparts increased DNA-binding specificity to the viral DNA polymerase and decreased dissociation from primer-template without reducing the elongation rate. J. Virol. 73:55-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weisshart, K., A. A. Kuo, G. R. Painter, L. L. Wright, P. A. Furman, and D. M. Coen. 1993. Conformational changes induced in herpes simplex virus DNA polymerase upon DNA binding. Proc. Natl. Acad. Sci. USA 90:1028-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong, I., and T. M. Lohman. 1993. A double-filter method for nitrocellulose binding: application to protein-nucleic acid interactions. Proc. Natl. Acad. Sci. USA 90:5428-5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuccola, H. J., D. J. Filman, D. M. Coen, and J. M. Hogle. 2000. The crystal structure of an unusual processivity factor, herpes simplex virus UL42, bound to the C terminus of its cognate polymerase. Mol. Cell 5:267-278. [DOI] [PubMed] [Google Scholar]