

Abstract
Although human immunodeficiency virus type 1 (HIV-1) recombinants have been found with high frequency, little is known about the forces that select for these viruses or their importance to pathogenesis. Here we document the emergence and dynamics of 11 distinct HIV-1 recombinants in a man who was infected with two subtype B HIV-1 strains and progressed rapidly to AIDS without developing substantial cellular or humoral immune responses. Although numerous frequency oscillations were observed, a single recombinant lineage eventually came to dominate the population. Numerical simulations indicate that the successive recombinant forms displaced each other too rapidly to be explained by any simple model of random genetic drift or sampling variation. All of the recombinants, including several resulting from independent recombination events, possessed the same sequence motif in the V3 loop, suggesting intense selection on this segment of the viral envelope protein. The outgrowth of the predominant V3 loop recombinants was not, however, associated with changes in coreceptor utilization. The final variant was instead notable for having lost 3 of 14 potential glycosylation sites. We also observed high ratios of synonymous-to-nonsynonymous nucleotide changes—suggestive of purifying selection—in all viral populations, with particularly high ratios in newly arising recombinants. Our study, therefore, illustrates the unusual and important patterns of viral adaptation that can occur in a patient with weak immune responses. Although it is hard to tease apart cause and effect in a single patient, the correlation with disease progression in this patient suggests that recombination between divergent viruses, with its ability to create chimeras with increased fitness, can accelerate progression to AIDS.
A striking characteristic of human immunodeficiency virus type 1 (HIV-1) infection is the development of substantial genetic diversity within the viral population, resulting from high replication and mutation rates and, perhaps, selection for altered cell type specificity and immunological escape (9, 56, 63; B. D. Walker and P. J. Goulder, Letter, Nature 407:313-314, 2000). Accumulating evidence suggests that the formation and spread of virus genome chimeras (recombinant viruses), are also important in the AIDS pandemic, accounting for a significant fraction of HIV transmissions (5, 6, 26, 37, 50, 53, 55), including recent outbreaks in China and Russia (33, 45, 59). These circulating recombinants were generated by recombining sequences from two HIV-1 major (M) group subtypes. Other recombinants have been reported to occur within a single HIV-1 subtype (15) and between HIV-1 groups M and O (43, 61). Since the formation of virus recombinants requires coinfection of the same target cell with two or more viruses and production and spread of heterodimeric virions (those containing one copy of viral RNA from each of two strains), it is remarkable that recombinants have been detected with such high frequency (10).
Previous studies of HIV-1 recombinants have focused mainly on the mosaic structures of the genomes, whereas the selective advantage or competitive selection for virus fitness (the relative ability to produce infectious progeny in a given environment) (16) associated with recombination in vivo has not been critically explored. While it is generally assumed that selection is responsible for the appearance of recombinant forms, other possibilities, such as neutral recombination and genetic drift, have not been rigorously evaluated. Despite evidence for selection in HIV-1 env across groups of human patients (65) and evidence for selection in specific cytotoxic T lymphocyte (CTL) epitopes encoded by gag and tat in simian immunodeficiency virus (SIV)-infected macaques (1), analyses of longitudinally sampled HIV-1 sequences from individual patients have thus far failed to reveal any statistically clear examples of natural selection acting during chronic infection in association with the development of AIDS (32; D. R. Shriner, R. Shankarappa, M. A. Jensen, D. C. Nickle, J. E. Mittler, J. B. Margolick, and J. I. Mullins, submitted for publication). Demonstrating that selection favors recombinants with higher fitness, as well as enumerating the type and magnitude of selective changes, will further our understanding of HIV-1 evolution in vivo and may help in rational vaccine design.
In this study we report on the successive changes in the infecting virus population within a patient in whom a series of recombinant viruses appeared. A previous study showed that the man described here was initially infected with two divergent subtype B HIV-1 strains, referred to as common (C) and unique (U) variants, that were distinguishable as a result of being phylogenetically unlinked and ∼15% divergent over the env C2-V5 coding region (34). This patient experienced a rapid disease course, developed no detectable neutralizing immune response against autologous virus, and had undetectable or low levels of cellular immune responses against heterologous viral antigens (34). Here we report on the evolutionary dynamics of viral recombinants formed between C and U variants in this patient and explore the potential role of selection in this process. Our data indicate that recombination performs an important function in creating chimeras with higher fitness, and that purifying, competitive selection can play an important role in the evolutionary dynamics of HIV-1.
MATERIALS AND METHODS
Study subjects and specimens.
Initial clinical progression and virologic and immunologic characterization of this patient were reported previously (34). This patient (patient B), a Caucasian male, was presumed to be infected on the same date as his homosexual partner (patient A), following contact with a third HIV-1-infected male. He progressed to AIDS within 2 years, was treated with multiple antiretroviral drugs (Fig. 1A) beginning at 27 months postinfection (p.i.), and died 62 months p.i. By contrast, his sexual partner, who was infected with only the common variant, maintained strong HIV-1-specific immune responses and did not develop AIDS before going onto highly active antiretroviral therapy 4 years after infection (34) (data not shown). Samples from 11 time points from patient B (from 5, 6, 21, 30, 33, 35, 38, 44, 47, 53, and 56 months p.i.) were available for this study.
FIG. 1.

Sequential analysis of immunological and virologic parameters in patient B. (A) CD4+- and CD8+-cell numbers, HIV-1 RNA copies per milliliter of plasma, and the use of antiretroviral therapies over time. Abbreviations: ZDV, zidovudine; ddI, didanosine; d4T, stavudine; 3TC, lamivudine; NVP, nevirapine; NFV, nelfinavir. (B) Viral lineage frequencies measured as the percentage of the total number of clones examined at each time point. (C) Log ratios of the frequency of parental C-derived sequences to that of parental U-derived sequences in V3 and C3-V5, respectively. A dashed line indicates the initial value of ln(C/U). (D and E) dS/dN (see Materials and Methods) over time in the V3 loop (D) and C3-V5 (E). The symbols for both panels are defined in panel E. The label “0/0” at the bottom of panel D indicates no synonymous mutations or nonsynonymous mutations. Lines connected to these points are dashed to signify that 0/0 is not interpretable. Asterisks indicate significance (P < 0.05) for the indicated time points at a dS/dN ratio of >1.
Nucleic acid preparation, PCR, and sequence analysis.
We examined 371 molecular clones of HIV-1 sequences derived from a total of 11 time points over the course of infection, including those in peripheral blood mononuclear cells (PBMC), plasma, and PBMC-derived viral isolates from almost every time point. Primary and cocultured PBMC viral DNAs were quantified and amplified by nested PCR as previously described (34) using first-round PCR primers ED5 and ED12 and second-round primers DR7 and DR8 to amplify the C2-V5 region of env. To avoid template resampling (S.-L. Liu, A. G. Rodrigo, R. Shankarappa, et al., Letter, Science 273:415-416, 1996), about 500 amplifiable copies of viral DNA (from PBMC and cocultured cells) were used for PCR in most experiments. PBMC viral DNA at 47 and 53 months p.i. had fewer than 20 templates; thus, individual endpoint PCR products were sequenced in order to exclude resampling. Viral RNA load in plasma was determined by the Quantiplex HIV-RNA assay (Chiron Corporation, Emeryville, Calif.) and by serial endpoint dilution and PCR of cDNA as previously described (34). About 500 plasma RNA templates were used in each PCR with a hot-start protocol (Roche Molecular System. Inc., Branchburg, N.J.). PCR products were cloned, sequenced, and analyzed as previously described (34).
Virus stocks, cloning of expression competent env genes, and virus pseudotype construction.
Viral isolates were obtained from samples taken at 30, 33, and 35 months p.i. using methods previously described (34). gp160 coding sequences were amplified from these infected cells using the Genamp XL PCR kit (Perkin-Elmer Cetus, Foster City, Calif.) with primers ED3 and Nef3 (57) for the first round and primers MP1 (5′-AAATATGgctagcAAGGGGATCAGGAAGAATTATCAG; NheI site shown in lowercase letters) and JA506 (5′-CGACggatccTTTGACCACTTGCCACCCAT [for HIV-1 HXB2 positions 8825 to 8796]; EcoRI site shown in lowercase letters) for the second round. DNA extracted from the parental virus isolate (obtained at 9 and 5 months p.i. for C and U variants, respectively) and those obtained at 33 and 35 months were used as templates for PCR amplification. PCR conditions were 94°C for 15 s and 68°C for 10 min for the first 16 cycles, followed by an additional 12 cycles of 94°C for 15 s and 68°C for 8 min, with increments of 15 s per cycle, with magnesium acetate at 1.10 mM. Second-round PCR products were cloned into pWR508 (64) to provide improved clone stability and then subcloned into pcDNA3.1(−) (Invitrogen, San Diego, Calif.). Max Efficiency STBL2 competent cells (Life Technologies, Gaithersburg, Md.) were used for transformation. Positive clones were screened by restriction enzyme digestion and confirmed by DNA sequencing of insert termini. For each virus, at least three clones were tested for gp160 expression by Western blotting with patient sera (1:1,000) and enhanced chemiluminescence detection (Amersham Life Science, Little Chalfont, Buckinghamshire, United Kingdom).
Generation of reporter virus pseudotypes with HIV-1 envelopes and coreceptor specificity determinations.
293T cells were cotransfected with 10 μg of the luciferase-producing NL4-3-Luc-R−E− (11) and 10 μg of each HIV-1 Env-expressing vector pcDNA3.1(−). Plasmids expressing HIV-1 HXB2 (20) and JRFL (29) env genes were used as positive controls. Virus-containing supernatants were collected 48 h posttransfection, passed through 0.2-μm-pore-size filters (Corning, Corning, N.Y.), stored at −80°C, and quantified by p24 enzyme-linked immunosorbent assay (Abbott Laboratory, Chicago, Ill.).
A panel of U87.MG CD4+ cells coexpressing CCR1, -2b, -3, or -5 or CXCR4 (kind gift of Dan R. Littman) were used to determine coreceptor use (14) of the molecular clone-derived Env, pseudotyped virus, and biological isolates. Cells were plated at 2 × 104 cells per well in a 24-well plate in the presence of selection medium. The following day, virus (50 to 100 ng of p24) was added and incubated for 4 h at 37°C, and then the plates were washed. Cells were lysed 4 days after infection and lysates assayed for luciferase activity in a Berthold (Bad Wilbad, Germany) Autolumat luminometer.
Statistical analyses.
To assess selection, estimates for dS and dN were derived from MEGA, version 2.1 (31). The method of Kumar separates sites into zerofold-, twofold-, and fourfold-degenerate categories. The twofold-degenerate sites are further subdivided into simple and complex twofold-degenerate sites, where simple twofold-degenerate sites are those at which a transitional change results in a synonymous substitution and the two transversional changes result in nonsynonymous substitutions. All other twofold-degenerate sites, including those for the three isoleucine codons, belong to the complex twofold-degenerate-site category. Standard errors were estimated from 1,000 bootstrap replicates. dS/dN ratios were evaluated using procedures described by Rodrigo et al. (51). A χ2 test was used to determine whether there were differences in the numbers of phylogenetically informative sites on either side of a putative breakpoint that supported an association with one or the other parental lineage. The expected values used in the χ2 test were estimated by taking the relative proportions of phylogenetically informative sites in which the recombinant lineage was associated with the C versus the U parent and multiplying these by the total length of the two regions on either side of the breakpoint. This procedure is equivalent to randomly permuting sites while holding constant the raw marginal totals in the two-by-two contingency table.
Estimation of selection coefficients.
The selective advantage, s, that one type has over another during the time interval [t1, t2] is given by the following equation: s = (ln[p(t2)/p(t1)] − ln[q(t2)/q(t1)])/g[t2 − t1], where p(t) and q(t) are the frequencies of the two types at time t (measured in days) and g is the number of days per generation (38). To get an approximate estimate for the variability in our estimates for s, we calculated 95% bootstrap ranges for each value of s by drawing Nt1 and Nt2 sequences (where Nt1 and Nt2 are total number of sequences obtained at times t1 and t2, respectively) at random from simulated multinomial distributions in which the probabilities of choosing the dominant type, the previously dominant type, and “any other type” were p(t), q(t), and 1 − p(t) − q(t), respectively, for t = t1 and t = t2. In cases when the dominant type was not detected at time t1 or when the previously dominant type was not detected at time t2, the corresponding probabilities for p or q were set to 0.5/Nt, to allow the calculation of an approximate lower bound for the 95% bootstrap range on s.
Simulation of genetic drift.
To simulate genetic drift in the patient we created a Monte Carlo program in which N sequences were selected at random with replacement each generation. The program recorded the change index (C), as defined in the text, for each simulation. The sampling scheme (number of sequences sampled at different time points) was designed to mimic the actual sampling scheme from the patient, assuming a viral generation time of 1.78 days (23). The initial genotypic frequencies were chosen to match those in the patient. To simulate the formation of new lineages by mutation or recombination and the reintroduction of previous ones from putative virus reservoirs, an average of rN individuals of each of 10 lineages (the original two, plus eight new ones) were introduced into the population each generation, where r is the reintroduction rate. These simulations were repeated 1,000 times for N = 50, 100, 150,…, 500 (values of N under which rapid frequency fluctuations sometimes occur) and r = 0, 0.001, 0.002, 0.004,…, 0.064 (values of r that span the range under which the change index is maximized). Copies of this simulation program (written in C) can be obtained from the authors by request.
Estimate for nucleotide diversity and Ne.
Sequence editing, assembly, and analysis were performed as previously described (34). Nucleotide alignments were generated using ClustalW (62) and then edited and gap stripped using MacClade (version 3; Sinauer Associates, Inc., Sunderland, Mass.). The best model of evolution for this data set was determined to be HKY+ 71, using the hierarchical likelihood ratio test as implemented in ModelTest (46). This model was used in the construction of a neighbor-joining phylogenetic tree (54) in PAUP∗ (version 4.0b2a; Sinauer Associates, Inc., Sunderland, Mass.). We used the formula π = 2Neμ to estimate effective population size from diversity data. Because this formula only applies to sequences that are derived from a single parent, we obtained a reasonable equivalent to the normal value for π by removing mutations from branches that connect the different recombinant lineages using TreeEdit (http://evolve.zoo.ox.ac.uk/software/TreeEdit/main.html). These branch-pruning procedures should yield a conservative estimate for π, which is desirable for our application since we have wanted to obtain a minimum estimate for Ne (see text for explanation).
Nucleotide sequence accession numbers.
Viral sequences were deposited in GenBank under accession numbers U79077 through U79087 and AF185294 through AF185566.
RESULTS
Emergence and evolutionary dynamics of chimeric viral populations.
To monitor the abundance of viral populations over time we sequenced a total of 371 molecular clones (9 to 55 clones from each time point) from three sources (plasma, PBMC, and PBMC-cocultured viral isolates) over a period of 4.6 years. No significant differences were observed among these sources for the parameters examined here (data not shown); hence, the frequencies of different variants from these populations were pooled for the following analyses.
The parental U lineage predominated early in infection; however, by month 30 p.i. it was no longer detected and was replaced by the C parent and a heterogeneous collection of recombinants (Fig. 1B). These replacements coincided with a five- to sevenfold increase in viral load and a substantial drop in both CD4+- and CD8+-T-cell counts. Between months 30 and 53 p.i., genotypic frequencies fluctuated, with the parental C lineage predominating at months 30 and 38 p.i., and three major recombinants—V3r, C2V3r, and X4r (Fig. 2; see details below)—predominating at months 33, 35, 44, 47, 53, and 56 p.i. (Fig. 1B). From month 53 p.i. and onward, 100% of the sequences sampled belonged to a single V3r sublineage: V3r-3 (Fig. 1B and 2; see details below).
FIG. 2.


Structures of recombinant viral env genes. (A) Schematic representation of the three major recombinants (V3r, C2V3r, and X4r) plus eight minor HIV-1 Env C2-V5 recombinants (MI-1 to MI-8). Each minor recombinant subpopulation consisted of at least three clones from different blood compartments and/or different time points. The breakpoints were assigned and tested as described in Materials and Methods. The number of informative sites is indicated for each segment. Segments derived from the C and U parental sequences and ambiguous regions are indicated by shading as shown at the bottom of the figure. Nucleotide coordinates from the HIV-1 HXB2 genome sequence are provided as reference at the top of the figure, along with approximate boundaries of the relatively conserved and variable regions. (B) Alignment of consensus sequences of HIV-1 env C2-V5 recombinants. Sequences include the two parental lineages U (when detected) and C (at 5, 6, 21, 30, and 38 months p.i.); the X4r, V3r-1, V3r-2, V3r-3, and C2V3r recombinants; and eight minor recombinants (MI-1 to MI-8). Consensus sequences were generated using the program MAJORITY implemented in the genetic data environment (GDE) (58). Sequences likely to have been derived from the C lineage are lightly shaded. The precise breakpoints between C and U lineages were not determined; thus, shading continues to the midpoint between adjacent informative sites from the two parental lineages. Potential N-linked glycosylation sites (NXT or NXS) are underlined. Glycosylation sites that were lost over time are shown with white type on a black background. New glycosylation sites are highlighted with dark shading. An amino acid change (Q to R) responsible for the X4 phenotype of X4r is shown at alignment position 54. Application of Fisher's exact test confirms that the C2V3r recombinants had one breakpoint (P < 0.0001) and that the V3r and X4r recombinants had two breakpoints (P < 0.0001 for the two breakpoints in V3r; P < 0.0005 for the two breakpoints in X4r).
The V3r recombinant lineage, with seven phylogenetically informative mutation sites in common with the C lineage flanked on both sides by U variant sequences (P < 0.0001 for the assigned breakpoints) (Fig. 2), was the first detected (at 21 months p.i. in plasma) and the most-abundant recombinant lineage identified in this patient. Three subgroups of the V3r lineage (termed V3r-1, -2, and -3) evolved sequentially (Fig. 1B). The V3r-1 subgroup was abundant through 47 months p.i. but was not detected thereafter. V3r-2 was present at relatively low levels through 44 months and was not detected thereafter. V3r-3 was first detected at 44 months p.i. and then dominated at 53 and 56 months p.i. Recombinants generated between the V3r-1 and V3r-3 lineages were also detected at 47 months p.i. (data not shown), when both subgroups were abundant.
C2V3r had one breakpoint (P < 0.0001) separating the C2-V5 region into two parts: the C2-V3 region derived from the C lineage and the C3-V5 region derived from the U lineage (Fig. 2). This recombinant was first identified at 33 months p.i. (Fig. 1B) and was continuously detected through 44 months, during which time 26% of the 140 sequences examined had the C2V3r structure. Phylogenetic analysis suggested that C2V3r was more likely to have evolved from a V3r parent than from the parental U variant found at 5, 6, and 21 months p.i. (data not shown).
Another recombinant, X4r, was found in the viral isolates from 30 and 35 months p.i. and in the PBMC from 44 months p.i. (Fig. 1B). As shown in Fig. 2B, the V3 loop of X4r was a hybrid of C and U lineages (P < 0.0001 for this breakpoint; P = 0.0008 for a breakpoint upstream of the V3 loop). Also distinguishing X4r from other subpopulations was the elimination of a potential N-linked glycosylation site spanning the first cysteine of the V3 loop (Fig. 2B). X4r sequences retained the GPGR motif found in all U parent sequences except for those at 21 months p.i. (the last time the U parent was detected) (Fig. 2). This, plus phylogenetic analysis of the nonrecombinant regions (data not shown), suggests that the X4r recombinant evolved before 21 months p.i. However, it never obtained high-level representation except transiently in the PBMC-culturable virus population.
In addition to the more abundant recombinants discussed above, eight minor recombinant subpopulations (consisting of at least two molecular clones with distinct mosaic structures) (Fig. 2A) and five unique recombinants (consisting of single molecular clones with distinct mosaic structures), were also identified in this patient (data not shown). Almost all of the minor recombinants were detected from multiple time points or from different sample sources at the same time, strongly arguing against the possibility of the recombinants forming during PCRs. However, we cannot exclude this possibility for the recombinants represented by only one molecular clone. Hence, the following analyses include only the 11 recombinants found at least twice.
Coreceptor specificity and phenotype of parental and recombinant viruses.
Based on the predicted amino acids of the V3 loop sequences, all of the foregoing viruses were predicted to be non-syncytium-inducing on MT-2 cells and to utilize the CCR5 coreceptor for virus entry, although both the C parent and the recombinant lineages had more basic amino acids than the U parent (Fig. 2B). Only one recombinant lineage, X4r, which was detected over the span from 30 to 44 month p.i. (Fig. 1B), was predicted to be syncytium inducing and utilize the alternative coreceptor CXCR4 (by virtue of a Q-to-R amino acid change at position 25 of the V3 loop) (12, 13, 22). As predicted, virus isolates derived from the parental U and C lineage viruses were found to be non-syncytium inducing in MT-2 cells (data not shown) and to use the CCR5 coreceptor, and neither used CXCR4, CCR2b, or CCR3 (Fig. 3A). Also as predicted, viral isolates from 30 and 35 months p.i. induced syncytia in MT-2 cells (data not shown), and those from 30, 33, and 35 months p.i. used both CCR5 and CXCR4 for entry (Fig. 3A). The viral isolate from 44 months p.i. did not induce syncytia, probably due to minor representation or absence of the X4r variant from culturable virus at this time (data not shown). To confirm these relationships we constructed virus pseudotypes containing envelopes derived from the C, U, and V3r molecular clones and found that each used CCR5, while X4r-derived pseudotypes used only CXCR4 (Fig. 3B). Taken together, these results demonstrate that the acquisition of the X4 phenotype was due to the transient outgrowth of the X4r recombinant.
FIG. 3.

Coreceptor specificity and SI phenotype of virus isolates and cloned env pseudotypes. A panel of U87.MG CD4+ cells coexpressing CCR1, -2b, -3, or -5 or CXCR4 (14) was used to determine coreceptor use (3) of molecularly cloned Env-pseudotyped virus and biological isolates. (A) Viral antigen p24 production in these cells infected with either two parental C and U viruses or with virus isolates at 30, 33, and 35 months p.i. O.D., optical density. (B) Results of virus pseudotype infection of parental U87.MG CD4+ cells and those coexpressing either CCR5 or CXCR4 using cloned viral env sequences to direct entry. Plasmids expressing HIV-1 HXB2 and JRFL env genes were used as positive controls. An empty expression vector with no envelope gene was used as a negative control.
Frequency fluctuations cannot be easily explained by genetic drift.
As shown in Fig. 1B, recombinant populations turned over rapidly. We hypothesize that these fluctuations reflect the outcome of competition between a succession of recombinant forms with differing within-host fitnesses. An alternative hypothesis is that these fluctuations are due to some combination of genetic drift and sampling variation. To distinguish between these two hypotheses, we performed Monte Carlo simulations of random genetic drift with different population sizes, N, assuming a generation time of 1.78 days (23). To mimic sampling variation, the simulated sequences were sampled according to the scheme used for collecting samples from the patient (i.e., 20 sequences at 5 months p.i., 29 sequences at 6 months p.i., etc.). To quantify the extent to which populations fluctuated, we calculated a change index, C, using the following formula:
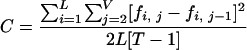 |
where L is the number of recombinant lineages, T is the number of time points, and fi,j is the frequency of lineage i at time point j. This index ranges from a minimum of 0 (no change) to a maximum of 1 (complete replacement of a single dominant lineage by another dominant lineage at every time point). Because frequency changes are squared, large shifts contribute more to the change index than small shifts. This is desirable, because we are interested in determining whether the large frequency shifts seen in Fig. 1B can be explained by drift.
When we applied this index to the data in Fig. 1B, we obtained a C of 0.157. In an initial set of simulations we obtained values for C of ≥0.15 in less than 0.1% of runs with an N of ≥50 (data not shown). However, these initial simulations do not account for the potential for recombinational and mutational input and the reintroduction of sequences from latently infected cells and other long-lived viral reservoirs. To account for such inputs we added a reintroduction term to simulate continuous low-level input of both new and existing recombinant forms. By preventing fixation of genotypes, these inputs increase the number of large frequency shifts that can occur. However, even for those reintroduction rates that maximized the change index (0.004 to 0.016 reintroduction per lineage per generation, depending on the population size), large frequency shifts were restricted to very low population sizes, and the frequency of runs for which C was ≥0.15 fell to negligible levels for N values of >350 (Fig. 4). This approximate cutoff of N = 350 is far below the estimated number of productively infected cells in major viral compartments (2.5 × 107 to 10 × 107 [25]) and is well below typical estimates for the effective population size of HIV-1 in vivo (which have ranged from a low of 512 to a high of >10,000 depending on the patient and method of analysis [32, 52]).
FIG. 4.

Percentage of Monte Carlo simulation runs in which the change index (C; a measure of how rapidly genotypic frequencies fluctuated, see text) was the same as or greater than that observed in the patient (0.157) as a function of population size. Each symbol gives the mean of 1,000 replicates. The dotted line indicates the 5% significance cutoff. Symbols connected with solid lines correspond to reintroduction rates of 0.001 (▵), 0.002 (⋄), 0.004 (□), and 0.008 (○), respectively. Symbols connected with dashed lines correspond to reintroduction rates of 0.016 (▵), 0.032 (⋄), and 0.064 (○), respectively.
To determine the effective population size for this patient, we applied the maximum-likelihood program COALESCE (30) to V3r sequences (the most-prevalent type) to all time points for which we obtained at least 10 V3r sequences. Assuming a point mutation rate of 2.6 × 10−5 mutations/site/generation (35, 36), we obtained an average Ne of 2,425 (range, 500 to 7,163); however, it is not clear that all of the assumptions of COALESCE are met for this patient. A second method is to estimate Ne from the formula π = 2Neμ (60), where μ is the mutation rate cited above and π is the nucleotide diversity when random genetic drift and neutral mutation reach an equilibrium (i.e., the peak nucleotide diversity). To distinguish new mutations from existing mutations (i.e., mutations separating the U and C lineages), we estimated π using phylogenetic trees in which mutations on branches connecting different recombinant lineages had been removed. Our procedure of completely removing branches connecting recombinant lineages is conservative in that branches of length zero would not ordinarily link lineages derived from a single infecting virus. This procedure yielded an estimated Ne of 476 ± 65 (mean ± standard deviation) for the samples (obtained at months 21, 33, and 38 p.i.) spanning the period of rapid frequency change, and for which we had enough sequence data to obtain a reliable estimate for π. We note that this method may underestimate Ne since diversity may not yet have reached equilibrium in this patient. Neither of our methods for estimating Ne accounts for inputs of viruses from other reservoirs and compartments, but these effects are difficult to quantify in the absence of HIV-1 sequence data from other tissues and organs. We note that both methods give estimates consistent with, or slightly lower than, estimates for Ne from other studies (32, 52). Both methods, nevertheless, yielded estimates for Ne that exceed the N = 350 cutoff from the genetic drift simulations mentioned above. Given the generous assumptions underlying this cutoff, we conclude that straightforward models of genetic drift cannot explain the rapid frequency changes observed in this patient.
Selection coefficients associated with frequency changes.
The above analyses suggest that selection played a role in the fluctuation of the various recombinant forms. Assuming a viral generation time of 1.78 days (23), we estimated that the parental C variant lineages were ∼0.3 and 3.3% more fit than the parental U lineages during the interval between 6 and 21 p.i. and the interval between 21 and 30 month p.i., respectively (Table 1). The subsequent frequency oscillations between months 30 and 53 p.i. can be explained by selection, assuming that each successive dominant lineage had a 2.5 to 9.1% fitness advantage over the previous dominant lineage (Table 1). Although it is tempting to speculate that these fluctuations were due to shifting selection pressures brought on by changes in HIV-1-specific immune responses, the extremely weak CTL, antibody, and lymphoproliferative responses in this patient (34) do not provide significant support for this hypothesis. It is also possible that drug treatments had some influence on these displacements; however, drug therapies had little-to-no effect on HIV-1 viral RNA load in plasma, suggesting that drug-mediated selection was weak in this patient.
TABLE 1.
Selection coefficients associated with shifts in dominant lineages
| Interval (mo. p.i.) | Shift | Mean selection coefficient (95% bootstrap range)a | Pb |
|---|---|---|---|
| 6-21 | U dominant, but C increasing | 0.003 (−0.001-0.010) | 0.144 |
| 21-30 | U→C | >0.033 (0.027-∞) | <0.001 |
| 30-33 | C→V3r-1 | >0.062 (0.046-∞) | <0.001 |
| 33-35 | V3r-1→C2V3r | 0.031 (0.006-0.063) | 0.055 |
| 35-38 | C2V3r→C | 0.071 (0.045-∞) | <0.001 |
| 38-44 | C→C2V3r | 0.025 (0.012-∞) | 0.001 |
| 44-47 | C2V3r→V3r-1 | 0.091 (0.065-∞) | <0.001 |
| 47-53 | V3r-1→V3r-3 | >0.042 (0.035-∞) | <0.001 |
| 53-56 | None (V3r-3 dominant) |
Selection coefficients and 95% bootstrap ranges were calculated as described in Materials and Methods. In cases in which one of the dominant lineages was not detected by DNA sequencing, we obtained a minimum estimate for the selection coefficient (denoted with “>”) by setting the frequency of the missing type to 1/N, where N is the total number of sequences at that time.
P gives the probability that the observed frequency changes [e.g., U(6) = 44, C(6) = 5, U(21) = 26, C(21) = 7] are significant by Fisher's exact test.
Presence of a common V3 motif in all different forms of recombinants.
The multiple breakpoints deduced from the sequence alignment in Fig. 2B indicated that many of the recombinants in this patient arose independently. The fact that all recombinants except X4r contained a V3 loop identical to that found in the parental C variant (Fig. 1C) (X4r is a hybrid of parental U and C in the V3 loop; Fig. 2), suggests that the V3 loop was under purifying selection. None of the recombination events led to alterations in coreceptor usage, because, as shown above, all of the major recombinants except for X4r (which used the CXCR4 coreceptor) were shown by virus reconstruction and propagation in tissue culture to have the same CCR5 coreceptor specificity as the parental U and C viruses. This suggests that selection is acting to preserve the common V3 sequence that had a relatively high fitness.
Rate of accumulation of synonymous and nonsynonymous substitutions and changes in the numbers of potential glycosylation sites.
To gain additional insight into viral evolution in this patient, we used standard phylogenetic procedures to estimate the number of synonymous (dS) and nonsynonymous (dN) substitutions per potential synonymous or nonsynonymous site, respectively. Interestingly, we observed dS/dN ratios of >1 (suggesting purifying selection) in most of the analyzed populations in both V3 loop (28/34) and C3-V5 (28/34) (Fig. 1D and E). Similar results were also obtained in the combined C2-V5 region (31/34) analyzed (data not shown). This contrasts sharply with ratios of dS/dN of <1 (suggesting diversifying selection) typically seen in HIV-1 envelope sequences (2, 66). Furthermore, a strong dS bias was observed in each variant population soon after it emerged (Fig. 1D and E). Although not reaching significance in most instances (P < 0.05), this bias appeared to be most pronounced in the V3 loop (Fig. 1D and E). The peak dS/dN ratio for the C variant V3 loop at 21 months (8.72 [P > 0.10]), compared to that of the U variant (2.70 [P > 0.10]), may also explain the timing of emergence (21 months p.i.) and the structure of V3r; i.e., it was the C variant V3 loop that was recombined into the backbone of the U variant but not vice versa.
Three of the 14 potential N-linked glycosylation sites that were present in all previous V3r lineages were lost in every clone of the V3r-3 subgroup that was most abundant near the time of the patient’s death (Fig. 2B). The loss of glycosylation sites, which coincided with a drop in CD4+ and CD8+ T-cell counts during months 47 to 56 p.i. (Fig. 1), may have reflected the absence of any appreciable antibody responses in this patient (34) since glycosylation can protect critical antibody recognition sites (49). The loss of these sites, together with the high dS/dN ratios and the rapid rate of progression to AIDS, bolsters our suggestion that immunological pressures played little role in driving viral diversification in this patient. We conclude, therefore, that the rapid turnover in recombinants discussed above can most simply be explained by selection for general increases in viral replication rates (i.e., increases in viral fitness) as opposed to selection for escape mutants or changes in coreceptor specificity.
DISCUSSION
Although there is general agreement that high rates of mutation and replication contribute to viral diversity, the extent to which selection contributes to viral diversity remains the subject of debate. High ratios of nonsynonymous-to-synonymous mutations in the V3 loop of Env (2, 66), together with evidence for CTL and antibody escape mutants (4, 24, 40, 41, 47), have led many to speculate that the diversification of HIV-1 in vivo is a reflection of selection for HIV-1 variants (40, 42). However, these assertions have been undercut by estimates of a low effective population size and the fact that standard population genetic tests provide surprisingly little support for the hypothesis that HIV is under selection during chronic infection (32, 39; Shriner et al., submitted). All of these studies, however, have focused on selection for mutational changes in nucleotides and amino acids. Here, we contribute to this debate by quantifying the magnitude and the form of selection for recombinant forms in vivo.
To test whether genetic drift could account for the frequency fluctuations observed in this patient, we performed numerical simulations of genetic drift with a range of population sizes. For realistic values of viral population sizes, our model of genetic drift and sampling variation could not account for the rapid turnover of recombinant forms. Adding a mutation-reintroduction rate to simulate mutation and/or input of viruses from latently infected cells or tissue or spatial compartments did not increase turnover rates enough to explain the observed fluctuations. Although we cannot rule out more complex models of genetic drift (for example, ones that invoke a combination of temporal and spatial variation), we believe that selection is the most-parsimonious explanation. A more complex model of genetic drift would require additional assumptions, such as temporal variation in the rate at which virus is released from different tissues that may not apply to our patient. We note in this regard that spatial heterogeneity can often reduce genetic turnover by preventing genotypes in the primary compartment from drifting to extinction (as illustrated for the high reintroduction rates in Fig. 4). Detailed longitudinal studies relating genetic turnover to the genetic makeup of specific virological compartments would, nonetheless, be highly valuable for future studies of viral pathogenesis.
The fact that all of the recombinants had the same sequence motif in the V3 loop provides independent support for our contention that selection was responsible for the emergence of these recombinants. To shed light on the selective forces that caused the same motif to appear in all of the recombinants, we performed additional experimental and phylogenetic studies. Since the V3 loop is recognized by antibodies and contains CTL epitopes, it is reasonable to speculate that the observed frequency fluctuations resulted from selection for escape mutants, but this hypothesis was undercut by the extremely weak-to-nonexistent CTL, antibody, and lymphoproliferative responses in this patient (34). Another selective factor thought to influence the evolution of the V3 loop is coreceptor specificity, but we obtained no evidence for such a change in this patient, although a transient outgrowth of an X4-using recombinant virus (X4r) in the PBMC-cocultured viral isolate was noted. Drug treatments are another potentially important selective force, but the therapy regimen was largely ineffective in this patient, with the viral load remaining above 5 × 104/ml throughout the study period. Even if these drugs were a selective force, the extensive recombination observed in this patient would have uncoupled the pol and env loci, so that changes in env would be independent of the action of drug-mediated selection at pol. The absence of evidence for selection for escape mutants, changes in coreceptor utilization, or strong drug effects, together with the eventual loss of glycosylation sites, leaves selection for replicative capacity as the most parsimonious explanation for the numerous frequency fluctuations in this patient.
To gain further insight into the nature of selection in this patient we performed longitudinal analyses of the ratio of synonymous site (dS) or nonsynonymous site changes (dN). These analyses revealed a strong preponderance of dS changes early after variant emergence and evidence that selective pressures changed over time. The predominance of dS is indicative of purifying selection (27) and contrary to the dN bias reported for env gene sequences (2, 66). These results suggest the existence of transient but strong purifying selection when new lineages emerged or came to dominate in the population. Stronger purifying selection appeared to act on the V3 loop of each variant as it emerged (compared to the C3-V5 region), possibly due to immunological escape or growth in a new cellular niche, although immune responses were extremely weak and no change in coreceptor specificity, one means of acquiring a new niche, accompanied these changes.
We also examined the possibility that selection could have been related to changes in glycosylation sites. Extensive glycosylation has been observed on envelope proteins of HIV and other lentiviruses (28), and glycosylation is important in modulating virus entry, infectivity, and host immune responses (17, 19). Studies employing SIV and simian/human immunodeficiency virus in the macaque model have shown that glycosylation sites in gp120 mask antibody binding and neutralization sites, leading to virus escape from host immune recognition (7, 8, 49). SIV Env N-linked glycosylation sites tend to be lost when the virus is grown in tissue culture; however, these sites are rapidly reacquired when grown in an animal host (18). Indeed, deglycosylated Env protein elicits a strong neutralizing antibody response in SIV-infected macaques that is largely specific to the deglycosylated Env (49). These studies suggest that glycosylation hinders optimal virus growth kinetics but plays the more critical role of shielding the virus from an intact immune response. We found that 3 of the 14 conserved potential N-linked glycosylation sites present in the C2-V5 region were lost in every clone of the V3r-3 subgroup found late in infection. Our results are consistent with the hypothesis that emergence and outgrowth of the relatively deglycosylated V3r-3 variant lineage resulted from competitive selection in the absence of a strong immune response. In other words, immunologic shielding provided by glycosylation was no longer required or strongly selected for in this patient. The loss of glycosylation sites, therefore, is consistent with our suggestion that selection was related to general increases in viral replicative capacity. Furthermore, other recent studies have demonstrated that ex vivo viral fitness correlates with disease state as quantified by HIV-1 RNA in plasma (48).
Our demonstration that a large number of recombinants can appear within a short period of time and that these recombinants had higher fitness than the parental viruses has important implications for AIDS pathogenesis. Theoretically, we expect variants with high fitness to be more virulent than variants with low fitness. Under a commonly cited model for viral pathogenesis (44) the steady-state density of CD4+ target cells is δc/pk, where δ is the death rate of infected cells, c is the clearance rate of free virus, p the rate at which infected cells release new virions, and k is the rate that viruses infect target cells. This model predicts that any change in viral kinetic parameters that increases viral fitness (i.e., increases in p or k or decreases in c or δ) will decrease steady-state CD4+ target cell densities. Indeed, it has been shown experimentally that relatively pathogenic viruses, e.g., the syncytium-inducing HIV-1 strains that are isolated from patients at the later stages of disease, have broader CD4+ T-cell tropisms and generally replicate more efficiently (21). In this study we found that the emergence of HIV-1 envelope recombinants between months 6 and 30 p.i. coincided with a dramatic decline in CD4+ and CD8+ T cells and a five- to sevenfold increase in plasma viral load (Fig. 1A). We are, of course, aware that it is hard to distinguish cause and effect from the study of a single patient. Our study, nevertheless, raises the possibility that infection from multiple sources, leading to recombination between distinct strains of HIV-1 in vivo, can exacerbate CD4+ T-cell decline and hasten progression to AIDS, as well as add to concerns for live vaccine approaches.
Acknowledgments
We thank Dan R. Littman and Vineet N. KewalRamani for providing the NL4-3-Luc-R−E− and the HXB2 and JRFL env plasmids and detailed methods for the coreceptor assays; Michelle Berrey, Timothy Schacker, and Theresa Shea for care of the patient; Eric Peterson for provision of samples; Matthew Czislowski for DNA sequencing; Lisa M. Frenkel and Raj Shankarappa for comments on the manuscript; and Katherine Davis for editorial assistance.
This work was supported by grants from the U.S. Public Health Service, including a New Investigator's Award to J.E.M. from the University of Washington Center for AIDS Research and support for S.-L.L. on training grant 5T32 CA09437.
REFERENCES
- 1.Allen, T. M., D. H. O'Connor, P. Jing, J. L. Dzuris, B. R. Mothe, T. U. Vogel, E. Dunphy, M. E. Liebl, C. Emerson, N. Wilson, K. J. Kunstman, X. Wang, D. B. Allison, A. L. Hughes, R. C. Desrosiers, J. D. Altman, S. M. Wolinsky, A. Sette, and D. I. Watkins. 2000. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407:386-390. [DOI] [PubMed] [Google Scholar]
- 2.Bagnarelli, P., F. Mazzola, S. Menzo, M. Montroni, L. Butini, and M. Clementi. 1999. Host-specific modulation of the selective constraints driving human immunodeficiency virus type 1 env gene evolution. J. Virol. 73:3764-3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Björndal, A., H. Deng, M. Jansson, J. R. Fiore, C. Colognesi, A. Karlsson, J. Albert, G. Scarlatti, D. R. Littman, and E. M. Fenyö. 1997. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 71:7478-7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borrow, P., H. Lewicki, X. Wei, M. S. Horwitz, N. Peffer, H. Meyers, J. A. Nelson, J. E. Gairin, B. H. Hahn, M. B. A. Oldstone, and G. M. Shaw. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3:205-211. [DOI] [PubMed] [Google Scholar]
- 5.Carr, J. K., M. Avila, M. Gomez Carrillo, H. Salomon, J. Hierholzer, V. Watanaveeradej, M. A. Pando, M. Negrete, K. L. Russell, J. Sanchez, D. L. Birx, R. Andrade, J. Vinoles, and F. E. McCutchan. 2001. Diverse BF recombinants have spread widely since the introduction of HIV-1 into South America. AIDS 15:F41-F47. [DOI] [PubMed] [Google Scholar]
- 6.Carr, J. K., J. N. Torimiro, N. D. Wolfe, M. N. Eitel, B. Kim, E. Sanders-Buell, L. L. Jagodzinski, D. Gotte, D. S. Burke, D. L. Birx, and F. E. McCutchan. 2001. The AG recombinant IbNG and novel strains of group M HIV-1 are common in Cameroon. Virology 286:168-181. [DOI] [PubMed] [Google Scholar]
- 7.Chackerian, B., L. M. Rudensey, and J. Overbaugh. 1997. Specific N-linked and O-linked glycosylation modifications in the envelope V1 domain of simian immunodeficiency virus variants that evolve in the host alter recognition by neutralizing antibodies. J. Virol. 71:7719-7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng-Mayer, C., A. Brown, J. Harouse, P. A. Luciw, and A. J. Mayer. 1999. Selection for neutralization resistance of the simian/human immunodeficiency virus SHIVSF33A variant in vivo by virtue of sequence changes in the extracellular envelope glycoprotein that modify N-linked glycosylation. J. Virol. 73:5294-5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coffin, J. M. 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis and therapy. Science 267:483-489. [DOI] [PubMed] [Google Scholar]
- 10.Coffin, J. M. 1996. Retroviridae: the viruses and their replication, p. 1767-1847. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology. Lippincott-Raven Publishers, Philadelphia, Pa.
- 11.Connor, R. I., B. K. Chen, S. Choe, and N. R. Landau. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935-944. [DOI] [PubMed] [Google Scholar]
- 12.de Jong, J. J., A. de Ronde, W. Keulen, M. Tersmette, and J. Goudsmit. 1992. Minimal requirements for the HIV-1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J. Virol. 66:6777-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Jong, J. J., J. Goudsmit, W. Keulen, B. Klaver, W. Krone, M. Tersmette, and A. de Ronde. 1992. Human immunodeficiency virus type 1 clones chimeric for the envelope V3 domain differ in syncytium formation and replication capacity. J. Virol. 66:757-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng, H., R. Liu, W. Ellmeier, S. Choe, D. Unutmaz, M. Burkhart, P. Di Marzio, S. Marmon, R. E. Sutton, C. M. Hill, C. B. Davis, S. C. Peiper, T. J. Schall, D. R. Littman, and N. R. Landau. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661-666. [DOI] [PubMed] [Google Scholar]
- 15.Diaz, R. S., E. C. Sabino, A. Mayer, J. W. Mosley, M. P. Busch, et al. 1995. Dual human immunodeficiency virus type 1 infection and recombination in a dually exposed transfusion recipient. J. Virol. 69:3272-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domingo, E., and J. J. Holland. 1997. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51:151-178. [DOI] [PubMed] [Google Scholar]
- 17.Douglas, N. W., G. H. Munro, and R. S. Daniels. 1997. HIV/SIV glycoproteins: structure-function relationships. J. Mol. Biol. 273:122-149. [DOI] [PubMed] [Google Scholar]
- 18.Edmonson, P., M. Murphey-Corb, L. N. Martin, C. Delahunty, J. Heeney, H. Kornfeld, P. R. Donahue, G. H. Learn, L. Hood, and J. I. Mullins. 1998. Evolution of a simian immunodeficiency virus pathogen. J. Virol. 72:405-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feizi, T., and M. Larkin. 1990. AIDS and glycosylation. Glycobiology 1:17-23. (Erratum, Glycobiology 1:315, 1991.) [DOI] [PubMed] [Google Scholar]
- 20.Fisher, A. G., E. Collalti, L. Ratner, R. C. Gallo, and F. Wong-Staal. 1985. A molecular clone of HTLV-III with biological activity. Nature 316:262-265. [DOI] [PubMed] [Google Scholar]
- 21.Fouchier, R. A., L. Meyaard, M. Brouwer, E. Hovenkamp, and H. Schuitemaker. 1996. Broader tropism and higher cytopathicity for CD4+ T-cells of a syncytium-inducing compared to a non-syncytium inducing HIV-1 isolate as a mechanism for accelerated CD4+ T cell decline in vivo. Virology 219:87-95. [DOI] [PubMed] [Google Scholar]
- 22.Fouchier, R. A. M., M. Groenink, N. A. Kootstra, M. Tersmette, H. G. Huisman, F. Miedema, and H. Schuitemaker. 1992. Phenotype-associated sequence variation in the third variable domain (V3) of the human immunodeficiency virus type 1 gp120 molecule. J. Virol. 66:3183-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu, Y.-X. 2001. Estimating mutation rate and generation time from longitudinal samples of DNA sequences. Mol. Biol. Evol. 18:620-626. [DOI] [PubMed] [Google Scholar]
- 24.Goulder, P. J. R., C. Brander, Y. Tang, C. Tremblay, R. A. Colbert, M. M. Addo, E. S. Rosenberg, T. Nguyen, R. Allen, A. Trocha, M. Altfeld, S. He, M. Bunce, R. Funkhouser, S. I. Pelton, S. K. Burchett, K. McIntosh, B. T. M. Korber, and B. D. Walker. 2001. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 412:334-338. [DOI] [PubMed] [Google Scholar]
- 25.Haase, A. T. 1999. Population biology of HIV-1 infection: viral and CD4+ T cell demographics and dynamics in lymphatic tissues. Annu. Rev. Immunol. 17:625-656. [DOI] [PubMed] [Google Scholar]
- 26.Hoelscher, M., B. Kim, L. Maboko, F. Mhalu, F. von Sonnenburg, D. L. Birx, and F. E. McCutchan. 2001. High proportion of unrelated HIV-1 intersubtype recombinants in the Mbeya region of southwest Tanzania. AIDS 15:1461-1470. [DOI] [PubMed] [Google Scholar]
- 27.Kimura, M. 1983. The neutral theory of molecular evolution. Cambridge University Press, Cambridge, United Kingdom.
- 28.Korber, B., C. Kuiken, B. Foley, B. Hahn, F. McCutchan, J. W. Mellors, and J. Sodroski (ed.). 1998. Human retroviruses and AIDS 1998. A compilation and analysis of nucleic acid and amino acid sequences. Theoretical Biology and Biophysics, Los Alamos National Laboratory, Los Alamos, N.Mex.
- 29.Koyanagi, Y., S. Miles, R. T. Mitsuyasu, J. E. Merrill, H. V. Vinters, and I. S. Y. Chen. 1987. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science 236:819-822. [DOI] [PubMed] [Google Scholar]
- 30.Kuhner, M. K., J. Yamato, and J. Felsenstein. 1995. Estimating effective population size and mutation rate from sequence data using Metropolis-Hastings sampling. Genetics 140:1421-1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar, S., K. Tamura, I. B. Jakobsen, and M. Nei. 2001. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17:1244-1245. [DOI] [PubMed] [Google Scholar]
- 32.Leigh Brown, A. J. 1997. Analysis of HIV-1 env gene sequences reveals evidence for a low effective number in the viral population. Proc. Natl. Acad. Sci. USA 94:1862-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liitsola, K., I. Tashkinova, T. Laukkanen, G. Korovina, T. Smolskaja, O. Momot, N. Mashkilleyson, S. Chaplinskas, H. Brummer-Korvenkontio, J. Vanhatalo, P. Leinikki, and M. O. Salminen. 1998. HIV-1 genetic subtype A/B recombinant strain causing an explosive epidemic in injecting drug users in Kaliningrad. AIDS 12:1907-1919. [DOI] [PubMed] [Google Scholar]
- 34.Liu, S. L., T. Schacker, L. Musey, D. Shriner, M. J. McElrath, L. Corey, and J. I. Mullins. 1997. Divergent patterns of progression to AIDS after infection from the same source: human immunodeficiency virus type 1 evolution and antiviral responses. J. Virol. 71:4284-4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mansky, L. M. 1996. Forward mutation rate of human immunodeficiency virus type 1 in a T lymphoid cell line. AIDS Res. Hum. Retrovir. 12:307-314. [DOI] [PubMed] [Google Scholar]
- 36.Mansky, L. M., and H. M. Temin. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69:5087-5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCutchan, F. E., J. K. Carr, M. Bajani, E. Sanders-Buell, T. O. Harry, T. C. Stoeckli, K. E. Robbins, W. Gashau, A. Nasidi, W. Janssens, and M. L. Kalish. 1999. Subtype G and multiple forms of A/G intersubtype recombinant human immunodeficiency virus type 1 in Nigeria. Virology 254:226-234. [DOI] [PubMed] [Google Scholar]
- 38.Nagylaki, T. 1992. Introduction to theoretical population genetics. Springer-Verlag, New York, N.Y.
- 39.Nijhuis, M., C. A. Boucher, P. Schipper, T. Leitner, R. Schuurman, and J. Albert. 1998. Stochastic processes strongly influence HIV-1 evolution during suboptimal protease-inhibitor therapy. Proc. Natl. Acad. Sci. USA 95:14441-14446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nowak, M. A., R. M. May, R. E. Phillips, S. Rowland-Jones, D. G. Lalloo, S. McAdam, P. Klenerman, B. Koppe, K. Sigmund, C. R. M. Bangham, and A. J. McMichael. 1995. Antigenic oscillations and shifting immunodominance in HIV-1 infections. Nature 375:606-611. [DOI] [PubMed] [Google Scholar]
- 41.O'Connor, D. H., T. M. Allen, T. U. Vogel, P. Jing, I. P. DeSouza, E. Dodds, E. J. Dunphy, C. Melsaether, B. Mothe, H. Yamamoto, H. Horton, N. Wilson, A. L. Hughes, and D. I. Watkins. 2002. Acute phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nat. Med. 8:493-499. [DOI] [PubMed] [Google Scholar]
- 42.Overbaugh, J., and C. R. Bangham. 2001. Selection forces and constraints on retroviral sequence variation. Science 292:1106-1109. [DOI] [PubMed] [Google Scholar]
- 43.Peeters, M., F. Liegeois, N. Torimiro, A. Bourgeois, E. Mpoudi, L. Vergne, E. Saman, E. Delaporte, and S. Saragosti. 1999. Characterization of a highly replicative intergroup M/O human immunodeficiency virus type 1 recombinant isolated from a Cameroonian patient. J. Virol. 73:7368-7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perelson, A. S., P. Essunger, Y. Cao, M. Vesanen, A. Hurley, K. Saksela, M. Markowitz, and D. D. Ho. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188-191. [DOI] [PubMed] [Google Scholar]
- 45.Piyasirisilp, S., F. E. McCutchan, J. K. Carr, E. Sanders-Buell, W. Liu, J. Chen, R. Wagner, H. Wolf, Y. Shao, S. Lai, C. Beyrer, and X. F. Yu. 2000. A recent outbreak of human immunodeficiency virus type 1 infection in southern China was initiated by two highly homogeneous, geographically separated strains, circulating recombinant form AE and a novel BC recombinant. J. Virol. 74:11286-11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Posada, D., and K. A. Crandall. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817-818. [DOI] [PubMed] [Google Scholar]
- 47.Price, D. A., P. J. Goulder, P. Klenerman, A. K. Sewell, P. J. Easterbrook, M. Troop, C. R. Bangham, and R. E. Phillips. 1997. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. USA 94:1890-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quinones-Mateu, M. E., S. C. Ball, A. J. Marozsan, V. S. Torre, J. L. Albright, G. Vanham, G. van Der Groen, R. L. Colebunders, and E. J. Arts. 2000. A dual infection/competition assay shows a correlation between ex vivo human immunodeficiency virus type 1 fitness and disease progression. J. Virol. 74:9222-9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reitter, J. N., R. E. Means, and R. C. Desrosiers. 1998. A role for carbohydrates in immune evasion in AIDS. Nat. Med. 4:679-684. [DOI] [PubMed] [Google Scholar]
- 50.Robertson, D. L., P. M. Sharp, F. E. McCutchan, and B. H. Hahn. 1995. Recombination in HIV-1. Nature 374:124-126. [DOI] [PubMed] [Google Scholar]
- 51.Rodrigo, A. G., and J. I. Mullins. 1996. HIV-1 molecular evolution and the measure of selection. AIDS Res. Hum. Retrovir. 12:1681-1685. [DOI] [PubMed] [Google Scholar]
- 52.Rouzine, I. M., and J. M. Coffin. 1999. Linkage disequilibrium test implies a large effective population number for HIV in vivo. Proc. Natl. Acad. Sci. USA 96:10758-10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sabino, E. C., E. G. Shpaer, M. G. Morgado, B. T. M. Korber, R. Diaz, V. Bongertz, S. Cavalcante, B. Galvao-Castro, J. I. Mullins, and A. Mayer. 1994. Identification of human immunodeficiency virus type 1 envelope genes recombinant between subtypes B and F in two epidemiologically linked individuals in Brazil. J. Virol. 68:6340-6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saitou, N., and M. Nei. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406-425. [DOI] [PubMed] [Google Scholar]
- 55.Salminen, M. O., J. K. Carr, D. L. Robertson, P. Hegerich, D. Gotte, C. Koch, E. Sanders-Buell, F. Gao, P. M. Sharp, B. H. Hahn, D. S. Burke, and F. E. McCutchan. 1997. Evolution and probable transmission of intersubtype recombinant human immunodeficiency virus type 1 in a Zambian couple. J. Virol. 71:2647-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scarlatti, G., E. Tresoldi, A. Björndal, R. Fredriksson, C. Colognesi, H. K. Deng, M. S. Malnati, A. Plebani, A. G. Siccardi, D. R. Littman, E. M. Fenyö, and P. Lusso. 1997. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat. Med. 3:1259-1265. [DOI] [PubMed] [Google Scholar]
- 57.Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch, H. Farzadegan, P. Gupta, C. R. Rinaldo, G. H. Learn, X. He, X. L. Huang, and J. I. Mullins. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73:10489-10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith, S. W., R. Overbeek, C. R. Woese, W. Gilbert, and P. M. Gillevet. 1994. The genetic data environment: an expandable GUI for multiple sequence analysis. Comput. Appl. Biol. Sci. 10:671-675. [DOI] [PubMed] [Google Scholar]
- 59.Su, L., M. Graf, Y. Zhang, H. von Briesen, H. Xing, J. Kostler, H. Melzl, H. Wolf, Y. Shao, and R. Wagner. 2000. Characterization of a virtually full-length human immunodeficiency virus type 1 genome of a prevalent intersubtype (C/B′) recombinant strain in China. J. Virol. 74:11367-11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tajima, F. 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takehisa, J., L. Zekeng, E. Ido, Y. Yamaguchi-Kabata, I. Mboudjeka, Y. Harada, T. Miura, L. Kaptu, and M. Hayami. 1999. Human immunodeficiency virus type 1 intergroup (M/O) recombination in Cameroon. J. Virol. 73:6810-6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wain-Hobson, S. 1992. Human immunodeficiency virus type 1 quasispecies in vivo and ex vivo. Curr. Top. Microbiol. Immunol. 176:181-193. [DOI] [PubMed] [Google Scholar]
- 64.Wang, R. F., and J. I. Mullins. 1995. Mammalian cell/vaccinia virus expression vectors with increased stability in Escherichia coli: production of feline immunodeficiency virus envelope protein. Gene 153:197-202. [DOI] [PubMed] [Google Scholar]
- 65.Yamaguchi-Kabata, Y., and T. Gojobori. 2000. Reevaluation of amino acid variability of the human immunodeficiency virus type 1 gp120 envelope glycoprotein and prediction of new discontinuous epitopes. J. Virol. 74:4335-4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang, L., R. S. Diaz, D. D. Ho, J. W. Mosley, M. P. Busch, and A. Mayer. 1997. Host-specific driving force in human immunodeficiency virus type 1 evolution in vivo. J. Virol. 71:2555-2561. [DOI] [PMC free article] [PubMed] [Google Scholar]