

Abstract
Varicella-zoster virus (VZV) open reading frame 17 (ORF17) is homologous to herpes simplex virus (HSV) UL41, which encodes the viral host shutoff protein (vhs). HSV vhs induces degradation of mRNA and rapid shutoff of host protein synthesis. An antibody to ORF17 protein detected a 46-kDa protein in VZV-infected cells. While HSV vhs is located in virions, VZV ORF17 protein was not detectable in virions. ORF17 protein induced RNA cleavage, but to a substantially lesser extent than HSV-1 vhs. Expression of ORF17 protein did not inhibit expression from a β-galactosidase reporter plasmid, while HSV type 1 vhs abolished reporter expression. Two VZV ORF17 deletion mutants were constructed to examine the role of ORF17 in virus replication. While the ORF17 VZV mutants grew to peak titers that were similar to those of the parental virus at 33°C, the ORF17 mutants grew to 20- to 35-fold-lower titers than parental virus at 37°C. ORF62 protein was distributed in a different pattern in the nuclei and cytoplasm of cells infected with an ORF17 deletion mutant at 37°C compared to 33°C. Inoculation of cotton rats with the ORF17 deletion mutant resulted in a level of latent infection similar to that produced by inoculation with the parental virus. The importance of ORF17 protein for viral replication at 37°C but not at 33°C suggests that this protein may facilitate the growth of virus in certain tissues in vivo.
Infection of cells with varicella-zoster virus (VZV) alters the expression and activities of a number of host proteins. For example, VZV down-regulates expression of major histocompatibility class I and class II proteins on the surfaces of cells (1, 2, 7). Infection of Vero cells with VZV induces apoptosis (38). VZV induces extensive formation of syncytia in cells, both in vitro and in vivo, but does not rapidly inhibit protein synthesis (3).
One herpesvirus protein that has a major effect on host cell proteins is the herpes simplex virus (HSV) virion host shutoff protein (vhs). HSV vhs is located in the tegument of virions and is released into the cytoplasm after virus entry, resulting in the rapid shutoff of host cellular protein synthesis (29, 36, 43, 48). HSV vhs preferentially induces cleavage of mRNA relative to other classes of RNA and may be targeted to mRNA by its interaction with a cellular translation factor, eIF4H (13, 36, 43). While an HSV type 1 (HSV-1) vhs mutant, obtained by chemical mutagenesis of the virus, showed impaired growth in cell culture (26), more recent studies using site-directed mutagenesis to construct HSV-1 vhs mutants have shown that viruses lacking a functional vhs are only marginally impaired in replication in cell culture (36, 46).
VZV open reading frame 17 (ORF17) encodes a homolog of the HSV vhs protein. ORF17 is predicted to encode a 455-amino-acid protein with 39% amino acid identity to HSV-1 vhs. The two proteins share four domains that are highly conserved (5). HSV-1 vhs has a deletion (corresponding to VZV ORF17 amino acids 170 to 189) relative to VZV ORF17 protein, while ORF17 protein has a deletion (corresponding to HSV-1 vhs amino acids 306 to 358) relative to HSV-1 vhs. HSV-1 vhs contains a domain that binds to HSV VP16 and regulates the activity of vhs (27, 41, 42). The VZV ORF17 protein is predicted to lack this binding domain (41), even though the virus encodes a homolog of VP16 (the VZV ORF10 protein). HSV-1 vhs contains a domain, LGYAYIN, that is postulated to bind to poly(A) sequences (45); this sequence is only partially conserved (MGYAYVE) in VZV ORF17.
Each of the alphaherpesviruses that has been sequenced has a vhs homolog. HSV-2, pseudorabies virus (PRV), equine herpesvirus 1 (EHV-1), and bovine herpesvirus 1 all express proteins that are highly conserved in relation to HSV-1 vhs (5, 14, 15, 17). Here we identify and characterize the VZV ORF17 protein and examine its importance in viral replication and latency.
MATERIALS AND METHODS
Cells and viruses.
Human melanoma (MeWo) cells, a gift from Charles Grose, were used for transfections and preparation of virus stocks. Human schwannoma cells (ST88-14) were a gift from Cynthia Morton (37). Human neuroblastoma cells (SK-N-SH), human fibroblasts (MRC-5), and African green monkey kidney cells (BSC-1 cells and Vero cells) were obtained from the American Type Culture Collection. Recombinant viruses were derived from cosmids corresponding to the attenuated Oka strain of VZV (47). An early clinical isolate of VZV (Molly strain) was used for purification of virions. Cell-free virus was prepared from MRC-5 or melanoma cells infected with VZV as described previously (40).
Plasmids and cosmids.
A maltose binding protein-ORF17 fusion protein was constructed by PCR amplification of a portion of VZV ORF17 (nucleotides 24149 to 25513) using VZV cosmid NotI A (9). The first primer contained an EcoRI site followed by VZV nucleotides 24296 to 24330 (TTTGAATTCGGTAAACGCGAGAATTTACACGGAC), and the second primer contained a PstI site followed by VZV nucleotides 25516 to 25492 (TTTCTGCAGTTAATTCCAATATTTTGTTAATACA). Amplified DNA was digested with EcoRI and PstI and inserted into plasmid pMAL-C2 (New England Biolabs, Beverly, Mass.). The resulting plasmid, pMAL-ORF17L, encodes the maltose binding protein fused to amino acids 50 to 455 of ORF17.
VZV ORF17 is located between VZV nucleotides 24149 and 25513 in cosmid VZV NotI A. VZV cosmids NotI A, NotI BD, MstII A, and MstII B, from the Oka vaccine strain, encompass the entire VZV genome and are used to generate recombinant Oka (ROka) virus (9) (Fig. 1). Two viruses containing different deletions in ORF17 were constructed by the RecA-assisted restriction endonuclease cleavage technique (16). Cosmid NotI A was incubated with two oligonucleotides, CACGACAAACTGACAAGATGATCTGCCTGCGCGACCAAAACGTATGCAGTCTTACCTCTA and TCATCCTCCCCATTGGATAATTTGGAACGCGCATTTGTTGAACATATAATCGCCGTGGTA, centered around the two HhaI sites at VZV nucleotides 24666 and 25284 in the presence of the Escherichia coli RecA protein. After methylation of the cosmid DNA with HhaI methylase and S-adenosylmethionine, the oligonucleotide-recA complexes were dissociated by heating, and the DNA was digested with HhaI. The large DNA fragment was ligated to itself to produce VZV cosmid NotI A-17DA. A second deletion in ORF17 was produced by incubating cosmid NotI A with two oligonucleotides, GATATTTGTATTGGAACGTTGTACAGACGGCCCATTATCACGTGGAGCCAAGGCAATTAT and ATCTGTTCAGCAAGTTATTCAGGATACCGGCCTGAAAATTCCACATCAAATGGACACTTC, centered around the two HaeIII sites at VZV nucleotides 24414 and 25059, and after methylation of the DNA and cleavage with HaeIII, cosmid NotI A-17DB was produced. NotI A-17DA is predicted to encode VZV ORF17 amino acids 1 to 173 followed by deletion of amino acids 174 to 378, two out-of-frame amino acids, and a stop codon, whereas NotI A-17DB is predicted to encode amino acids 1 to 89, followed by deletion of amino acids 90 to 304, and in-frame amino acids 305 to 455.
FIG. 1.

Construction of recombinant viruses with deletions in ORF17. The VZV genome is 124,884 bp long (top) and consists of unique long (UL), unique short (US), terminal repeat (TR), and internal repeat (IR) DNA segments. NotI and MstII DNA restriction fragments used to construct parental VZV are diagrammed. Cosmid NotI A-17DA has codons 174 to 378 of ORF17 deleted, whereas cosmid NotI A-17DB has codons 90 to 304 deleted. Numbers in diagrams of cosmids NotI A-17DA and NotI A-17DB indicate the sites of the deletions. Cosmids NotI A-17DA and NotI A-17DB were used to construct viruses ROka17DA and ROka17DB, respectively.
To generate a rescued virus in which the deletion in ORF17 is restored, a 5.0-kbp EcoRI-EcoRV fragment containing VZV nucleotides 22664 to 27684 was isolated from cosmid NotI A and subcloned into pGEM-2. The resulting plasmid was named pORF17. To construct a plasmid containing full-length ORF17 for in vitro transcription, ORF17 was amplified from cosmid NotI A by PCR using primers CCTAGACCATGGGGCTCTTTGGACTGACAC (which contains an NcoI site followed by VZV nucleotides 24149 to 24179) and CGTGAATTCGTCGCTCTTATGTGTGTTTTAATTCC (which contains an EcoRI site followed by VZV nucleotides 25509 to 25534). The PCR product was digested with NcoI and EcoRI and inserted into the NcoI and EcoRI sites of pSPUTK (11) to create plasmid pSP6VZV17. Plasmids pSPUTK, pSP6vhs, pSPPRVvhs, pSPSR19N, and pCITE-1 (Novagen), used in RNA cleavage assays, were kindly provided by James Smiley (11, 12).
To construct a plasmid expressing full-length ORF17 for expression in mammalian cells, the Eco57I-TfiI DNA fragment corresponding to VZV nucleotides 24116 to 25670 from cosmid NotI A was isolated, the ends were blunted with T4 DNA polymerase, and the DNA was inserted into the HindIII site (after it was blunted with the Klenow fragment of DNA polymerase I) of pCMV (19) to create plasmid pCMV17. Plasmid pCMV17DA, which has a deletion in ORF17, was constructed from pCMV17 by using the RARE procedure as described above. Plasmids pCMV, pCMV vhs, pCMV vhs1, and pRSVβ-gal, used for the β-galactosidase cotransfection assays, were provided by James Smiley (19).
Production of an antibody to ORF17 protein, immunoblotting, and Southern blotting.
E. coli (strain BL21) containing pMAL-ORF17L was propagated and lysed, and the maltose binding protein-ORF17 fusion protein was bound to an amylose resin column. The column was washed extensively, and the fusion protein was eluted with 10 mM maltose and dialyzed with phosphate-buffered saline containing 10 mM maltose. Mice were immunized once with the maltose binding protein-ORF17 fusion protein in complete Freund's adjuvant and four additional times in incomplete Freund's adjuvant. An antiserum from the mice was absorbed with an E. coli lysate containing the maltose binding protein and with uninfected melanoma cells.
Purified virions were prepared as described previously (21, 22, 39). VZV-infected and uninfected cell lysates and purified virions were boiled, fractionated on sodium dodecyl sulfate-polyacrylamide gels, and transferred to nitrocellulose membranes. The membranes were incubated either with a mouse antibody to ORF17 or VZV gE (Chemicon Corp., Temucula, Calif.), or with a rabbit antibody to ORF10 (31) or ORF61 (20).
VZV DNA was purified from nucleocapsids (9), cut with AflII, fractionated on agarose gels, and transferred to nylon membranes. The membranes were hybridized to a [32P]dCTP-radiolabeled probe corresponding to the NotI A cosmid.
Transfections and growth of VZV mutants.
Melanoma cells were transfected with plasmid pCMV62 and VZV cosmid NotI BD, MstII A, MstII B, and NotI A, NotI A-17DA, or NotI-17DB by the calcium phosphate method (9). Transfected cells were passaged until a cytopathic effect (CPE) was detected. To repair the deletion in the ORF17 deletion mutant, pORF17 was linearized and melanoma cells were transfected with 0.5 μg of virion DNA from the ORF17 deletion mutant, 1.5 μg of pORF17, and 50 ng of pCMV62. The transfected cells were passaged in melanoma cells at 37°C (nonpermissive temperature for the deletion mutant), cell-free virus was prepared, and the virus was plaque purified.
The β-galactosidase cotransfection assay was performed as previously described (19) with the following modifications. Melanoma cells in 12-well plates were cotransfected with 0.11 pmol (0.5 μg) of pRSVβ-gal per well and various amounts of pCMV, pCMV vhs, pCMV vhs1, pCMV17, or pCMV17DA by using Lipofectamine 2000 (Invitrogen, Carlsbad, Calif.). The total amount of DNA was maintained at 0.61 pmol per well by adding pCMV to the transfections. At 48 h after transfection, cell lysates were prepared and β-galactosidase activity was assayed by incubating the lysates with o-nitrophenyl-β-d-galactopyranoside at 37°C for 60 min. A410 was determined by using an enzyme-linked immunosorbent assay (ELISA) reader.
Growth curves for VZV mutants were performed by inoculating melanoma or Vero cells with virus-infected melanoma cells and, on consecutive days after infection, treating the cells with trypsin and titering them on uninfected melanoma cells.
Viral DNA synthesis was assayed in Vero cells. Vero cells were infected with cells containing about 200 PFU of VZV, and cells were harvested at days 1 to 5 after infection. Total cellular and viral DNAs were prepared as described previously (8), applied to a slot blot along with serial dilutions of control VZV DNAs, and hybridized to a [32P]dCTP probe corresponding to the four cosmids that encompass the VZV genome. The signal was quantified by using a phosphorimager (Storm System; Amersham Biosciences, Sunnyvale, Calif.).
RNA cleavage assay.
Cleavage of capped mammalian RNA and RNA containing a picornavirus internal ribosomal entry site (IRES) sequence was performed as described previously (11, 12, 44). Effector constructs pSP6vhs, pSP6PRVvhs, and pSP6VZV17 were linearized by digestion with EcoRI and transcribed using the Riboprobe in vitro transcription system (Promega). One microgram of each linearized effector plasmid was transcribed in a 20-μl reaction volume containing 40 mM Tris-Cl (pH 7.9), 6 mM MgCl2, 2 mM spermidine, 10 mM NaCl, 10 mM dithiothreitol, 2.5 mM (each) ATP, GTP, TTP, and CTP, 0.5 mM 7-methylguanosine 5′ nucleotide, 50 U of RNasin (RNase inhibitor), and 20 U of SP6 RNA polymerase. Reaction mixtures were incubated at 37°C for 1 h, and transcribed RNA was extracted with phenol-chloroform, precipitated, and resuspended in 12 μl of water. Two microliters of each transcribed RNA was translated in a 50-μl reaction volume containing 40 μCi of [35S]methionine and rabbit reticulocyte lysate (Promega). After incubation for 1 h at 30°C, aliquots of the products were fractionated on sodium dodecyl sulfate-polyacrylamide gels.
A radiolabeled reporter construct was derived from the canine recognition particle α subunit (SRPα) mRNA (11). A 2.4-kb runoff transcript containing SRPα mRNA was generated using SP6 polymerase and EcoRV-linearized pSPSR19N plasmid DNA as a template. The in vitro transcription reaction was performed as described above, except that 7-methylguanosine 5′ nucleotide was omitted. The reaction product was subjected to electrophoresis on a 1% agarose gel, and the full-length RNA was excised from the gel, electroeluted, extracted with phenol-chloroform, precipitated, and resuspended in water. The uncapped runoff transcript was used to generate capped radiolabeled reporter RNA by using vaccinia virus guanylyltransferase (Life Technologies, Gaithersburg, Md.) in the presence of [α-32P]GTP. RNA (500 ng) was incubated with 50 mM Tris-HCl (pH 7.9), 1.25 mM MgCl2, 6 mM KCl, 2.5 mM dithiothreitol, 0.1 mg of RNase-free bovine serum albumin/ml, 50 U of RNasin, 0.1 mM S-adenosyl-l-methionine, 1 U of guanylyltransferase, and 50 μCi of [α-32P]GTP in a reaction volume of 30 μl for 45 min at 37°C. The reaction mixture was extracted once with phenol-chloroform, and the RNA was recovered by ethanol precipitation.
A radiolabeled capped reporter RNA, containing a picornavirus IRES sequence, was produced by linearizing pCITE-1 (12) with EcoNI and performing in vitro transcription as described above with following modifications: the concentration of GTP was reduced to 25 μM and 1 μCi of [α-32P]GTP was added. The radiolabeled RNA was gel purified as described above.
The RNA cleavage assay was performed by incubating the radiolabeled (∼5,000 Cerenkov counts per min) reporter RNA with in vitro-translated effector proteins in reticulocyte buffer (1.6 mM Tris-acetate [pH 7.8], 80 mM KCl, 2 mM Mg acetate, 0.25 mM ATP, 0.1 mM dithiothreitol, 10 U of RNasin) at 30°C. Aliquots (5 μl) of the reaction were collected at various time points, 20 μg of carrier tRNA was added, and RNA was extracted with Trizol according to the manufacturer's instructions (Invitrogen). The RNA was subjected to electrophoresis on a 1% agarose-6% formaldehyde gel and transferred to a nylon membrane, and autoradiography was performed. The signal from full-length RNA was quantified by using a phosphorimager to estimate the rate of RNA degradation.
Immunofluorescence microscopy.
Cell-free virus was prepared from MRC-5 cells infected with an ORF17 deletion mutant and the ORF17 rescued virus and was used to infect melanoma cells on glass coverslips. The cells were incubated either at 33°C for 48 h, at 37°C for 48 h, or at 33°C for 3 h and then 37°C for 45 h; they were then fixed with methanol-acetone (1:1) at −20°C for 15 min. Cells were then incubated with a murine anti-gE or anti-ORF62 monoclonal antibody (Chemicon), followed by a fluorescein isothiocyanate-conjugated goat anti-mouse antibody, and were examined by immunofluorescence microscopy. In some experiments, DAPI (4′,6′-diamidino-2-phenylindole; 1 μg/ml) was added with the secondary antibody to visualize the nuclei.
Animal experiments.
Seven-week-old female or male cotton rats were anesthetized and inoculated intramuscularly with 50 μl containing 3 × 105 PFU of VZV-infected melanoma cells at six sites on each side of the thoracic and lumbar spine as described previously (39). As a control some animals were inoculated with uninfected melanoma cells, while other animals received VZV ROka-infected melanoma cells that had been heated to 95°C for 10 min (heat-inactivated ROka). One month after inoculation, the animals were sacrificed, and thoracic and lumbar dorsal root ganglia were removed, rinsed in phosphate-buffered saline, and frozen at −80°C.
DNA pooled from left dorsal root ganglia of mock- or VZV-infected animals was used in PCRs. The frozen tissue was triturated with a Kontes pestle, and nucleic acid was extracted using the Puregene DNA isolation kit (Gentra Systems, Minneapolis, Minn.). The treated tissue was incubated at 56°C overnight in lysis solution containing 0.2 μg of proteinase K (Invitrogen)/ml. Samples were treated with RNase A (20 μg/ml) for 30 min at 37°C, proteins were removed by precipitation, and the DNA was precipitated, washed in 70% ethanol, and resuspended in DNA hydration buffer.
RNA was extracted from pooled right dorsal root ganglia by addition of Trizol (Invitrogen) to the frozen tissue, followed by disruption of the tissue using a Turrax T8 tissue homogenizer (IKA Works, Inc., Wilmington, N.C.). RNA was purified according to the manufacturer's instructions and was resuspended in water.
RNA was treated with RNase-free DNase I (Invitrogen) for 30 min at 37°C, followed by incubation at 65°C for 15 min to inactivate the DNase. cDNA was obtained by incubating 1 μg of RNA, 50 U of Superscript II reverse transcriptase (Invitrogen), and 0.5 μg of oligo(dT)12-18 (Invitrogen) for 50 min at 42°C in a 20-μl reaction volume.
DNA (500 ng) from dorsal root ganglia, cDNA (5 μl) prepared from ganglion RNA, or serial dilutions of cosmid NotI A in 500 ng of DNA from ganglia of uninfected animals were amplified in a 50-μl PCR mixture containing 2.5 U of Taq DNA polymerase (Roche), 50 mM KCl, 2.5 mM MgCl2, 20 mM Tris (pH 8.3), 200 μM each deoxynucleoside triphosphate, 5% glycerol, and 0.5 μM oligonucleotides. Primers AGCGTTGTAGCAGACGAGCAT and ATGACAGCTTCCAACCCTGT were used to amplify ORF21, and primers TTATCTAATGGGTCGCACC and GTATATTCCGCGGTTTCTGC were used to amplify ORF63. PCR was performed by denaturing the DNA at 94°C for 3 min, followed by 35 cycles of 1 min at 94°C, 1 min at 55°C, and 1 min at 72°C, with a final incubation for 7 min at 72°C. Southern blot analysis was performed as described previously, using ORF21 and ORF63 probes (39).
RESULTS
ORF17 encodes a 46-kDa nonstructural protein.
To characterize VZV ORF17 protein, we generated a mouse antiserum against the protein by immunizing animals with a maltose binding protein-ORF17 fusion protein (containing amino acids 50 to 455 of ORF17). Sequence analysis of ORF17 in the fusion protein (derived from the vaccine strain of Oka) showed six nucleotide changes, which resulted in four amino acid changes relative to the sequence of the Dumas strain of VZV (10). VZV nucleotides 24516 to 24518 were deleted in the Oka vaccine strain (relative to the Dumas strain), resulting in deletion of Ser 123 in ORF17 protein. Nucleotides 24578, 24654, and 25067 were changed (from the Dumas strain to the Oka vaccine strain) from A to G, C to T, and G to A, respectively. This changed amino acids 144 (Thr to Ala), 169 (Thr to Met), and 307 (Val to Ile). The amino acid sequence of ORF17 in the fusion protein was identical to that found in another sequence of the Oka vaccine strain and in the parental Oka virus (Y. Gomi and K. Yamanishi, unpublished data). Immunoblotting of lysates from VZV (ROka)-infected cells with an antibody to ORF17 protein detected a 46-kDa protein that was not present in uninfected cells (Fig. 2A).
FIG. 2.

Immunoblot of ORF17 protein from VZV-infected cells. (A) Murine antiserum to ORF17 protein detects a 46-kDa protein from cells infected with ROka or ORF17 rescued virus (ROka17DAR) but not from uninfected cells or cells infected with an ORF17 deletion mutant (ROka17DA or ROka17DB). (B) Cells infected with ROka, ORF17 rescued virus, or ORF17 deletion mutants express 68- to 100-kDa proteins that react with a monoclonal antibody to VZV gE. Molecular sizes (in kilodaltons) are indicated at the right of each gel.
Since the HSV vhs protein is present in the virion tegument, we isolated VZV virions and performed immunoblotting for ORF17 and other viral proteins. Surprisingly, ORF17 protein was not detected in virions (Fig. 3). The purity of the virion preparation was verified by the presence of ORF10 tegument protein and gE in virions, but not ORF61 nonstructural protein (20).
FIG. 3.

ORF17 protein is not detected in virions. Cell lysates from VZV-infected cells, uninfected cells, or purified VZV virions were run on polyacrylamide gels, and immunoblotting was performed using antibodies to VZV ORF17 protein (A), ORF10 protein (B), gE (C), or ORF61 protein (D). ORF17 protein is detected in VZV-infected cells, but not in virions. Molecular sizes (in kilodaltons) are given to the right of each gel.
ORF17 protein cleaves capped RNA to a lesser extent than does HSV-1 vhs.
To determine whether ORF17 protein cleaves RNA to a similar degree as the HSV-1 vhs protein, we performed an RNA cleavage assay (11, 12). Plasmids containing wild-type ORF17, HSV-1 vhs, and PRV vhs were used for in vitro transcription and translation to generate [35S]methionine-labeled proteins. The resulting proteins were of the expected sizes of 46, 56, and 41 kDa for VZV ORF17 protein, HSV-1 vhs, and PRV vhs protein, respectively (Fig. 4A). These proteins were used as effectors in the RNA cleavage assay.
FIG. 4.



ORF17 protein has RNA cleavage activity. (A) In vitro translation of VZV ORF17, PRV vhs, and HSV-1 vhs proteins. Molecular sizes (in kilodaltons) are indicated at the right of the gel. (B) A 2.4-kb radiolabeled capped RNA was incubated with the in vitro-translated proteins, and at various time points, aliquots of reaction products were obtained and RNA was extracted. (C) Relative densities of signal obtained from RNA after incubation with in vitro-translated vhs proteins over various times. The signal obtained from full-length radiolabeled RNA was quantified using a phosphorimager. The relative density of the signal was expressed as the percentage of the signal remaining compared with that obtained at time zero.
A target RNA was transcribed from pSPSR19N by using guanylyl transferase, yielding a transcript of 2.4 kb with a radiolabeled cap at its 5′ terminus (11). Incubation of the target RNA with in vitro-translated HSV-1 vhs resulted in degradation of 95% of the full-length RNA within 10 min of incubation (Fig. 4B and C). In contrast, incubation of the RNA with VZV ORF17 protein or PRV vhs resulted in much slower degradation of full-length RNA. The proportion of full-length RNA remaining after incubation with VZV ORF17 protein, PRV vhs, or HSV-1 vhs for 30 min was 25, 15, or 0.7%, respectively. Degradation of full-length RNA in the presence of an in vitro-translated empty vector served as a control; 54% of full-length RNA remained after 30 min of incubation with the empty-vector control.
Radiolabeled capped RNA was also transcribed from a plasmid containing a picornavirus IRES sequence (11), and incubation of the target RNA with VZV ORF17 protein resulted in degradation of most, but not all, of the target RNA after 30 min (unpublished data). Incubation with HSV-1 vhs resulted in complete degradation of the full-length target RNA within 5 min of incubation, and incubation of the RNA with PRV vhs resulted in nearly complete digestion of the target RNA at 30 min. Thus, VZV ORF17 protein induced partial degradation of both a cap-dependent and an IRES-containing RNA, and the effect of the VZV protein more closely resembled the pattern of RNA degradation seen with PRV vhs than that seen with HSV-1 vhs.
ORF17 protein does not inhibit expression of a reporter gene in mammalian cells.
Coexpression of HSV-1 vhs with a reporter plasmid has been shown to inhibit expression of the plasmid (19, 34). To determine whether VZV ORF17 protein also inhibits expression of a reporter gene, we cotransfected melanoma cells with a β-galactosidase expression plasmid and a plasmid expressing either ORF17, an ORF17 deletion mutant, HSV-1 vhs, or an HSV-1 vhs mutant. While VZV ORF17 was expressed in the cells (Fig. 5A), neither VZV ORF17 protein nor an ORF17 deletion mutant (VZV ORF17DA) inhibited expression of β-galactosidase (Fig. 5B and C). In contrast, expression of HSV-1 vhs resulted in a dose-dependent decrease in β-galactosidase activity, while the HSV-1 vhs mutant (HSV-1 vhs1) had little activity in the assay.
FIG. 5.

ORF17 protein does not shut off expression of a β-galactosidase reporter plasmid. (A) Transfection of melanoma cells with pCMV17, followed by immunoblotting with an antibody to ORF17 protein, results in expression of a 46-kDa protein. (B and C) Transfection of cells with a plasmid expressing VZV ORF17 or ORF17DA does not inhibit expression from a β-galactosidase reporter plasmid, while a plasmid expressing HSV-1 vhs markedly reduces expression and a plasmid expressing an HSV-1 vhs mutant (HSV-1 vhs1) has little activity. The low level of activity seen with the plasmid expressing HSV vhs1 was not noted in other studies (19, 34) but may be due to differences in the cell line, transfection method, or total amount of DNA used in our experiments and the prior studies.
ORF17 is critical for growth of VZV at 37°C but not at 33°C.
To determine the role of ORF17 in viral replication, two different ORF17 deletion mutants were constructed. Cosmids NotI A-17DA and NotI A-17DB have deletions in more than 40% of ORF17. Transfection of melanoma cells with pCMV62, NotI BD, MstII A, MstII B, and NotI A resulted in CPE 9 to 10 days later, while transfections in which NotI A-17DA was substituted for NotI A yielded CPE 18 to 21 days after transfection; when NotI A-17DB was used, CPE was detected 12 to 17 days after transfection. VZV derived from cosmid NotI A-17DA, termed ROka17DA, has codons 174 to 378 of ORF17 deleted, while ROka17DB (derived from NotI A-17DB) has a deletion of codons 90 to 304. To verify that phenotypic changes were due solely to the deletion in ORF17, the deletion was restored in one of the viruses, by cotransfecting ROka17DA virion DNA with a DNA fragment containing ORF17 and plaque purifying the rescued virus (ROka17DAR).
Southern blotting was performed to verify that DNAs from VZV ROka, ROka17DA, and ROka17DB had the expected configurations. Digestion of virion DNA with AflII, followed by hybridization with a probe containing the NotI A fragment of VZV, showed a 4.8-kb band with VZV ROka or ROka17DAR but a 4.2-kb band with ROka17DA, due to the deletion in ORF17 (Fig. 6). A similar band of 4.2 kb was seen with ROka17DB (data not shown).
FIG. 6.
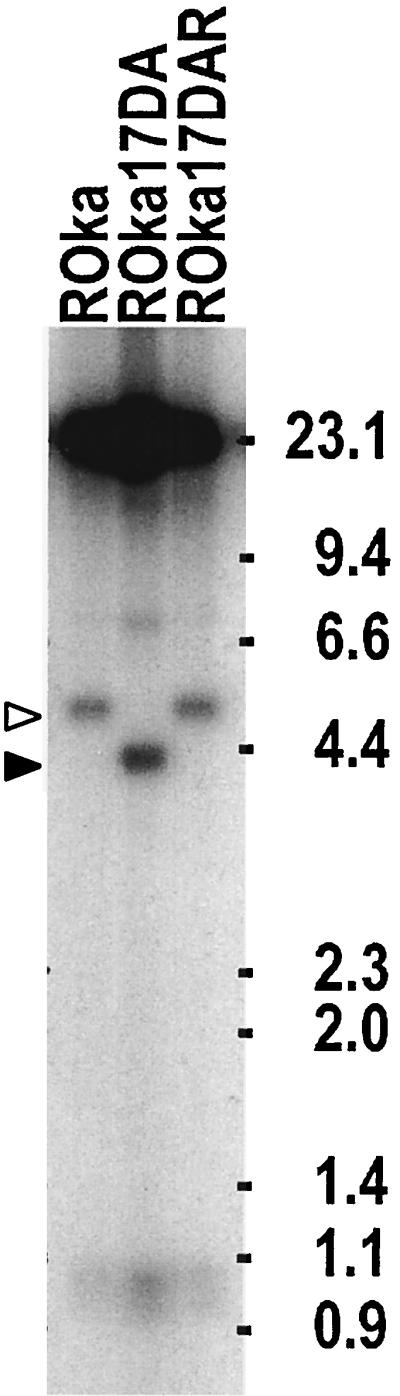
Southern blot of virion DNAs from cells infected with the parental VZV strain (ROka), an ORF17 deletion mutant, or the ORF17 rescued virus. Digestion of ROka or ROka17DAR DNA with AflII resulted in a fragment of 4.8 kb, while digestion of DNA from ROka17DA resulted in a 4.2-kb band. Molecular sizes (in kilobase pairs) are indicated at the right.
Immunoblotting of lysates from cells infected with the ORF17 deletion mutants (ROka17DA and ROka17DB) by use of an antibody to ORF17 protein showed that ORF17 protein was not detected (Fig. 2A). ORF17 protein was expressed in the ORF17 rescued virus (ROka17DAR). Lysates from the same cells expressed VZV gE, indicating that the cells were infected with virus (Fig. 2B).
When ROka17DA was grown at 33°C, the mutant grew to nearly the same peak titers (within twofold) as the parental virus in melanoma cells (Fig. 7A). Surprisingly, when the same virus was grown at 37°C, the ORF17 mutant grew to 34-fold-lower peak titers than the parental virus. Similar results were seen with ROka17DB (data not shown). To verify that the temperature-sensitive phenotype was due to the deletion in ORF17, and not due to a mutation elsewhere, the ORF17 rescued virus was tested for growth at different temperatures. The rescued virus (ROka17DAR) had a phenotype similar to that of the parental virus (ROka) in melanoma cells (Fig. 7A). Similar results were seen when the viruses were grown in Vero cells (Fig. 7B). The ORF17 deletion mutant also showed a temperature-sensitive phenotype in primary human fibroblasts (MRC-5 cells), human schwannoma cells, human neuroblastoma cells (SK-N-SH), and BSC-1 monkey kidney cells (data not shown). Thus, ORF17 is important for virus replication at 37°C but not at 33°C.
FIG. 7.

Growth curve of VZV ORF17 deletion mutants in cell culture. Melanoma (A) or Vero (B) cells were inoculated with cells containing approximately 100 PFU of either the parental virus (ROka), an ORF17 deletion mutant (ROka17DA), or the ORF17 rescued mutant (ROka17DAR). At various times after infection, the cells were harvested and the titer of virus was determined in melanoma cells.
To determine whether ORF17 affects the rate of viral DNA synthesis, total DNA was prepared from Vero cells infected with VZV ROka or ROka17DA at 33 or 37°C for various times after infection, and the amount of VZV DNA was quantified. Infection of Vero cells with VZV ROka at either temperature resulted in the synthesis of increasing amounts of viral DNA (Fig. 8). In contrast, infection of cells with VZV ROka17DA at 33°C resulted in a sevenfold-lower peak level of viral DNA than infection with ROka at 33°C. Infection with ROka17DA at 37°C resulted in no detectable increase in viral DNA synthesis. These results indicate that viral DNA synthesis is partially impaired in cells infected with the ORF17 deletion mutant at 33°C and is undetectable at 37°C.
FIG. 8.

Viral DNA synthesis in Vero cells infected with VZV ROka or ROka17DA. Cells were infected at a multiplicity of infection of approximately 0.001 and maintained at 33 or 37°C for 1 to 5 days, after which total DNA was prepared and hybridized to a VZV probe. The signal was quantified by using a phosphorimager, and the amount of VZV DNA was determined by using signals from control viral DNA.
ORF17 alters the intracellular localization of the ORF62 protein, but not gE, at 37°C.
To examine why ORF17 is critical for growth of VZV at the higher temperature, we infected melanoma cells with cell-free ROka17DA or ROka17DAR, incubated the cells at 33 or 37°C, and stained the cells with antibodies to ORF62 protein or gE. As expected, infection of cells with the ORF17 rescued virus (ROka17DAR) resulted in expression of ORF62 protein and gE in foci of 20 to 100 infected cells. Similarly, infection with the ORF17 deletion mutant at 33°C showed large clusters of cells that expressed the two proteins. However, when cells were infected with the ORF17 mutant at 37°C, foci of only 5 to 20 infected cells were noted, and both proteins were expressed in these foci.
Intracellular localization of ORF62 protein was markedly different in cells infected with the ORF17 deletion mutant at the two temperatures (Fig. 9). Cells infected with the ORF17 deletion mutant and incubated at 33°C, or infected with the ORF17 rescued virus and incubated at 33 or 37°C, showed expression of ORF62 protein in the nuclear membrane and scattered in fine granules in the nucleus (Fig. 9A, B, and D). In contrast, cells infected with the ORF17 deletion mutant and incubated at 37°C showed ORF62 protein in large clumps predominantly in the nucleus and not in the nuclear membrane (Fig. 9C). The localization of gE did not vary with the virus or the temperature (Fig. 9E through H). To determine whether the change in the intracellular localization of ORF62 protein occurred early after infection, cells were infected with the ORF17 deletion mutant, incubated at 33°C for 3 h, and then shifted to 37°C. ORF62 protein still showed aberrant localization in cells infected with the mutant (Fig. 9K). ORF62 protein appeared to localize to both the cytoplasm and the nucleus in cells infected with the ORF17 mutant at 37°C (Fig. 9L), while ORF62 protein was largely confined to the nuclei of cells infected with the ORF17 rescued virus (Fig. 9J).
FIG. 9.

ORF17 is responsible for normal distribution of ORF62 protein at 37°C. Melanoma cells were infected with cell-free VZV, incubated at 37 or 33°C for 48 h, fixed in cold acetone-methanol, and stained with an antibody to ORF62 protein (A through D) or gE (E through H). Cells were infected either with VZV ROka17DAR at 37°C (A and E) or 33°C (B and F) or with ROka17DA at 37°C (C and G) or 33°C (D and H). Melanoma cells were also infected with cell-free VZV, incubated at 33°C for 3 h followed by 37°C for 48 h, and stained with an antibody to ORF62 protein either alone (I and K) or with DAPI (J and L). Cells were infected with either VZV ROka17DAR (I and J) or ROka17DA (K and L). ORF62 protein shows nuclear-membrane and punctate nuclear staining in cells infected with ROka17DA at 33°C or with ROka17DAR at 33 or 37°C. ORF62 protein is present in coarse clumps in the nuclei and cytoplasm, but not in the nuclear membranes, of cells infected with ROka17DA at 33°C. Magnification, ×504 for panels A through H and ×320 for panels I through L.
An ORF17 deletion mutant is not impaired in its ability to establish latent infection.
The reduction in replication of the ORF17 deletion mutant at 37°C suggested that it might also be impaired in its ability to establish latency. Therefore, cotton rats were inoculated along both sides of the thoracic and lumbar spine with cells containing VZV ROka or ROka17DA, and 1 month later DNA was isolated from pooled thoracic and lumbar dorsal root ganglia. A PCR assay followed by Southern blotting could reliably detect 10 copies of VZV cosmid DNA mixed with 500 ng of DNA from uninfected dorsal root ganglia. Positive signals (≥10 copies of VZV DNA/500 ng of ganglion DNA) were detected in ganglia from 13 of 19 VZV ROka-infected and 8 of 20 ROka17DA-infected cotton rats, an insignificant difference. As expected, DNA extracted from ganglia of cotton rats inoculated with uninfected cells, or with heat-inactivated ROka-infected cells, showed no detectable VZV DNA.
To estimate the copy number of VZV DNA in ganglia from latently infected cotton rats, densitometry was performed using autoradiograms from Southern blots of PCR products corresponding to 500 ng of ganglion DNA obtained from animals infected with VZV ROka or ROka17DA. The densitometry signals were compared to those obtained by serial dilutions of VZV cosmid DNA mixed with 500 ng of ganglion DNA from uninfected animals. The geometric mean number of VZV copies from PCR-positive ganglia of animals infected with ROka17DA (43 copies) was similar to that in animals infected with ROka (30 copies) (Fig. 10).
FIG. 10.

Infection of cotton rats with cells containing ROka or ROka17DA results in similar mean copy numbers of VZV DNA in dorsal root ganglia. Filled circles represent copy numbers for animals considered positive for VZV, and open circles correspond to animals that had copy numbers below the threshold (10 copies of VZV DNA per 500 ng of ganglion DNA) for reliable detection. Horizontal bars indicate the geometric mean copy numbers for PCR-positive ganglia. Animals infected with cells containing heat-inactivated (HI) ROka or with uninfected cells had no detectable viral DNA in their ganglia.
To determine the number of animals expressing a VZV transcript during latency, RNA was isolated from dorsal root ganglia of cotton rats and was reverse transcribed, and the resulting cDNA was amplified by PCR using ORF63 primers followed by Southern blotting. ORF63 transcripts were present in ganglia from 7 of 9 VZV ROka-infected and 6 of 10 ROka17DA-infected cotton rats (Fig. 11). No VZV transcripts were detected in animals inoculated with uninfected cells or heat-inactivated ROka-infected cells.
FIG. 11.

Infection of cotton rats with cells containing ROka or ROka17DA results in ORF63 transcripts expressed in dorsal root ganglia. RNA was isolated from dorsal root ganglia, cDNA was amplified, and PCR was performed, followed by Southern blotting for ORF63. ORF63 transcripts were detected in animals 1, 2, 3, 5, 6, 8, and 9 infected with VZV ROka and in animals 1, 3, 4, 5, 6, and 9 infected with ROka17DA. Animals inoculated with uninfected cells or with cells containing heat-inactivated (HI) ROka had no detectable ORF63 transcripts. ORF63 RNA was present in VZV-infected cells. cDNA was prepared in the presence (+) or absence (−) of reverse transcriptase (RT).
DISCUSSION
We have shown that ORF17 encodes a 46-kDa nonstructural protein that induces cleavage of RNA and is not detectable in virions. In contrast, HSV-1 vhs is located in the tegument of virions and is released into the cytoplasm after entry, resulting in rapid shutoff of host cellular synthesis, even in the absence of new viral protein synthesis (25). While HSV-1 spreads rapidly in cell culture, VZV infection occurs more slowly, and therefore rapid shutoff of host protein synthesis could be detrimental for virus replication. Since ORF17 protein is not located in virions, and the protein is predicted to be expressed late in the viral replicative cycle, it might be expected that infection of cells with VZV would not result in rapid shutoff of host protein synthesis. In fact, infection of melanoma cells with the ORF17 rescued virus did not result in a detectable difference in host protein synthesis 30 h after infection, compared with cells infected with an ORF17 deletion mutant or uninfected cells (unpublished data). An earlier study showed that host protein synthesis in VZV-infected cells is very gradually diminished over 48 h (3). Thus, unlike HSV-1, VZV does not rapidly shut off host protein synthesis.
We found that VZV ORF17 had modest RNA cleavage activity for cap-dependent and IRES-containing RNAs but was unable to inhibit protein synthesis in a cotransfection assay. Similarly, an HSV-1 vhs point mutant retains partial cleavage activity for the same picornavirus IRES sequence and very weak activity for the cap-dependent RNA (28) but has lost the ability to inhibit expression of a reporter gene in mammalian cells and to shut off host protein synthesis during infection. Taken together, these results suggest that the reduced RNA cleavage activity of vhs may not be sufficient for inhibition of protein synthesis. While deletion of ORF17 had no detectable effect on host protein synthesis (unpublished data), the modest RNA cleavage activity of ORF17 protein might still serve to shut off expression of certain viral proteins in the replication cycle and coordinate an orderly cascade of gene expression, as has been shown for HSV-1 (33).
Other alphaherpesviruses encode homologs of HSV-1 vhs. The bovine herpesvirus 1 vhs homolog has host shutoff activity and induces degradation of RNA resulting in inhibition of protein synthesis in infected cells (17, 24). The EHV-1 vhs protein has a shutoff function similar to that of HSV when it is cotransfected with an expression vector into cells, and EHV-1 vhs is present in virions. However, infection of cells with EHV-1 does not result in shutoff of host protein synthesis (14). The PRV vhs protein has weak RNase activity (11), and infection of cells with PRV results in delayed shutoff of host protein synthesis that requires new viral protein synthesis (4, 18). Thus, the substantially weaker RNA cleavage activity of the VZV ORF17 protein and the prolonged delay in host shutoff by VZV suggests that the activity of the VZV protein more closely resembles that of the PRV vhs homolog than those of its other alphaherpesvirus homologs.
The ORF17 deletion mutant was impaired in viral DNA replication at 33°C compared to the parental virus, and no VZV DNA replication was detected at 37°C. Thus, ORF17 protein has an important role in the replication of viral DNA. The aberrant localization of the ORF62 protein seen in cells infected with the ORF17 deletion mutant and grown at 37°C might contribute to the effect of ORF17 deletion on VZV DNA replication. ORF62 protein transactivates putative immediate-early, early, and late VZV genes (35) and interacts with the USF transcription factor to activate the ORF28 and ORF29 promoters (30). ORF28 encodes the single stranded DNA binding protein, and ORF29 encodes the viral DNA polymerase (10); by analogy with their homologs in HSV-1, both of these proteins are likely to be essential for VZV DNA replication (6). Therefore, the impairment in viral DNA replication seen in the ORF17 deletion mutant at 37°C may be due to aberrant localization of ORF62 protein resulting in reduced expression of viral DNA replication proteins.
The distribution of ORF62 protein in the nuclei of cells infected with the ORF17 deletion mutant at 37°C was abnormal. Multiple attempts to demonstrate an interaction of ORF17 protein with ORF62 protein by coimmunoprecipitation assays were unsuccessful (data not shown). ORF17 protein might interact with ORF62 indirectly, or ORF17 might alter the activity of another protein that could affect localization of ORF62. Recently, the ORF66 protein kinase was shown to phosphorylate ORF62 protein and inhibit its localization in the nucleus (23). Thus, ORF17 is the second viral protein that affects the localization of ORF62. ORF62 protein is a virion-associated transactivator (21) that enhances the infectivity of VZV DNA (32). The abnormal localization of ORF62 protein in infected cells might limit its incorporation into the virion and reduce its ability to initiate virus replication.
We found that an ORF17 deletion mutant established a latent infection in cotton rats at a level similar to that established by the parental virus. As noted above, the ORF17 deletion mutant replicated to lower titers in vitro at 37°C; however, despite the fact that cotton rats have a core body temperature of 39°C (G. Prince, personal communication), the mutant was not impaired in its ability to establish latency. Strelow and Leib (46) found that mice inoculated with HSV-1 with a nonsense linker in vhs had lower replication of virus in the cornea, trigeminal ganglia, and brain than mice inoculated with the parental virus. Furthermore, they showed that the level of latent viral DNA in trigeminal ganglia was reduced 30-fold from that with the parental virus. The impairment in establishment of latency by the HSV-1 mutant compared with the VZV mutant may be due to an inherent difference between the role of HSV-1 vhs in neural pathogenesis and that of its VZV homolog, or it may be due to differences in the two animal models.
Growth of the VZV ORF17 deletion mutant was severely impaired in melanoma and Vero cells at 37°C but minimally impaired at 33°C. ORF17 protein might accelerate the degradation of mRNAs encoding a certain protein(s) that otherwise inhibits viral replication at 37°C (but not at 33°C). The difference between the growth of the VZV ORF17 deletion mutant at 33°C and its growth at 37°C suggests that ORF17 might not be required for growth in cooler temperatures such as those in distal portions of the skin or upper respiratory tract but might be important for viral replication in the central nervous system, lung, or other organs.
Acknowledgments
We thank Yasuyuki Gomi and Koichi Yamanishi for sharing their unpublished data, James Smiley and Holly Saffran for plasmids and advice, Paul Kinchington for antibody, Greg Prince for advice about cotton rats, and Stephen Straus for review of the manuscript.
H. Sato was the recipient of a Japan Society for the Promotion of Science (JSPS) fellowship in biomedical and behavioral research at NIH, and L. D. Callanan was a Howard Hughes Medical Institute-National Institutes of Health Research Scholar.
REFERENCES
- 1.Abendroth, A., B. Slobedman, E. Lee, E. Mellins, M. Wallace, and A. M. Arvin. 2000. Modulation of major histocompatibility class II protein expression by varicella-zoster virus. J. Virol. 74:1900-1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abendroth, A., I. Lin, B. Slobedman, H. Ploegh, and A. M. Arvin. 2001. Varicella-zoster virus retains major histocompatibility complex class I proteins in the Golgi compartment of infected cells. J. Virol. 75:4878-4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asano, Y., and M. Takahashi. 1980. Studies on the polypeptides of varicella-zoster (V-Z) virus. II. Syntheses of viral polypeptides in infected cells. Biken J. 23:95-106. [PubMed] [Google Scholar]
- 4.Ben-Porat, T., T. Rakusanova, and A. S. Kaplan. 1971. Early functions of the genome of herpesvirus. II. Inhibition of the formation of cell-specific polysomes. Virology 46:890-899. [DOI] [PubMed] [Google Scholar]
- 5.Berthomme, H., B. Jacquemont, and A. Epstein. 1993. The pseudorabies virus host-shutoff homolog gene: nucleotide sequence and comparison with alphaherpesvirus protein counterparts. Virology 193:1028-1032. [DOI] [PubMed] [Google Scholar]
- 6.Challberg, M. D. 1986. A method for identifying the viral genes required for herpesvirus DNA replication. Proc. Natl. Acad. Sci. USA 83:9094-9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen, J. I. 1998. Infection of cells with varicella-zoster virus down-regulates surface expression of class I major histocompatibility complex antigens. J. Infect. Dis. 177:1390-1393. [DOI] [PubMed] [Google Scholar]
- 8.Cohen, J. I., and H. Nguyen. 1997. Varicella-zoster virus glycoprotein I is essential for growth of virus in Vero cells. J. Virol. 71:6913-6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen, J. I., and K. E. Seidel. 1993. Generation of varicella-zoster virus (VZV) and viral mutants from cosmid DNAs: VZV thymidylate synthetase is not essential for replication in vitro. Proc. Natl. Acad. Sci. USA 90:7376-7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davison, A. J., and J. E. Scott. 1986. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 67:1759-1816. [DOI] [PubMed] [Google Scholar]
- 11.Elgadi, M. M., C. E. Hayes, and J. R. Smiley. 1999. The herpes simplex virus vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. J. Virol. 73:7153-7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elgadi, M. M., and J. R. Smiley. 1999. Picornavirus internal ribosome entry site elements target RNA cleavage events induced by the herpes simplex virus virion host shutoff protein. J. Virol. 73:9222-9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng, P., D. N. Everly, Jr., and G. S. Read. 2001. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J. Virol. 75:10272-10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng, X., Y. G. Thompson, J. B. Lewis, and G. B. Caughman. 1996. Expression and function of the equine herpesvirus 1 virion-associated host shutoff homolog. J. Virol. 70:8710-8718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fenwick, M. L., and R. D. Everett. 1990. Transfer of UL41, the gene controlling virion-associated host cell shutoff, between different strains of herpes simplex virus. J. Gen. Virol. 71:411-418. [DOI] [PubMed] [Google Scholar]
- 16.Ferrin, L. J., and R. D. Camerini-Otero. 1991. Selective cleavage of human DNA: RecA-assisted restriction endonuclease (RARE) cleavage. Science 254:1494-1497. [DOI] [PubMed] [Google Scholar]
- 17.Hinkley, S., A. P. Ambagala, C. J. Jones, and S. Srikumaran. 2000. A vhs-like activity of bovine herpesvirus-1. Arch. Virol. 145:2027-2046. [DOI] [PubMed] [Google Scholar]
- 18.Ihara, S., L. Feldman, S. Watanabe, and T. Ben-Porat. 1983. Characterization of the immediate-early functions of pseudorabies virus. Virology 131:437-454. [DOI] [PubMed] [Google Scholar]
- 19.Jones, F. E., C. A. Smibert, and J. R. Smiley. 1995. Mutational analysis of the herpes simplex virus virion host shutoff protein: evidence that vhs functions in the absence of other viral proteins. J. Virol. 69:4863-4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinchington, P. R., D. Bookey, and S. E. Turse. 1995. The transcriptional regulatory proteins encoded by varicella-zoster virus open reading frames (ORFs) 4 and 63, but not ORF 61, are associated with purified virus particles. J Virol. 69:4274-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinchington, P. R., J. K. Hougland, A. M. Arvin, W. T. Ruyechan, and J. Hay. 1992. The varicella-zoster virus immediate-early protein IE62 is a major component of virus particles. J. Virol. 66:359-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinchington, P. R., K. Fite, A. Seman, and S. E. Turse. 2001. Virion association of IE62, the varicella-zoster virus (VZV) major transcriptional regulatory protein, requires expression of the VZV open reading frame 66 protein kinase. J. Virol. 75:9106-9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kinchington, P. R., K. Fite, and S. E. Turse. 2000. Nuclear accumulation of IE62, the varicella-zoster virus (VZV) major transcriptional regulatory protein, is inhibited by phosphorylation mediated by the VZV open reading frame 66 protein kinase. J. Virol. 74:2265-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koppers-Lalic, D., F. A. Rijsewijk, S. B. Verschuren, J. A. van Gaans-Van den Brink, A. Neisig, M. E. Ressing, J. Neefjes, and E. J. Wiertz. 2001. The UL41-encoded virion host shutoff (vhs) protein and vhs-independent mechanisms are responsible for down-regulation of MHC class I molecules by bovine herpesvirus 1. J. Gen. Virol. 82:2071-2081. [DOI] [PubMed] [Google Scholar]
- 25.Krikorian, C. R., and G. S. Read. 1991. In vitro mRNA degradation system to study the virion host shutoff function of herpes simplex virus. J. Virol. 65:112-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwong, A. D., J. A. Kruper, and N. Frenkel. 1988. Herpes simplex virus virion host shutoff function. J. Virol. 62:912-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lam, Q., C. A. Smibert, K. E. Koop, C. Lavery, J. P. Capone, S. P. Weinheimer, and J. R. Smiley. 1996. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J. 15:2575-2581. [PMC free article] [PubMed] [Google Scholar]
- 28.Lu, P., H. A. Saffran, and J. R. Smiley. 2001. The vhs1 mutant form of herpes simplex virus virion host shutoff protein retains significant internal ribosome entry site-directed RNA cleavage activity. J. Virol. 75:1072-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLauchlan, J., C. Addison, M. C. Craigie, and F. J. Rixon. 1992. Noninfectious L-particles supply functions which can facilitate infection by HSV-1. Virology 190:682-688. [DOI] [PubMed] [Google Scholar]
- 30.Meier, J. L., X. Luo, M. Sawadogo, and S. E. Straus. 1994. The cellular transcription factor USF cooperates with varicella-zoster virus immediate-early protein 62 to symmetrically activate a bidirectional viral promoter. Mol. Cell. Biol. 14:6896-6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moriuchi, H., M. Moriuchi, and J. I. Cohen. 1995. Proteins and cis-acting elements associated with transactivation of the varicella-zoster virus (VZV) immediate-early gene 62 promoter by VZV open reading frame 10 protein. J. Virol. 69:4693-4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moriuchi, M., H. Moriuchi, S. E. Straus, and J. I. Cohen. 1994. Varicella-zoster virus (VZV) virion-associated transactivator open reading frame 62 protein enhances the infectivity of VZV DNA. Virology 200:297-300. [DOI] [PubMed] [Google Scholar]
- 33.Oroskar, A. A., and G. S. Read. 1989. Control of mRNA stability by the virion host shutoff function of herpes simplex virus. J. Virol. 63:1897-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pak, A. S., D. N. Everly, K. Knight, and G. S. Read. 1995. The virion host shutoff protein of herpes simplex virus inhibits reporter gene expression in the absence of other viral gene products. Virology 211:491-506. [DOI] [PubMed] [Google Scholar]
- 35.Perera, L. P., J. D. Mosca, W. T. Ruyechan, and J. Hay. 1992. Regulation of varicella-zoster virus gene expression in human T lymphocytes. J. Virol. 66:5298-5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Read, G. S., B. M. Karr, and K. Knight. 1993. Isolation of a herpes simplex virus type 1 mutant with a deletion in the virion host shutoff gene and identification of multiple forms of the vhs (UL41) polypeptide. J. Virol. 67:7149-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reynolds, J. E., J. A. Fletcher, C. H. Lytle, L. Nie, C. C. Morton, and S. R. Diehl. 1992. Molecular characterization of a 17q11.2 translocation in a malignant schwannoma cell line. Hum. Genet. 90:450-456. [DOI] [PubMed] [Google Scholar]
- 38.Sadzot-Delvaux, C., P. Thonard, S. Schoonbroodt, J. Piette, and B. Rentier. 1995. Varicella-zoster virus induces apoptosis in cell culture. J. Gen. Virol. 76:2875-2879. [DOI] [PubMed] [Google Scholar]
- 39.Sato, H., L. Pesnicak, and J. I. Cohen. 2002. Varicella-zoster virus ORF2 encodes a membrane phosphoprotein that is dispensable for viral replication and for establishment of latency. J. Virol. 76:3575-3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sato, H., S. Kageyama, M. Imakita, M. Ida, J. Yamamura, M. Kurokawa, and K. Shiraki. 1998. Immune response to varicella-zoster virus glycoproteins in guinea pigs infected with Oka varicella vaccine. Vaccine 16:1263-1269. [DOI] [PubMed] [Google Scholar]
- 41.Schmelter, J., J. Knez, J. R. Smiley, and J. P. Capone. 1996. Identification and characterization of a small modular domain in the herpes simplex virus host shutoff protein sufficient for interaction with VP16. J. Virol. 70:2124-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smibert, C. A., B. Popova, P. Xiao, J. P. Capone, and J. R. Smiley. 1994. Herpes simplex virus VP16 forms a complex with the virion host shutoff protein vhs. J. Virol. 68:2339-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smibert, C. A., D. C. Johnson, and J. R. Smiley. 1992. Identification and characterization of the virion-induced host shutoff product of herpes simplex virus gene UL41. J. Gen. Virol. 73:467-470. [DOI] [PubMed] [Google Scholar]
- 44.Smiley, J. R., M. M. Elgadi, and H. A. Saffran. 2001. Herpes simplex virus vhs protein. Methods Enzymol. 342:440-451. [DOI] [PubMed] [Google Scholar]
- 45.Sorenson, C. M., P. A. Hart, and J. Ross. 1991. Analysis of herpes simplex virus-induced mRNA destabilizing activity using an in vitro mRNA decay system. Nucleic Acids Res. 19:4459-4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strelow, L. I., and D. A. Leib. 1995. Role of the virion host shutoff (vhs) of herpes simplex virus type 1 in latency and pathogenesis. J. Virol. 69:6779-6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi, M., Y. Okuno, T. Otsuka, J. Osame, and A. Takamizawa. 1975. Development of a live attenuated varicella vaccine. Biken J. 18:25-33. [PubMed] [Google Scholar]
- 48.Zelus, B. D., R. S. Stewart, and J. Ross. 1996. The virion host shutoff protein of herpes simplex virus type 1: messenger ribonucleolytic activity in vitro. J. Virol. 70:2411-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]