

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) is a human-oncogenic herpesvirus. Cells from KSHV-associated tumors, such as Kaposi's sarcoma (KS) and primary effusion lymphoma (PEL), are of endothelial and B-cell origin, respectively. KSHV persists indefinitely in these cell lineages during latent infection. Indeed, cellular latency is a hallmark of all herpesviruses that is intimately linked to their pathogenesis. We previously characterized the promoter for the KSHV latency-associated nuclear antigen LANA/orf73. LANA is required for latent episome maintenance and has also been implicated in oncogenesis. Hence, regulation of LANA expression is critical to KSHV persistence. We find that a region extending to bp −1299 upstream of the LANA transcription start site is able to drive lacZ-reporter gene expression in several lines of transgenic mice. In agreement with KSHV's natural tropism, we detected reporter gene expression in CD19-positive B cells but not in CD3-positive T cells. We also detected expression in the kidney and, at a lower level, in the liver. In contrast to KS tumors, transgene expression was localized to kidney tubular epithelium rather than vascular endothelial cells. This suggests that our promoter fragment contains all cis-regulatory elements sufficient for B-cell specificity but not those required for endothelial specificity. Alternatively, while the trans-acting factors required for LANA expression in B cells are evolutionarily conserved, those that regulate endothelial cell-specific expression are unique to humans. Our in vivo studies address a conundrum in KSHV biology: in culture, KSHV is able to infect a variety of cell types indiscriminately, while in healthy latent carriers KSHV is found in B lymphocytes. The transgenic-mouse experiments reported here suggest that tissue-restricted LANA gene expression could explain B-cell-specific viral persistence.
Using representational difference analysis, Chang et al. (8) demonstrated the presence of a novel human virus in Kaposi's sarcoma (KS) biopsy specimens: Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8). KSHV has since been detected in two B-lineage lymphoproliferative disorders as well: primary effusion lymphoma (PEL) and multicentric Castleman's disease (6, 44). On the basis of the complete sequence of the 137-kbp unique region, KSHV is considered a gamma-2-herpesvirus, a member of the lymphotropic subgroup of the Herpesviridae. Consistent with this classification, KSHV is found in CD19+ B cells of KS patients (10, 23, 28). However, monocytes, circulating endothelial cells, endothelial cells in KS tumors, and in some instances even CD4 T cells have also been shown to harbor KSHV genomes (2, 3, 30).
KSHV, like all herpesviruses, displays two modes of replication: lytic replication, during which the host cell is destroyed and viral progeny are released, and latent replication, during which the viral genome persists indefinitely and no viral progeny are released. In KS, KSHV persists latently in virtually all tumor cells, while fewer than 5% show evidence of lytic mRNAs or protein (3, 45). Only a subset of viral genes is transcribed during KSHV latency (41, 51), while lytic gene expression and replication are induced in response to outside stimuli (7, 30). We and others have previously shown that the mRNAs for three latent proteins (v-FLIP, v-cyclin, and latency-associated nuclear antigen [LANA]) originate from the same promoter (11, 37, 41, 48). A larger, 5,400-nucleotide (nt) mRNA contains all three open reading frames (ORFs), while a smaller, spliced 1,700-nt mRNA contains the ORFs for v-cyclin and v-FLIP. No mRNA encoding just v-FLIP has been described, but v-FLIP can be translated by internal ribosome entry. These latent mRNAs can be detected in every KS tumor cell by in situ hybridization (11, 37). The same transcripts were also identified after experimental infection of SCID-hu Thy/Liv mice (12), indicating that their pattern of transcription typifies authentic KSHV latent infection in vivo rather than that of a particular tumor cell line in culture.
In the related Epstein-Barr virus (EBV), latency-associated genes are essential for episome maintenance and host cell transformation. The KSHV latent proteins LANA (orf73), v-cyclin (orf72), and v-FLIP (orf71), likewise, have been implicated in KSHV episome persistence and oncogenesis (1, 16, 18, 26). v-FLIP (orf71) has sequence homology to the FLICE-inhibitory proteins Equine herpesvirus-2 E8 and Herpesvirus saimiri (HVS) orf71. These proteins, in turn, are known to repress CD95/FAS and TRAIL/tumor necrosis factor alpha-mediated apoptosis. KSHV v-FLIP, as well, inhibits FAS-induced apoptosis by blocking caspase-3, -8, and -9 (13). v-cyclin (orf72) exhibits sequence and functional homology to the human cyclin-D/Prad oncogene. In general, cyclin-D proteins (D1, D2, and D3) associate with specific cyclin-dependent kinases (CDKs), and these complexes phosphorylate Rb family members. Unlike human cyclin-D, k-cyclin/cdk6-mediated phosphorylation of Rb is resistant to inhibition by the CDK inhibitors (CDKIs) p16INK4, p21CIP1, and p27KIP1 (46). As such it represents an activated allele. However, v-cyclin transfection studies in culture resulted in apoptosis (32), which implies that v-cyclin protein levels need to be tightly regulated in vivo. LANA/orf73 is the key latent protein expressed by KSHV. Although LANA shows no homology at the sequence level, its phenotype and structural features are reminiscent of EBV EBNA-1. Consistent with this resemblance, LANA can sustain KSHV episomes in culture (1, 9), and it functions as a transcriptional regulator of cellular and viral gene expression (22, 38). Together, LANA, v-FLIP, and v-cyclin are crucial for KSHV oncogenesis and latent persistence, and all three ORFs are controlled by the same cis-regulatory region, LANAp. Therefore, an in vivo analysis of their common promoter, LANAp, will elucidate the mechanism by which KSHV can establish and maintain latency.
In asymptomatic carriers KSHV is found in CD19-positive peripheral blood mononuclear cells (10, 28, 30). In culture, however, KSHV enters various human cell lines such as primary fibroblasts, 293 cells, and human primary endothelial cell preparations with low efficiency, as well as animal cells such as baby-hamster kidney (BHK) cells (39). The KSHV LANA protein is invariably expressed in this context, while lytic genes (e.g., orf29) are infrequently transcribed and mature capsids are rarely seen. We found similarly that the LANA promoter (LANAp) is active in a wide range of cell lines after transient transfection in culture (22), while in a SCID-hu Thy/Liv mouse model of de novo KSHV infection, LANA mRNA is transcribed in CD19-positive B cells but not in the much more abundant T-cell subsets (12). To address this discrepancy, we decided to examine the tissue specificity of LANAp in vivo by using transgenic mice.
MATERIALS AND METHODS
Transgenic mice.
All clones were based on a KSHV library from a KS lesion (51). All nucleotide sequence positions are given according to the numbering of Russo et al. (40). First, we amplified a 1,979-bp fragment from lambda-4 by PCR using Ready-To-Go beads (Amersham-Pharmacia, Piscataway, N.J.), by adding 100 ng of DNA and 100 pmol of each primer for a 25-μl total reaction volume. Cycling parameters were 94°C for 30 s, 57°C for 1 min, and 72°C for 2 min over 35 cycles. Primer 7326 (5′-TCGGGAAAgCTtGTCTGACA) spans the LANA AUG (positions 127300 to 127319) and introduces a HindIII site through T→G and G→T mutations (mutated nucleotides are lowercased). Primer 7327 (AGTCCCCTCACTGTTCGATCGCCGGCGAGCTC-5′) (positions 129161 to 129179) adds restriction sites for NheI, NotI, and XhoI at position −1299 relative to the transcription start site for LANA mRNA at position 127880 (11). The PCR product was cloned into pCR2.1 (Invitrogen, Carlsbad, Calif.) to yield pDD130B and was sequenced. To obtain pDD124, we cloned a 1,979-bp HindIII-NotI fragment from pDD130b into pβgeo (pDD216). Plasmid pβgeo contains a promoterless Escherichia coli lacZ ORF fused to the ORF for neomycin/G418 resistance, followed by a simian virus 40 (SV40) polyadenylation site and a unique XhoI site in a puc18 background. The transgene DNA was excised with NotI-XhoI and gel purified according to standard procedures (20). Transgenic mice were generated by the National Institute of Child Health and Human Development Transgenic Mouse Development Facility operated by the University of Alabama at Birmingham (contract NO1-HD-5-3229). All animal experiments were performed in accordance with IACUC regulations.
Flow cytometry.
A total of 106 cells were washed and stained with 4 μl of 20 mM 12C-FDG (Molecular Probes, Eugene, Oreg.) for 1 h at 37°C according to the manufacturer's recommendations. Next, cells were stained in 200 μl of fluorescence-activated cell sorter (FACS) buffer and 1 μl of a phycoerythrin-conjugated antibody against CD19, CD3, or CD45 (Caltag, Burlingame, Calif.) for 1 h on ice. Finally, cells were washed twice and analyzed on a FACSscan. A total of 40,000 events were recorded for each experiment.
β-Galactosidase staining.
The heart, liver, lung, brain, kidney, skeletal muscle, and testis were removed, placed in 12-well plates, and rinsed in 2 ml of ice-cold phosphate-buffered saline (PBS). Organs were sliced sagittally with a razor blade to facilitate penetration. PBS was aspirated, and the second half was fixed by addition of 2 ml of fixing buffer (1% formaldehyde, 0.2% glutaraldehyde, 2 mM MgCl2, 5 mM EGTA [100 mM stock, pH 8.0], 0.02% NP-40) for 15 min at room temperature. Tissues were rinsed twice in 2 ml of ice-cold PBS-0.02% NP-40 and stained in the dark overnight at 25°C in PBS supplemented with 1 mg of 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside/ml, 6 mM potassium ferricyanide, 6 mM potassium ferrocyanide, 2 mM MgCl2, 0.02% NP-40, and 0.01% sodium deoxycholate. After staining, organs were washed three times, for 10 min each time, in PBS, postfixed in 1 ml of fixing buffer at 4°C overnight (this also enhances the blue color), and counterstained with Orange G (1% [wt/vol] in 2% tungstophosphoric acid) for 1 min at 25°C. Tissues were washed three times in PBS, dehydrated through varying concentrations of ethanol (25, 50, and 70% in PBS; 5 min each), and stored at 4°C. Where indicated, organs were embedded and sectioned on a cryostat.
Immunohistochemistry.
Sections were heated to 65°C for 5 min, incubated in xylene for 5 min, washed through 70% ethanol, and rinsed extensively in water. To quench endogenous peroxidase activity, sections were incubated for 30 min in 0.3% peroxidase-water and for 10 min in 20% sucrose-PBS. To block nonspecific staining, sections were incubated for 30 min with diluted normal serum from the same species as the secondary antibody, washed in PBS-0.4% Tween 20 for 1 min, and incubated with the primary antibody (Factor VIII; Dako, Carpintera, Calif.) at a 1:200 dilution for 2 h at room temperature. Slides were washed twice in PBS and processed using a peroxidase Vectastain ABC kit (PK-4001; Vector Laboratories, Burlingame, Calif.). Peroxidase was visualized using diaminobenzidine (SK-4100; Vector Laboratories). Positive- and negative-control slides were developed in parallel, and the reactions were stopped at the same time by immersing the slides in water. Slides were dehydrated through graded ethanol and mounted for microscopy.
RNA isolation, analysis, and quantitative real-time PCR.
RNA was isolated and analyzed by using the primers listed in Table 1 and real-time reverse transcriptase PCR (RT-PCR) as previously described (15, 19).
TABLE 1.
Real-time PCR primers used in this studya
| Name | Primer (5′ to 3′) |
|---|---|
| lacZ-1f | CCGCTGATCCTTTGCGAAT |
| lacZ-1r | CAGTATTTAGCGAAACCGCCA |
| lacZ-1p | FAM-CGCCCACGCGATGGGTAACAGT-TAMRA |
| lacZ-245f | GCGTTACCCAACTTAATCGCC |
| lacZ-311r | GCCTCTTCGCTATTACGCCA |
| lacZ-408f | GGAGTGCGATCTTCCTGAGG |
| lacZ-477r | CGCATCGTAACCGTGCATC |
| lacZ-2887f | GGGCCGCAAGAAAACTATCC |
| lacZ-2957r | TCTGACAATGGCAGATCCCA |
| mu-apoBf | TCACCAGTCATTTCTGCCTTTG |
| mu-apoBr | CACGTGGGCTCCAGCATT |
| mu-apoBp | FAM-CCAATGGTCGGGCACTGCTCAA-TAMRA |
| mu-TBPf | ACGGACAACTGCGTTGATTTT |
| mu-TBPr | ACTTAGCTGGGAAGCCCAAC |
| mu-TBPp | FAM-TGTGCACAGGAGCCAAGAGTGAAGA-TAMRA |
RESULTS
Generation of LANAp-lacZ transgenic animals.
A minimal cis-regulatory region of LANAp extending to −279 is necessary and sufficient for high-level reporter gene expression in tissue culture (22, 43, 48). However, LANAp activity in the BJAB B-cell line increased if more-distal sequences were included (22). Therefore, the entire 5′ untranslated region (positions 127300 to 127880) and the upstream sequence up to −1299 (position 129161) were PCR amplified and cloned in front of a β-galactosidase-neomycin fusion gene (βgeo) and SV40 poly(A) site, in effect replacing the LANA initiation codon with the reporter initiation codon (Fig. 1A and B). The reporter was sequenced, and lacZ expression was confirmed by transient transfection into 293 and human umbilical vein endothelial cells (data not shown).
FIG. 1.

LANAp transgenic founder mice. (A) Location of LANAp in the KSHV genome. (B) Transgene fragment, isolated from plasmid pDD124 and used for microinjection. Arrows indicate the location of primer lacZf1:r1. (C) Tail DNA analysis of 10 independent founder mice by use of lacZf1:r1. (D) Specificity and quantitative real-time PCR performance parameters for the primer pairs used in this study. (E) Dot blot of mouse tail DNAs in the colony.
We obtained several founder mice that carried the β-galactosidase-neomycin fusion gene (βgeo) under the control of LANAp. Figure 1C depicts results of an analysis of the first 10 microinjected mice. PCR of genomic DNA showed that 3 of 10 animals contained the transgene (lines 63, 66, and 77). We later obtained additional transgenic founder animals (lines 54, 55, 50, 51, and 44). Founder 63 proved infertile, but the others transmitted the transgene in accordance with Mendelian rules. To verify the PCR results, all mice in our colony were routinely rescreened by DNA dot blot analysis using the entire transgene (pDD124) as a probe (these data are shown in Fig. 1E). Only mice that were positive by PCR and dot blot analysis were included for further breeding and for analysis.
Organ survey of LANAp activity by real-time quantitative RT-PCR.
To verify LANAp activity, tissue-specific RNA was isolated and subjected to quantitative real-time RT-PCR as previously described (19, 22). We employed several independent primer pairs specific for the lacZ transgene (Table 1). Quantitative real-time RT-PCR for the single-copy murine apoB or TATA-binding protein (TBP) gene was used to normalize the results. Figure 1D shows that quantitative real-time PCR is linear over 4 orders of magnitude and that all lacZ primer pairs amplified with similar efficiencies, as evidenced by their similar slopes. Figure 2A shows results of a real-time quantitative RT-PCR experiment for various organs. We analyzed three independent lines (lines 51, 54, and 44). For each line, mRNA was isolated from several animals, and transgene and TBP mRNA levels were determined by previously developed methods and calculations (15). The lacZ-specific Ct signal was compared to the TBP mRNA-specific signal (ΔCt), and mRNA levels were calculated as 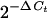 and expressed as a percentage of the TBP mRNA signal. As a positive control, we used mRNA from a transgenic mouse (ROSA [50]) that transcribes extremely high levels of lacZ mRNA in almost all tissues. In the LANAp transgenic animals all tissues exhibited low but significant levels of transgene mRNA. Average transgene mRNA levels ranged from 0.01 to 10% of TBP mRNA levels, suggesting that either the transgene was expressed at relatively low levels or transcription was restricted to a subset of cells in the organ. Figure 2B shows, in detail, the results of a real-time quantitative RT-PCR for heart mRNA. Here transgene RNA (lacZ) from heart tissue of two transgenic animals was amplified. In this case apoB amplification served as the positive control, while no signal was observed in the absence of reverse transcriptase. Consistent with the analysis in other organs, lacZ mRNA in the heart was significantly less abundant than the endogenous apoB message (approximately 60- to 100-fold, based on Ct differences). Such low levels of transgene expression could stem from a specialized cell which resides within the organ, or it could be the result of trapped B lymphocytes which express the transgene (see below). This prompted us to analyze transgene expression by in situ methods.
and expressed as a percentage of the TBP mRNA signal. As a positive control, we used mRNA from a transgenic mouse (ROSA [50]) that transcribes extremely high levels of lacZ mRNA in almost all tissues. In the LANAp transgenic animals all tissues exhibited low but significant levels of transgene mRNA. Average transgene mRNA levels ranged from 0.01 to 10% of TBP mRNA levels, suggesting that either the transgene was expressed at relatively low levels or transcription was restricted to a subset of cells in the organ. Figure 2B shows, in detail, the results of a real-time quantitative RT-PCR for heart mRNA. Here transgene RNA (lacZ) from heart tissue of two transgenic animals was amplified. In this case apoB amplification served as the positive control, while no signal was observed in the absence of reverse transcriptase. Consistent with the analysis in other organs, lacZ mRNA in the heart was significantly less abundant than the endogenous apoB message (approximately 60- to 100-fold, based on Ct differences). Such low levels of transgene expression could stem from a specialized cell which resides within the organ, or it could be the result of trapped B lymphocytes which express the transgene (see below). This prompted us to analyze transgene expression by in situ methods.
FIG. 2.

(A) Transgene mRNA analysis. Shown are lacZ transgene mRNA levels as percentages of TBP mRNA levels in the kidney, liver, and spleen. Each circle represents one mouse. The diamond represents mRNA levels in a ROSA transgenic mouse, which expresses high, constitutive levels of lacZ mRNA and serves as a positive control. (B) Real-time RT-PCR analysis of lacZ (shaded squares) and apoB (solid squares) mRNA levels from heart tissue. Relative fluorescence (ΔRn) is recorded on a logarithmic scale on the vertical axis, and cycle number is shown on the horizontal axis. T indicates the threshold setting used for calculating individual Ct values. RT-negative samples showed no amplification.
LANAp activity in kidney and liver. Figure 3C shows a whole-mount image of a kidney from a transgene-positive animal that was stained for β-galactosidase activity. In contrast to the human organ, the mouse (and rat) kidney is unilobular with a single, pyramid-shaped papilla. Staining followed a diffuse pattern involving the renal cortex and the outer and the inner stripes of the outer medulla, but not the inner medulla, calyx, or renal pelvis. Figure 3D shows the kidney of a transgene-positive mouse with a less exacerbated phenotype. Here transgene expression was confined to the outer medulla involving the inner stripe, presumably the distal tubules. A control specimen from a transgene-negative littermate exhibited no staining at all (Fig. 3A). In comparison, a Tie-2p-lacZ transgenic animal showed a very different staining pattern (Fig. 3B). Here the staining was restricted to the major vasculature, since the Tie-2 promoter is active only in the major vascular endothelial cells (24). Next, we sectioned the organ. LANAp directed weak β-galactosidase expression predominantly to the collecting ducts (Fig. 4A). Staining was significant compared to that in a nontransgenic animal (Fig. 4B), but its level and pattern differed from those for tie-2 transgenic mice (Fig. 4C). To further delineate the cell lineage of LANAp expression, we performed costaining with Factor VIII, a marker for endothelial cells. As expected, Tie-2-directed β-galactosidase activity was concentrated in the highly vascularized renal corpuscles (Fig. 4D and G [higher magnification]) as well as the efferent and afferent arterioles, but not in the collecting ducts. It colocalized with Factor VIII expression. Even in the well-established Tie-2 transgenic mice, not all glomeruli stained, as in instances when the section did not cut through Bowman's capsule and the substrate did not have access. In contrast, LANAp directed reporter gene expression primarily to the collecting ducts and distal tubules in the loop of Henle (Fig. 4E, F, and H). We never found β-galactosidase activity in the glomeruli of LANAp transgenic mice (Fig. 4E). Kidney tubules are extremely convoluted. They are heterogeneous with regard to the diameter and thickness of the inner epithelial layer and are enmeshed with vasculature of the vasa recta. The interstitial capillary endothelial cells of the vasa recta stained positive for Factor VIII in all panels, in particular Fig. 4F, but they did not express the transgene.
FIG. 3.

Transgene expression in the kidney, as observed by a coronal view through the center of the organ. Shown is whole-mount X-Gal staining of a kidney from a nontransgenic mouse (A), a Tie-2p-lacZ transgene-positive kidney (B), a LANAp-lacZ transgenic-mouse kidney (C), and a kidney of a littermate with more restrictive staining (D).
FIG. 4.

Factor VIII and X-Gal costaining of kidney sections from transgenic animals. (A through C) Section through the outer medulla from a LANAp-lacZ transgenic mouse (A), a nontransgenic littermate (B), and a tie-2p-lacZ transgenic mouse (C). Sections are counterstained with hematoxylin and eosin. (D through H) Factor VIII and X-Gal costaining of kidney sections from either tie-2p-lacZ (D and G) or LANAp-lacZ (E, F, and H) mice. (I through K) X-Gal staining of liver sections, counterstained with hematoxylin and eosin, from transgenic animals. Sections are from a LANAp-lacZ transgenic mouse (I), a nontransgenic littermate (J), and a tie-2p-lacZ transgenic mouse (K). Blue staining, β-galactosidase activity; brown staining, Factor VIII expression. Black arrowheads indicate areas of intense staining; white arrowheads indicate the absence of staining.
Transgene expression was more diffuse in the liver, with generalized low-level expression but some concentration in the endothelial cells lining the portal tracts (Fig. 4I). Again, LANAp-dependent reporter gene activity (Fig. 4I) was low, diffuse, and not as tightly localized as Tie-2-mediated lacZ expression (Fig. 4K) but significantly higher than that of a nontransgenic control (Fig. 4J). Low-level, sporadic transgene expression was similarly evident in the heart and the capillaries located in muscle endomysium, but not in muscle fibers (data not shown). The brain was consistently negative. LANAp activity in the various organs is summarized in Table 2. Animals from all transgenic lines exhibited similar staining patterns, which argues that the pattern was a result of LANAp specificity as opposed to integration site effects. However, as the whole-mount image in Fig. 3 illustrates, we observed various degrees of persistence even between littermates from the same founder. This is presumably due to endogenous modifiers (e.g., methylation modifiers [14]), which segregate in the 129 × C57BL/6 hybrid background. This is expected and can even be seen in tissue from tie-2p-lacZ transgenic animals, in which not every renal corpuscle stains positive β-galactosidase (see Fig. 3D). In sum, the in situ analysis corroborated the mRNA quantification and demonstrated that LANAp directed low-level expression of the lacZ transgene in transgenic mice.
TABLE 2.
Summary of LANAp transgene expression
| Organ | Expressiona of:
|
|
|---|---|---|
| LANAp | tie-2p | |
| Brain | − | NA |
| Eye | − | + |
| Lung | − | + |
| Liver | + | + |
| Kidney | +++ | + |
| Testes | − | + |
| Ovary | − | + |
| Skin (ear) | − | + |
| Smooth muscle | +/− | − |
| Heart | +/− | + |
| Spleen | ||
| B cells | +++ | − |
| T cells | − | − |
| Bone marrow | ||
| B cells | +++ | − |
| T cells | − | − |
In three independent lines, totaling > 20 animals. +++, high; +, moderate; +/−, variable; −, negative; NA, not analyzed
LANAp activity is restricted to CD19 lymphocytes.
To test the hypothesis that LANAp activity was restricted to B lymphocytes, single-cell suspensions of spleen and bone marrow cells were incubated with phycoerythrin-conjugated anti-CD3 or anti-CD19 antibodies to mark T and B cells, respectively. Next, a fluorescent substrate for β-galactosidase (12C-FDG) was added and cells were analyzed by flow cytometry. Figure 5 compares splenic lymphocytes (gated using forward and side scatter) of a transgene-positive animal (Fig. 5A through C) to those of a negative-control animal (Fig. 5D through F). Although there was some background staining associated with 12C-FDG, in the single staining the transgenic splenocytes (Fig. 5D) exhibited significantly brighter (1 log unit) mean 12C-FDG fluorescence than the control (Fig. 5A). The two animals exhibited similar levels of CD19-positive B lymphocytes (Fig. 5B and E). Combined staining with phycoerythrin-conjugated anti-CD19 and 12C-FDG revealed that the majority of 12C-FDG-bright-cells (mean fluorescence, >102) in the transgenic animal were CD19 positive (Fig. 5C), whereas there were no CD19-positive cells of a similar 12C-FDG brightness in the transgene-negative control animal (Fig. 5F). Analysis of splenocytes from the transgene-positive animal also detected the presence of a CD19-negative, 12C-FDG-dull population, as well as cells with no 12C-FDG activity at all (Fig. 5C). These could be macrophages, which would fit the reported tropism of KSHV. Since macrophages and NK cells have very high endogenous galactosidase activity, which could account for the 12C-FDG-dull staining in the absence of any transgene expression, this issue cannot be resolved by using a lacZ reporter (data not shown). This demonstrates that LANAp directs lacZ transgene expression specifically to the CD19 B-cell compartment, rather than being active in all lymphocyte lineages.
FIG. 5.

Transgene expression in B lymphocytes. (A through F) Representative two-color FACS plots of spleen cells from a transgene-positive animal (A, B, and C) compared to those for a transgene-negative animal (D, E, and F). CD19-phycoerythrin fluorescence is shown along the vertical axis, and 12C-FDG (fluorescein isothiocyanate) fluorescence is shown along the horizontal axis. Cells were incubated either with 12C-FDG only (A and D), with phycoerythrin-conjugated CD19 only (B and E), or with both (C and F). (G through I) Representative two-color FACS plots of spleen cells of a transgene-positive animal. 12C-FDG (fluorescein isothiocyanate) fluorescence is shown along the horizontal axis, and phycoerythrin fluorescence is shown along the vertical axis. Cells were incubated with 12C-FDG and either CD3 (G), CD19 (H), or CD45Rα/B220 (I) conjugated with phycoerythrin.
To corroborate the B-cell specificity of LANAp, we repeated our analysis in a LANAp transgene-positive animal using a marker specific for T cells (CD3) as well as an additional, independent marker for B cells (CD45Rα/B220). Figure 5G through I show a representative analysis of spleen lymphocytes. CD3-positive T cells showed background levels of 12C-FDG staining in the spleen (Fig. 5G), demonstrating that LANAp is inactive in T cells. As before, CD19 cells (Fig. 5H) stained uniformly bright for 12C-FDG, as did CD45Ra/B220 cells (Fig. 5I). Essentially similar results were observed in bone marrow (data not shown). A minor 12C-FDG-bright, CD19- and CD45-negative population was again in evidence. Table 3 quantifies our flow cytometric analysis from several lines of transgenic mice. Independently of transgene integration or founder effects, >80% of CD19 cells expressed the transgene, while <5% of CD3 cells did. This result is significant to a P value of ≤0.005 by a χ2 test of means. In sum, LANAp activity in transgenic mice is present in CD19 B cells but not in mature T cells, which parallels the tropism of KSHV in latently infected patients.
TABLE 3.
LANAp activity as measured by FDG fluorescence in CD3-positive T cells compared to CD19 positive B cells isolated from the spleena
| Tg line.offspring | LANAp activityb in:
|
|||||
|---|---|---|---|---|---|---|
| CD3-pos. cells
|
CD19-pos. cells
|
|||||
| FDG neg. | FDG pos. | Total spleen cells | FDG neg. | FDG pos. | Total spleen cells | |
| F44.181 | 95 | 5 | 20 | 19 | 81 | 54 |
| F50.142 | 93 | 7 | 27 | 12 | 88 | 66 |
| F54.231 | 95 | 5 | 21 | 13 | 88 | 64 |
| F51.150.5 | 95 | 5 | 19 | 27 | 73 | 67 |
| Total ± SD | 94.5 ± 1.0 | 5.5 ± 1.0 | 21.8 ± 3.6 | 17.8 ± 6.9 | 82.5 ± 7.2 | 62.8 ± 6.0 |
Independently of the founder line (F44, F50, F54, F51) or individual animal, FDG fluorescence is associated with the CD19 compartment but not the CD3 compartment. Tg, transgenic; pos., positive; neg., negative.
Expressed as percentage of positive cells.
DISCUSSION
KSHV persists in KS and PEL tumor cells, which are of endothelial and B-cell lineage, respectively. In latently infected asymptomatic carriers, KSHV has been detected in circulating CD19 B cells (10, 28). Macrophages have also been shown to harbor KSHV genomes (2). In ≥90% of infected tumor cells, KSHV is locked into the latent mode of infection and protein expression is restricted to only a few viral genes (21, 34, 41, 51). Most prominent among them is LANA, which is involved in transcriptional regulation (25, 27, 35, 36, 38), required for episome maintenance (1, 9, 47), and associated with viral oncogenesis (16, 36). LANA mRNA transcription is under the control of LANAp. LANAp is active in the absence of other viral gene products in transient assays and in latent PEL and KS tumor cells in vivo (11, 22, 43). In contrast to all other KSHV transcription units, neither LANA mRNA nor LANAp is induced upon KSHV lytic reactivation in PEL cells (11, 22). This implies that LANAp activity is shielded from other transcriptional events on the viral genome but is subject to cellular regulation, which justifies an analysis of LANAp in isolation.
Here we present an in vivo analysis of LANAp’s tissue-specific activity in several independently derived lines of transgenic mice. While overall LANAp activity was low compared to those of murine housekeeping genes (apoB and TBP), it was clearly detectable by quantitative real-time RT-PCR analysis. In the spleen, LANAp-directed transgene expression was consistently observed in CD19-positive B lymphocytes but never in CD3-positive, mature T lymphocytes. In addition to B cells, we also detected a CD19- and CD3-double-negative population which supports LANAp activity. While one expects these to be macrophages or NK cells, we have been unable to obtain an unambiguous answer, since those cells have extremely high endogenous β-galactosidase activity. These data demonstrate that all cis elements, which are sufficient for authentic B-cell-specific LANA expression in vivo, are contained within the proximal 1,299 bp relative to the transcription start site (and the 5′ untranslated region). We cannot exclude the possibility that sequence elements further upstream modulate tissue specificity further. This conclusion could not have been drawn from tissue culture experiments, since cell lines only approximate the assembly of transcription factors, which determines authentic B-cell function, and it is currently not feasible to construct recombinant KSHV strains.
Of the major organs, only the kidney and, to a lesser degree, the liver showed appreciable transgene expression by whole-mount analysis. Other organs (brain and skin) either were consistently negative in every animal we investigated or showed scattered lacZ-positive cells associated with the blood vessels (heart and muscle) upon closer inspection. In the latter cases, we cannot exclude the possibility that these stem from migrating cells or represent low-level activity in the target organ. Since circulating endothelial-cell precursors are recognized as a source of tissue vasculature (and potentially disseminate KS and KSHV), further experiments are needed to shed light on this issue. To exclude background effects from the analysis, we examined tie-2p-lacZ transgenic and mock-transgenic mice side by side. In the kidney, LANAp activity localized to the distal and proximal collecting tubules rather than the vasculature, i.e., LANAp activity was observed in kidney epithelial cells. The extent of transgene expression varied, which is consistent with this organ's ontogeny: The convoluted tubes arise from individual nephrogenic mesenchymal stem cells (approximately 1,000 to 2,000), which are induced to vascularize by proximity to the ureteric epithelium and subsequently fuse with it to form individual nephrons. Hence, LANAp activity is subject to varying degrees of induction in kidney development. This result seems at first contradictory, since endothelial lineage cells after extravascularization, rather than mesenchymal cells, constitute the bulk of KS tumor cells. A conservative interpretation of the data would conclude either that the cis elements required for the endothelial-cell activity of LANAp are not present in the transgene or that LANAp endothelial-cell specificity is mediated by trans-acting factors which are not evolutionarily conserved. Alternatively, the kidney specificity of LANAp seen in our transgenic-mouse study suggests a new target cell population for KSHV latent persistence. The shedding of KSHV in saliva (33, 49) and prepubescent transmission in regions of endemicity (4) argue that epithelial cells should not be excluded as a natural reservoir of this virus.
Based on sequence similarity, several Sp1 binding motifs have been identified in LANAp (11, 43, 48), some of which bind purified Sp1 in vitro and are essential for LANAp activity (J. H. Jeong and D. P. Dittmer, unpublished data). OCT-1 and GATA sites have also been located in LANAp, and these could mediate promoter activity in B lymphocytes, though their functional significance has not been established. The transgenic mice developed in this study mimic an important aspect of the KSHV replication cycle. They establish the starting point and in vivo validation for in-depth mutagenesis studies, which will identify tissue-specific cellular transcription factors that regulate LANAp activity and thereby LANA, v-cyclin, and v-FLIP protein levels. Conversely, we are using LANAp to direct the expression of other viral oncogenes in an effort to investigate their transforming ability in the relevant tissue context and the appropriate expression level.
Why should KSHV promoter elements respond to murine trans-activating factors? A great number of transcriptional regulators are conserved between mice and humans. One of the earliest transgenic models, that of the SV40 promoter/enhancer driving large T antigen, exhibited brain-specific expression, thus mirroring the observed tropisms of SV40 and human papovaviruses (BC virus and JC virus) for kidney and neuronal cells, respectively (5). This provided a model for JC virus- and SV40-associated malignant mesotheliomas. In contrast, alpha- and betaherpesvirus regulatory elements have proven to have less tissue specificity in transgenic mice. The fundamental importance of tissue specificity for gammaherpesviruses was recognized early on, as these viruses were grouped together based on their tissue tropism prior to any sequence analysis.
Finally, the tissue specificity required for long-term LANAp activity is consistent with the observed loss of KSHV episomes from explanted KS tumor material (17) or infected primary endothelial cell cultures. Our experiments show that LANAp activity is tissue specific rather than constitutive. As primary KS cells are selected for long-term growth in culture, they generally dedifferentiate. Presumably, LANAp activity cannot be maintained, LANA protein expression ceases, and the KSHV episome is lost during later cell division cycles. Consistent with this model, primary cells whose life is extended by human papillomavirus E6/E7 or ectopic telomerase expression maintain a more differentiated state and support extended LANAp activity and KSHV maintenance (31). This suggests a scenario in which KSHV entry into cells is minimally restricted, but the tissue-specific activity of LANAp ensures latent KSHV persistence only in specialized cell lineages.
Acknowledgments
We thank D. Ganem for critical reading, Z. Laszik for pathology advice, S. Hinds for technical assistance, J. Fowler for imaging, and J. Henthorn for expert flow cytometry. D.P.D. particularly thanks A. Teresky for introduction to mouse husbandry and transgenic analysis.
This work was supported by grants from the Presbyterian Health Foundation, the American Heart Association (AHA0060374Z), and NIH (RR1555777) to D.P.D. and by a National Eye Institute core grant (EY12190). Additional support was provided by Research to Prevent Blindness and the Presbyterian Health Foundation (to J.D.A.).
REFERENCES
- 1.Ballestas, M. E., P. A. Chatis, and K. M. Kaye. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641-644. [DOI] [PubMed] [Google Scholar]
- 2.Blasig, C., C. Zietz, B. Haar, F. Neipel, S. Esser, N. H. Brockmeyer, E. Tschachler, S. Colombini, B. Ensoli, and M. Sturzl. 1997. Monocytes in Kaposi's sarcoma lesions are productively infected by human herpesvirus 8. J. Virol. 71:7963-7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boshoff, C., T. F. Schulz, M. M. Kennedy, A. K. Graham, C. Fisher, A. Thomas, J. O. McGee, R. A. Weiss, and J. J. O'Leary. 1995. Kaposi's sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1:1274-1278. [DOI] [PubMed] [Google Scholar]
- 4.Bourboulia, D., D. Whitby, C. Boshoff, R. Newton, V. Beral, H. Carrara, A. Lane, and F. Sitas. 1998. Serologic evidence for mother-to-child transmission of Kaposi sarcoma-associated herpesvirus infection. JAMA 280:31-32. [DOI] [PubMed] [Google Scholar]
- 5.Brinster, R. L., H. Y. Chen, A. Messing, T. van Dyke, A. J. Levine, and R. D. Palmiter. 1984. Transgenic mice harboring SV40 T-antigen genes develop characteristic brain tumors. Cell 37:367-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cesarman, E., Y. Chang, P. S. Moore, J. W. Said, and D. M. Knowles. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186-1191. [DOI] [PubMed] [Google Scholar]
- 7.Chang, J., R. Renne, D. Dittmer, and D. Ganem. 2000. Inflammatory cytokines and the reactivation of Kaposi's sarcoma-associated herpesvirus lytic replication. Virology 266:17-25. [DOI] [PubMed] [Google Scholar]
- 8.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 9.Cotter, M. A., II, and E. S. Robertson. 1999. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254-264. [DOI] [PubMed] [Google Scholar]
- 10.Decker, L. L., P. Shankar, G. Khan, R. B. Freeman, B. J. Dezube, J. Lieberman, and D. A. Thorley-Lawson. 1996. The Kaposi sarcoma-associated herpesvirus (KSHV) is present as an intact latent genome in KS tissue but replicates in the peripheral blood mononuclear cells of KS patients. J. Exp. Med. 184:283-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dittmer, D., M. Lagunoff, R. Renne, K. Staskus, A. Haase, and D. Ganem. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 72:8309-8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dittmer, D., C. Stoddart, R. Renne, V. Linquist-Stepps, M. E. Moreno, C. Bare, J. M. McCune, and D. Ganem. 1999. Experimental transmission of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) to SCID-hu Thy/Liv mice. J. Exp. Med. 190:1857-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Djerbi, M., V. Screpanti, A. I. Catrina, B. Bogen, P. Biberfeld, and A. Grandien. 1999. The inhibitor of death receptor signaling, FLICE-inhibitory protein defines a new class of tumor progression factors. J. Exp. Med. 190:1025-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engler, P., D. Haasch, C. A. Pinkert, L. Doglio, M. Glymour, R. Brinster, and U. Storb. 1991. A strain-specific modifier on mouse chromosome 4 controls the methylation of independent transgene loci. Cell 65:939-947. [DOI] [PubMed] [Google Scholar]
- 15.Fakhari, F. D., and D. P. Dittmer. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 76:6213-6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friborg, J., Jr., W. Kong, M. O. Hottiger, and G. J. Nabel. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889-894. [DOI] [PubMed] [Google Scholar]
- 17.Gallo, R. C. 1998. The enigmas of Kaposi's sarcoma. Science 282:1837-1839. [DOI] [PubMed] [Google Scholar]
- 18.GoddenKent, D., S. J. Talbot, C. Boshoff, Y. Chang, P. Moore, R. A. Weiss, and S. Mittnacht. 1997. The cyclin encoded by Kaposi's sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J. Virol. 71:4193-4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heid, C. A., J. Stevens, K. J. Livak, and P. M. Williams. 1996. Real time quantitative PCR. Genome Res. 6:986-994. [DOI] [PubMed] [Google Scholar]
- 20.Hogan, B., and L. Lacy. 1994. Manipulating the mouse embryo. Cold Spring Harbor Laboratory Press, Plainview, N.Y.
- 21.Jenner, R. G., M. M. Alba, C. Boshoff, and P. Kellam. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 75:891-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeong, J., J. Papin, and D. Dittmer. 2001. Differential regulation of the overlapping Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) vGCR (orf74) and LANA (orf73) promoter. J. Virol. 75:1798-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kliche, S., E. Kremmer, W. Hammerschmidt, U. Koszinowski, and J. Haas. 1998. Persistent infection of Epstein-Barr virus-positive B lymphocytes by human herpesvirus 8. J. Virol. 72:8143-8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korhonen, J., I. Lahtinen, M. Halmekyto, L. Alhonen, J. Janne, D. Dumont, and K. Alitalo. 1995. Endothelial-specific gene expression directed by the tie gene promoter in vivo. Blood 86:1828-1835. [PubMed] [Google Scholar]
- 25.Krithivas, A., D. B. Young, G. Liao, D. Greene, and S. D. Hayward. 2000. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 74:9637-9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li, M., H. Lee, D. W. Yoon, J. C. Albrecht, B. Fleckenstein, F. Neipel, and J. U. Jung. 1997. Kaposi's sarcoma-associated herpesvirus encodes a functional cyclin. J. Virol. 71:1984-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim, C., H. Sohn, Y. Gwack, and J. Choe. 2000. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) binds ATF4/CREB2 and inhibits its transcriptional activation activity. J. Gen. Virol. 81:2645-2652. [DOI] [PubMed] [Google Scholar]
- 28.Mesri, E. A., E. Cesarman, L. Arvanitakis, S. Rafii, M. A. Moore, D. N. Posnett, D. M. Knowles, and A. S. Asch. 1996. Human herpesvirus-8/Kaposi's sarcoma-associated herpesvirus is a new transmissible virus that infects B cells. J. Exp. Med. 183:2385-2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milliancourt, C., S. Barete, A. G. Marcelin, C. Mouquet, N. Dupin, C. Frances, H. Agut, M. O. Bitker, and V. Calvez. 2001. Human herpesvirus-8 seroconversions after renal transplantation. Transplantation 72:1319-1320. [DOI] [PubMed] [Google Scholar]
- 30.Monini, P., S. Colombini, M. Sturzl, D. Goletti, A. Cafaro, C. Sgadari, S. Butto, M. Franco, P. Leone, S. Fais, G. Melucci-Vigo, C. Chiozzini, F. Carlini, G. Ascherl, E. Cornali, C. Zietz, E. Ramazzotti, F. Ensoli, M. Andreoni, P. Pezzotti, G. Rezza, R. Yarchoan, R. C. Gallo, and B. Ensoli. 1999. Reactivation and persistence of human herpesvirus-8 infection in B cells and monocytes by Th-1 cytokines increased in Kaposi's sarcoma. Blood 93:4044-4058. [PubMed] [Google Scholar]
- 31.Moses, A. V., K. N. Fish, R. Ruhl, P. P. Smith, J. G. Strussenberg, L. Zhu, B. Chandran, and J. A. Nelson. 1999. Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J. Virol. 73:6892-6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ojala, P. M., K. Yamamoto, E. Castanos-Velez, P. Biberfeld, S. J. Korsmeyer, and T. P. Makela. 2000. The apoptotic v-cyclin-CDK6 complex phosphorylates and inactivates Bcl-2. Nat. Cell Biol. 2:819-825. [DOI] [PubMed] [Google Scholar]
- 33.Pauk, J., M. L. Huang, S. J. Brodie, A. Wald, D. M. Koelle, T. Schacker, C. Celum, S. Selke, and L. Corey. 2000. Mucosal shedding of human herpesvirus 8 in men. N. Engl. J. Med. 343:1369-1377. [DOI] [PubMed] [Google Scholar]
- 34.Paulose-Murphy, M., N. K. Ha, C. Xiang, Y. Chen, L. Gillim, R. Yarchoan, P. Meltzer, M. Bittner, J. Trent, and S. Zeichner. 2001. Transcription program of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 75:4843-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Platt, G. M., G. R. Simpson, S. Mittnacht, and T. F. Schulz. 1999. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 73:9789-9795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Radkov, S. A., P. Kellam, and C. Boshoff. 2000. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene hras transforms primary rat cells. Nat. Med. 6:1121-1127. [DOI] [PubMed] [Google Scholar]
- 37.Rainbow, L., G. M. Platt, G. R. Simpson, R. Sarid, S. J. Gao, H. Stoiber, C. S. Herrington, P. S. Moore, and T. F. Schulz. 1997. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J. Virol. 71:5915-5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Renne, R., C. Barry, D. Dittmer, N. Compitello, P. Brown, and D. Ganem. 2001. The latency-associated nuclear antigen (LANA/orf73) of KSHV modulates cellular and viral gene expression. J. Virol. 75:458-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Renne, R., D. Blackbourn, D. Whitby, J. Levy, and D. Ganem. 1998. Limited transmission of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 72:5182-5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Russo, J. J., R. A. Bohenzky, M. C. Chien, J. Chen, M. Yan, D. Maddalena, J. P. Parry, D. Peruzzi, I. S. Edelman, Y. Chang, and P. S. Moore. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 93:14862-14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarid, R., O. Flore, R. A. Bohenzky, Y. Chang, and P. S. Moore. 1998. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72:1005-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarid, R., G. Pizov, D. Rubinger, R. Backenroth, M. M. Friedlaender, F. Schwartz, and D. G. Wolf. 2001. Detection of human herpesvirus-8 DNA in kidney allografts prior to the development of Kaposi's sarcoma. Clin. Infect. Dis. 32:1502-1505. [DOI] [PubMed] [Google Scholar]
- 43.Sarid, R., J. S. Wiezorek, P. S. Moore, and Y. Chang. 1999. Characterization and cell cycle regulation of the major Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) latent genes and their promoter. J. Virol. 73:1438-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soulier, J., L. Grollet, E. Oksenhendler, P. Cacoub, D. Cazals-Hatem, P. Babinet, M. F. d'Agay, J. P. Clauvel, M. Raphael, L. Degos, et al. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276-1280. [PubMed] [Google Scholar]
- 45.Staskus, K. A., W. Zhong, K. Gebhard, B. Herndier, H. Wang, R. Renne, J. Beneke, J. Pudney, D. J. Anderson, D. Ganem, and A. T. Haase. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71:715-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swanton, C., D. J. Mann, B. Fleckenstein, F. Neipel, G. Peters, and N. Jones. 1997. Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature 390:184-187. [DOI] [PubMed] [Google Scholar]
- 47.Szekely, L., C. Kiss, K. Mattsson, E. Kashuba, K. Pokrovskaja, A. Juhasz, P. Holmvall, and G. Klein. 1999. Human herpesvirus-8-encoded LNA-1 accumulates in heterochromatin-associated nuclear bodies. J. Gen. Virol. 80:2889-2900. [DOI] [PubMed] [Google Scholar]
- 48.Talbot, S. J., R. A. Weiss, P. Kellam, and C. Boshoff. 1999. Transcriptional analysis of human herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology 257:84-94. [DOI] [PubMed] [Google Scholar]
- 49.Vieira, J., M. L. Huang, D. M. Koelle, and L. Corey. 1997. Transmissible Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in saliva of men with a history of Kaposi's sarcoma. J. Virol. 71:7083-7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zambrowicz, B. P., A. Imamoto, S. Fiering, L. A. Herzenberg, W. G. Kerr, and P. Soriano. 1997. Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc. Natl. Acad. Sci. USA 94:3789-3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong, W., H. Wang, B. Herndier, and D. Ganem. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. USA 93:6641-6646. [DOI] [PMC free article] [PubMed] [Google Scholar]