

Abstract
The cyclin-dependent kinase (CDK) inhibitor p27Kip1 (p27) is an important regulator of cell cycle progression controlling the transition from G to S-phase. Low p27 levels or accelerated p27 degradation correlate with excessive cell proliferation and poor prognosis in several forms of cancer. Phosphorylation of p27 at Thr187 by cyclin E–CDK2 is required to initiate the ubiquitination-proteasomal degradation of p27. Protecting p27 from ubiquitin-mediated proteasomal degradation may increase its potential in cancer gene therapy. Here we constructed a non-phosphorylatable, proteolysis-resistant p27 mutant containing a Thr187-to-Ala substitution (T187A) which is not degraded by ubiquitin-mediated proteasome pathway, and compared its effects on cell growth, cell-cycle control, and apoptosis with those of wild-type p27. In muristerone A-inducible cell lines over-expressing wild-type or mutant p27, the p27 mutant was more resistant to proteolysis in vivo and more potent in inducing cell-cycle arrest and other growth-inhibitory effects such as apoptosis. Transduction of p27(T187A) in breast cancer cells with a doxycycline-regulated adenovirus led to greater inhibition of proliferation, more extensive apoptosis, with a markedly reduced protein levels of cyclin E and increased accumulation of cyclin D1, compared with wild-type p27. These findings support the potential effectiveness of a degradation-resistant form of p27 in breast cancer gene therapy.
Keywords: p27, Proteolysis, Breast cancer, T187A, Apoptosis
Abbreviations: CDK, cyclin-dependent kinase; FITC, fluorescein isothiocyanate; Mur A, muristerone A; PARP, poly(ADP-ribose) polymerase
1. Introduction
The G1-S transition of the cell cycle in mammalian cells is controlled by cyclins, cyclin-dependent kinases (CDKs), and their inhibitors [1]. Disruption of cell-cycle control by deregulation of CDK inhibitors is a common feature in tumor cells [2].
p27, a member of the Kip/Cip family of CDK inhibitors, is a putative tumor suppressor gene [3] that causes G1 arrest by inhibiting cyclin E–CDK2 [4,5]. p27 knockout mice develop generalized hyperplasia and pituitary tumors [6–8]. Furthermore, mice that are haploinsufficient for p27 are more sensitive to tumor development caused by radiation and chemical carcinogens [3]. The absence or reduction of p27 expression has been associated with aggressive behavior in several types of malignancies in humans, including breast, gastric, prostate, colon, and lung carcinomas [9–14]. However, no homozygous deletions and only rare point mutations have been found in the human p27 gene [15].
Gene therapy approaches to restoring p27 expression using adenoviral vectors [16–19] have been promising. These agents have induced cell-cycle arrest and loss of cyclin E–CDK2 activity in cell lines and xenograft models and have triggered apoptosis in cancer cells [20–23,17,24]. The concentration of p27 is thought to be regulated predominantly by the ubiquitin-dependent proteolytic pathway [25]. Degradation of p27 is triggered by its phosphorylation on Thr187 by the cyclin E-CDK2 complex [26–28]. The phosphorylation of Thr187 is required for the binding of p27 to Skp2, the F-box protein component of an SCF ubiquitin ligase (E3) complex, and such interaction in turn results in the polyubiquitylation and degradation of p27 [28,25,29,30]. Reduction of p27 levels in various types of malignant tumors results from accelerated proteolytic degradation by this pathway [31].
In this study, our goal was to determine whether a non-degradable p27 mutant containing a Thr187-to-Ala substitution (T187A), which is not influenced by ubiquitin-mediated degradation would be a better therapeutic agent than wild-type p27. We established two inducible systems to overexpress mutant and wild-type p27 and compared their effects on inhibition of cell growth and induction of apoptosis in breast cancer cells.
2. Materials and methods
2.1. Cell culture
The human embryonic kidney epithelial cell line HEK-293T (293T) was maintained in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum. The breast cancer cell lines MDA-MB-468, BT-549 and MDA-MB-231, were maintained in RPMI 1640 supplemented with 10% fetal bovine serum.
2.2. Plasmid construction, site-directed mutagenesis, and isolation of stable clones
Human cDNAs encoding wild-type p27 (p27wt) or a non-phosphorylatable mutant with a threonine-to-alanine substitution at position 187 [p27(T187A)] were provided by M. Pagano (Department of Pathology, New York University, New York, NY).
For the transient expression experiments, HindIII and XbaI digests from p27wt and p27(T187A) cDNAs were subcloned into the pcDNA3.1/myc-His expression vector (Invitrogen, Carlsbad, CA). A phosphomimetic mutation, T187E, was generated in the pcDNA3.1-p27 plasmid by oligonucleotide-directed mutagenesis and polymerase chain reaction using a QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The mutation was confirmed by sequencing.
For the stable ecdysone-inducible system experiments, p27 digests from the pcDNA3.1-p27wt and pcDNA3.1-p27(T187A) plasmids were inserted into the HindIII/PreI sites of the pIND vector (Invitrogen).
To obtain tightly regulated p27 expression, we established an ecdysone-inducible mammalian system in which expression of p27 transgenes containing ecdysone response elements was controlled by an ecdysone analog such as muristerone A (Mur A), a homolog of the ecdysone receptor ligand. First, 293T cells stably expressing the Drosophila ecdysone receptor (EcR) were selected with Zeocin (Invitrogen) and screened by immunoblotting. The DNA constructs pIND-p27wt-Myc, pIND-p27(T187A)-Myc, and pIND were then transfected into the cells, and clones were selected by dual selection using Zeocin and puromycin for 2–3 weeks. Clones were screened by immunoblotting with monoclonal anti-Myc antibodies (Invitrogen) for expression of p27-Myc tagged proteins upon induction with 2 μg Mur A.
2.3. Pulse-chase and protein stability experiments
For the pulse-chase experiments, stable clones were synchronized by serum starvation for 24 h, incubated in a methionine- and cysteine-free medium for 2 h, and then metabolically labeled with 200 μCi/ml of [35S]methionine and [35S]cysteine for 1 h. Proliferation was stimulated by the addition of 10% serumfor a chase period of 0, 4, 8, or 12 h. Cells were then lysed in radio-immunoprecipitation assay buffer and subjected to immunoprecipitation with anti-Myc antibodies. Proteins were separated by SDS–PAGE on a 10% gel, detected by autoradiography, and subjected to quantitative analysis with a Bio-Rad FX Pro molecular imager.
For the protein stability assay, cells were transfected with p27wt-Myc or p27(T187A)-Myc for 36 h and then treated with 20 μg/ml cycloheximide for the indicated times to inhibit new protein synthesis. Cell lysates were subjected to anti-Myc western blotting to measure their protein content. The 26S proteasome inhibitors, MG132 (Sigma–Aldrich, St. Louis, MO) and LLnL (Bio Molecular Research, Plymouth, PA) were dissolved in dimethyl sulfoxide.
2.4. In vitro ubiquitination and degradation assays
For the ubiquitination assay [32], 0.5 μl of 35S-labeled p27 (the substrate) translated in vitro was incubated with 10 μl of ubiquitination reaction mix (40 mM Tris–HCl [pH 7.6], 5 mM MgCl2 1 mM dithiothreitol, 10% glycerol, 1 mg/ml methylated ubiquitin, 1 μM ubiquitin aldehyde, 10 mM creatine phosphate, 100 μg/ml creatine phosphokinase, and 0.5 mM ATP) along with 30 μg of extract from HeLa cells that had been synchronized in S phase. After incubation at 30 °C for 60 min, ubiquitination was stopped with Laemmli sample buffer, and the products were subjected to SDS–PAGE. Polyubiquitinated forms of p27 were identified by autoradiography.
In vitro degradation assays were performed as described elsewhere [32]. Briefly, the degradation reaction mix (20 μl) was the same as the ubiquitination reaction mix except that the methylated ubiquitin was omitted and 10 μg of purified recombinant cyclin E–CDK2, immobilized on agarose beads, was added. The mixtures were incubated with radiolabeled substrate at 30 °C for 0, 10, 30, or 60 min. Proteins were resolved on 10% SDS–PAGE gels and detected by autoradiography.
2.5. Cell growth and colony formation assays, cell cycle analysis, and annexin V staining
For the cell growth assay, 1 × 104 cells were seeded on six-well plates overnight, incubated with Mur A, trypsinized, collected, and counted every day for 6 days. Colony formation assays were performed using standard methods [33]. Briefly, stable 293T clones were seeded in 60-mm dishes in triplicate and cultured at 37 °C with or without Mur A for 6 and 9 days. Colonies were then fixed and stained with 0.25% crystal violet in 50% ethanol for 20–30 min, air-dried, and counted. Cell cycle progression was analyzed using a FACScan apparatus (Thermo-LabSystems, Helsinki, Finland) as described [34]. Apoptosis was assessed using an annexin V–fluorescein isothiocyanate (FITC) staining kit according to the manufacturer’s instructions (BD PharMingen, San Diego, CA), and cells were analyzed by flow cytometry on an EPICS XL-MCL (Beckman-Coulter, Fullerton, CA).
2.6. In vitro kinase assay
Stable 293T clones were induced to express p27wt or mutant p27 by Mur A for 24 or 48 h, and whole-cell extracts were prepared as previously described [34]. Briefly, endogenous cyclin E was precipitated from 200 μg of cell lysate with anti-cyclin E antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and protein A–agarose beads for 2 h at 4 °C. The immunoprecipitates were washed twice in lysis buffer and twice in kinase buffer and processed for the kinase assay as previously described [34]. The activity of cyclin E-CDK2 was measured using 1 μg of histone H1 as the substrate. Samples were analyzed by SDS–PAGE and PhosphorImager (BioRad, Hercules, CA).
2.7. Construction of recombinant adenovirus vectors, Infection, and Immunoblot analysis
To overexpress p27 in breast carcinoma cells, a recombinant adenovirus vector expressing a doxycycline-regulated (Tet-O3 system) form of p27 was constructed according to the manufacturer’s recommendations (Clontech, Palo Alto, CA). We amplified human p27wt and p27(T187A) cDNAs from the pcDNA3.1-Myc constructs described above using polymerase chain reaction with a pair of primers containing a BamHI restriction site at the 5′ end and an AflII restriction site at the 3′ end. The amplified products were subcloned into the BamHI/AflII site of the adenoviral vector and transfected into HEK-293 cells to produce the recombinant virus. Recipient breast cancer cells were then transduced with the doxycycline-regulatory virus and either Myc-Ad-p27wt or Myc-Ad-p27(T187A) at a multiplicity of infection of 50. The cells were incubated in the presence or absence of the tetracycline analog doxycycline (1 μg/ml) in a tetracycline-free serum medium for 48 h and then harvested. All immunoblottings were performed following standard biochemical techniques.
3. Results
3.1. p27(T187A) is resistant to proteasome-mediated degradation
To determine whether phosphorylation of p27 at T187 affected its stability, we performed pulse-chase analysis by transiently transfecting p27wt, the non-phosphorylatable mutant p27(T187A), and the phosphomimetic mutant p27(T187E) separately into 293T cells. The half-lives of the three proteins were 4.5 ± 0.7, 11 ± 0.8, and 3.8 ± 0.8 h, respectively (Fig. 1A and B), indicating that p27(T187A) was much more stable than p27wt.
Fig. 1.
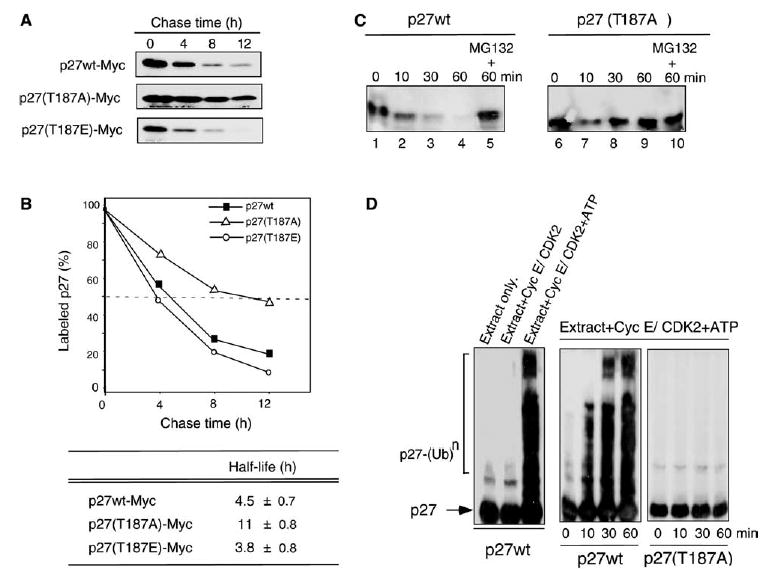
Effects of T187A mutation on stability and ubiquitination of p27. (A) Pulse-chase analysis of 293T cells transfected with Myc-tagged p27wt or its T187A and T187E mutants. The T187A mutation stabilized p27 and increased its half-life. (B) The intensities of the bands shown in (A) were quantified and expressed as percentages of labeled p27. Data shown are representative of two independent experiments with similar results. (C) In vitro proteolysis assay of p27wt and p27(T187A) in S-phase HeLa cell extracts. The proteasome inhibitor MG132 was used as a control. (D) In vitro ubiquitination assay of p27wt and p27(T187A) in S-phase HeLa cell extracts, with purified recombinant cyclin E–CDK2 complex and ATP added as indicated. Reactions were stopped at the indicated times. The bracket at left indicates ladder bands larger than 27 kDa corresponding to polyubiquitinated p27 [p27-(Ub)n].
We next assessed the degradation and ubiquitination of p27 in S-phase HeLa cell extracts. In the in vitro degradation assay, the cell extracts rapidly degraded p27wt, with most of the protein being proteolyzed within 60 min (Fig. 1C). The proteolytic degradation of p27wt was completely inhibited by addition of the 26S proteasome inhibitor MG132 (Fig. 1C). In contrast, the p27(T187A) mutant showed substantial resistance to degradation, even at 60 min. The in vitro p27 ubiquitination assay [28] showed that p27 was not ubiquitinated in the absence of cyclin E–CDK2 or/and ATP (Fig. 1D). The addition of ATP and recombinant cyclin E–CDK2 restored ubiquitination of p27wt but not p27(T187A), indicating that the proteolytic degradation of p27 was stimulated by its phosphorylation at T187. The p27(T187E) mutant was ubiquitinated and proteolyzed in our in vitro extract system to the same extent as p27wt (data not shown), confirming that T187E is a mimetic of phosphorylated p27. These results suggested that the activity of cyclin E-CDK2 and the presence of T187 were essential for the ubiquitination and subsequent degradation of p27 in vivo.
3.2. p27(T187A) is a more potent inhibitor of cell growth than p27wt
Several independent clones stably expressing Mur A-inducible p27 transgenes were selected and screened by immunoblotting. Much of the subsequent work was done with p27wt-Myc clones 11 and 14 and p27(T187A)-Myc clones 5 and 14 because of their high and equal expression of the p27-Myc fusion protein (Fig. 2A).
Fig. 2.
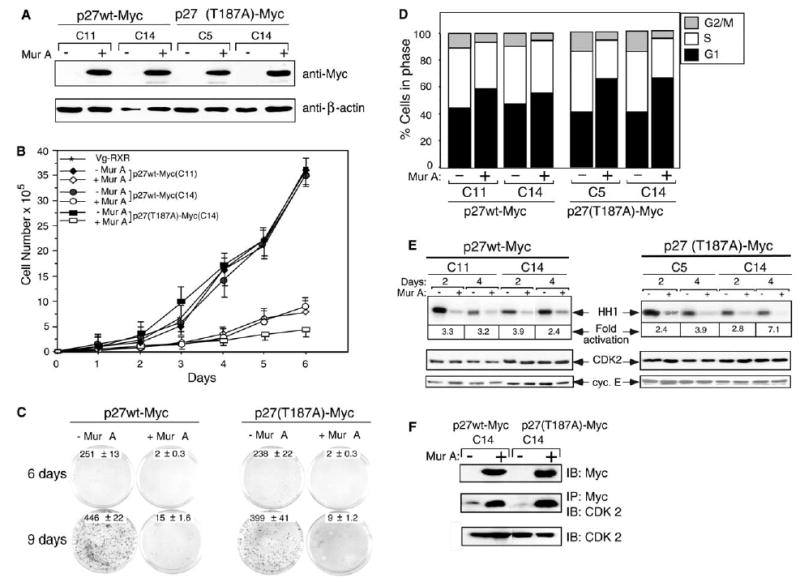
Growth inhibition and cell-cycle arrest induced by p27wt and p27(T187A). (A)Anti-Myc immunoblotting of total protein lysates from stable clones expressing p27wt-Myc (C11, C14) or p27(T187A)-Myc (C5, C14) with (+) or without (−) Mur A. β-Actin was used as a loading control. (B) Growth curves for the above clones. Total cells were counted for 6 days. Results shown are averages of three independent experiments. (C) Colony formation assay for the above clones, seeded at the same density in triplicate with or without Mur A for 6 or 9 days. Results represent three independent experiments. (D) Flow cytometric analysis of cell-cycle distribution. Results are means from three independent experiments. (E) Inhibition of CDK2 activity by induction of wild-type or mutant p27. Lysates of cells seeded with or without Mur A were immunoprecipitated with anti-CDK2 and anti-cyclin E antibodies to assess histone H1 (HH1) phosphorylation by cyclin E–CDK2. (F) Analysis of CDK2 bound to wild-type or mutant p27. Equal amounts of proteins were directly immunoprecipitated (IP) and immunoblotted (IB) with the indicated antibodies.
We used this system to evaluate the effects of p27wt and p27(T187A) on proliferation in a cell growth assay. Two days after induction with Mur A, cell growth was no different between control cells and the p27-expressing cells. However, by 4–6 days, the p27-overexpressing cells showed apoptotic morphology (data not shown), and their proliferation was markedly inhibited compared with that of control cells (P < 0.01). This growth suppression was greater in the p27(T187A)-expressing cells than in the p27wt-expressing cells (Fig. 2B). Colony formation of p27-overexpressing 293T cells was also substantially inhibited (Fig. 2C). Flow cytometric analysis confirmed that the proportion of cells in G1 arrest increased and the proportion of cells in S phase decreased 48 h after induction of p27 expression (Fig. 2D). p27(T187A) clones showed more extensive G1 arrest (44–59% and 47–56% in p27wt clones 11 and 14, respectively, and 41–62% and 41–64% in p27 mutant clones 5 and 14, respectively). These findings suggest that p27(T187A) was a more powerful CDK inhibitor, although both the wild-type and mutant forms of p27 were effective.
3.3. Both p27wt and p27(T187A) inhibit cyclin E–CDK2 kinase activity
An in vitro kinase assay showed that both p27wt and p27(T187A) inhibited cyclin E-associated kinase activity (Fig. 2E). p27wt reduced kinase activity up to 3.9-fold at 2 and 4 days after induction with Mur A, and p27(T187A) reduced kinase activity by 7.1-fold 4 days after induction. Neither total cyclin E nor CDK2 protein levels changed after induction of p27, but the decrease in CDK2-associated kinase activity was associated with an increased amount of p27 bound to cyclin E-CDK2 complexes (Fig. 2F). These results indicate that the decrease in cyclin E-CDK2 activities contributed to the G1 arrest induced by mutant p27.
3.4. p27(T187A) induces apoptosis more effectively than does p27wt
Overexpression of p27 has been shown to trigger apoptosis in mammalian cells [20,23]. We evaluated the pattern and extent of apoptotic events induced by overexpression of p27wt and p27(T187A). Flow cytometry showed that 2 days after induction by Mur A, the percentage of cells in sub-G1 phase increased from 2.1% to 5.1% with p27wt overexpression and from 2.8% to 10% with p27(T187A) overexpression. By 4 days after induction, the percentage of apoptotic cells increased from 6.1% to 10% for p27wt and from 8.6% to 23.3% for p27(T187A). These findings were confirmed by an annexin V assay in which both forms of p27 induced apoptosis, but p27(T187A) was more potent than p27wt [61.6% with p27wt and 71.5% with p27(T187A) (Fig. 3B and C)].
Fig. 3.
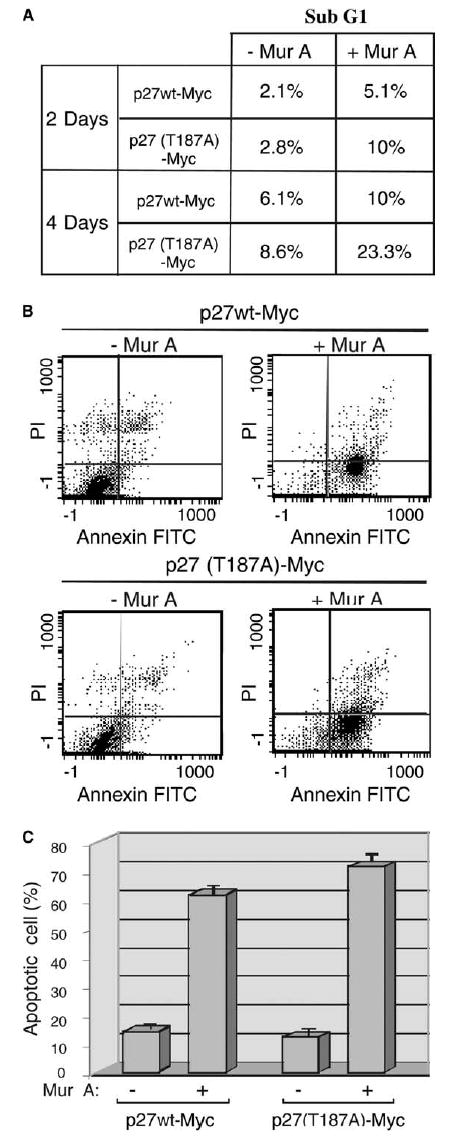
The degradation-resistant p27(T187A) mutant induces apoptosis more effectively than does p27wt. (A) Stable clones (C14) were induced to express p27wt and p27(T187A) upon addition of A (+) for 2 and 4 days. Percentage of apoptotic cells (Sub-G1 fraction) was calculated at the indicated times by using propidium iodide (PI) staining followed by FACS analysis. (B) flow cytometry analysis of apoptosis by Annexin V and propidium iodide staining. Same treatment of the cells as in (A), stable clones were induced by Mur A to express p27wt and p27(T187A), labeled with propidium iodide (PI) and annexin V–FITC. (C) Quantitative analysis of apoptotic cells from (B). The average percentages distribution of FITC-positive cells, corresponding to late (D2) and early (D4) apoptosis, are shown from three independent experiments.
3.5. p27 stability in breast cancer cells is regulated by the ubiquitin–proteasome system
Low expression of p27 protein is associated with excessive cell proliferation and has been linked to many types of human tumors, including breast cancers [35]. We screened a number of breast cancer cells and found that most of them had very low expression of p27. Treatment of breast carcinoma cell lines MDA-MB-468, BT-549 and MDA-MB-231 with two specific proteasome inhibitors, LLnL and MG132 resulted in a significant increase in total p27 levels (Fig. 4A), suggesting that p27 is primarily regulated by ubiquitin-mediated proteasomal degradation in vivo. Using cycloheximide chase assays, we determined the half-lives of both forms of p27 and found that the mutant was considerably more stable than the wild-type form (Fig. 4B). The half-life of p27wt was 10 h, whereas only 20% of p27(T187A) was degraded by that time (Fig. 4B).
Fig. 4.
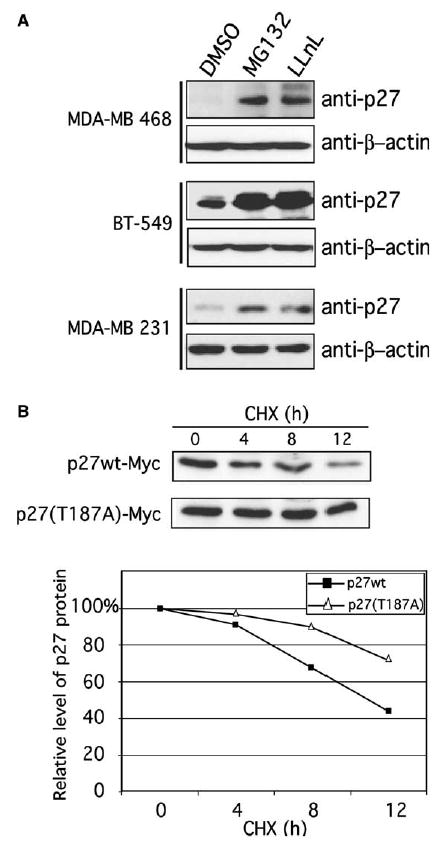
Degradation of p27 in breast cancer cells is regulated by the ubiquitin–proteasome system. (A) The proteasome inhibitors MG132 and LLnL increase basal p27 levels in the breast cancer cell lines MDA-MB-468, BT-549, and MDA-MB-231. Total cell lysates were immunoblotted with anti-p27 antibody. Dimethyl sulfoxide (DMSO) was used as a control. β-Actin was used as a loading control. (B) The stability of p27wt and p27(T187A) was analyzed using cycloheximide (CHX) chase experiments of BT-549 cells transfected with p27wt-Myc or p27(T187A)-Myc. Cell lysates were analyzed by western blotting with anti-Myc antibody. Band intensities were quantified by image analysis and expressed as percentages of the corresponding value from time zero.
3.6. Transduction with Ad-p27wt or Ad-p27(T187A) causes cell growth arrest and apoptosis
To further investigate the use of p27(T187A) as a means of stabilizing p27 protein level, we generated a doxycycline-regulated adenovirus vector to overexpress p27wt and p27(T187A) in two breast cancer cell lines. Ad-p27(T187A)-Myc had a longer half-life and increased the relative percentages of cells in G0/G1 more than Ad-p27wt-Myc (data not shown). Adenoviral transduction of MDA-MB-468 and BT-549 cells with either form of p27 in the absence of doxycycline increased the percentage of apoptotic cells, as detected by annexin V staining (Fig. 5A). To confirm that transduction of p27 induced apoptosis in breast carcinoma cells, we analyzed poly-(ADP-ribose) polymerase (PARP) cleavage. Adenoviral transduction with either form of p27 resulted in the appearance of the PARP cleavage products p85 and p25 from the intact form (p112), but no efficient cleavage was observed in the cells transduced with p27wt (Fig. 5B). Thus, the inducible expression of p27 by adenoviral vectors arrested breast carcinoma cells in G1 and led to apoptosis, while the mutant Ad-p27(T187A)-Myc had the strongest effect.
Fig. 5.
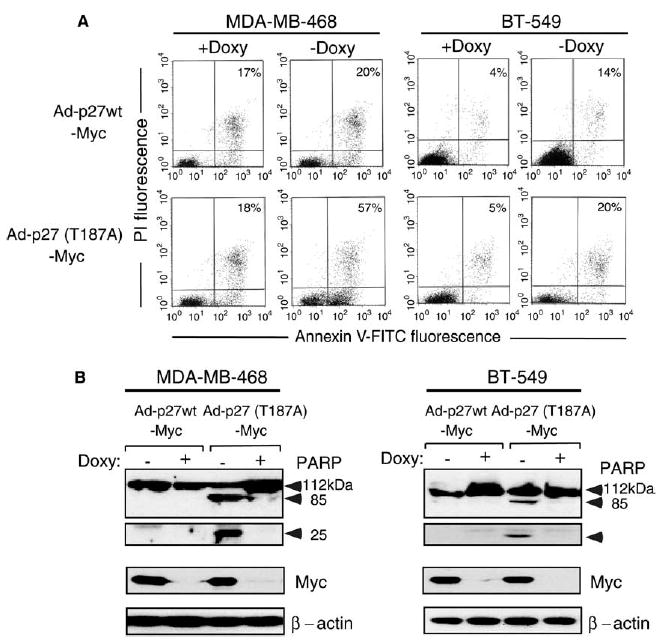
Doxycycline-regulatable adenovirus-mediated overexpression of p27(T187A) induces apoptosis more effectively than does p27wt in breast carcinoma cells. (A) Flow cytometric analysis of MDA-MB-468 and BT-549 breast cancer cells transduced with Ad-p27wt-Myc or Ad-p27(T187A)-Myc with (+) or without (−) doxycycline (Doxy). Apoptotic cells were identified by dual staining for propidium iodide (PI) and annexin V–FITC. Numbers indicate the percentage distributions of FITC-positive cells in late and early apoptosis. Similar results were seen in three independent experiments. (B) Total proteins were extracted from MDA-MB-468 and BT-549 cells transduced with Ad-p27wt-Myc and Ad-p27(T187A)-Myc and immunoblotted with antibodies against PARP, Myc, and β-actin.
3.7. Expression of cancer biomarkers after transduction of Ad-p27wt and Ad-p27(T187A) in breast cancer cells
We further determined the effects of p27wt and p27(T187A) on the levels of the cancer biomarkers cyclin E, cyclin D1, and Skp2. The products of these genes are considered to be oncoproteins [9,36,37]. High levels of cyclin E correlate strongly with poor outcome in patients with breast cancer [9,38]. Cyclin D1 binds to p27 and sequesters it from inhibiting cyclin E/CDK2 activity, which thereby contributes indirectly to the promotion of cell cycle progression. Skp2 is specifically involved in ubiquitin pathway-mediated degradation of p27 as an E3 ligase. We observed that overexpression of p27(T187A) significantly reduced cyclin E levels in all three breast cancer cell lines (MDA-MB-468, BT-549, and MDA-MB-231) compared with p27wt. Accumulation of cyclin D1 caused by overexpression of p27wt or the mutant form was present in all three cell lines. However, expression of the p27 mutant resulted in greater accumulation of cyclin D1 (Fig. 6). Levels of Skp2 decreased when both p27wt and p27(T187A) were transduced and overexpressed in these three cell lines. High expression levels of p27(T187A) were associated with greater reductions in Skp2 than high expression of p27wt in MDA-MB-468 and MDA-MB-231 cells. Furthermore, overexpression of p27 resulted in a reduction of retino-blastoma protein (pRb) phosphorylation in BT-549 and MDA-MB-231 cells, which was not seen in the pRb-null cell line MDA-MB-468 (data not shown). These results suggest that a reduction in pRb phosphorylation is not the only pathway involved in p27-mediated cell cycle arrest and apoptosis in breast cancer cells.
Fig. 6.
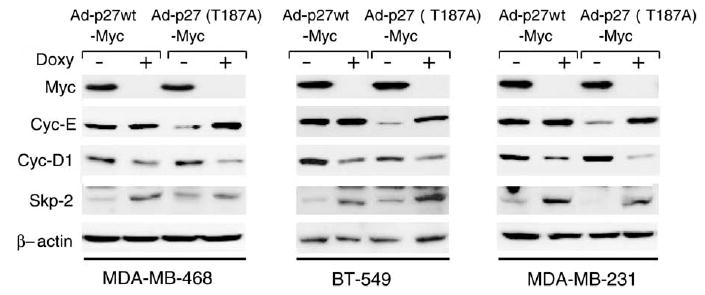
Expression levels of oncoproteins in p27wt and p27(T187A)-overexpressing breast cancer cells. The MDA-MB-468, BT-549 and MDA-MB-231, breast cancer cell lines were transduced with Ad-p27wt-Myc or Ad-p27(T187A)-Myc in the presence (+) or absence (−) of doxycycline for 48 h. Total cell lysates were prepared and expression levels of cyclin E, cyclin D1, and Skp2 were analyzed by immunoblotting.
4. Discussion
In this study, we constructed two inducible systems to overexpress a p27 mutant in which a T187A substitution prevented ubiquitin-mediated degradation. We showed that this non-phosphorylatable p27 mutant was less prone to degradation in pulse-chase experiments and was resistant to proteolysis in an in vitro degradation assay. We also confirmed that the mutant was significantly more resistant to ubiquitination and subsequent degradation than wild-type p27 was (Figs. 1 and 4). The growth of stable transfectants of the p27 mutant was more strongly suppressed than that of p27wt was (Fig. 2). The role of the CDK inhibitor for both forms of p27 was confirmed by its suppression of CDK2 activity. Finally, we demonstrated that overexpression of the degradation-resistant mutant encoded by the Ad-p27(T187A) vector caused greater inhibition of cell growth and stronger induction of apoptosis than did overexpression wildtype p27, as confirmed by annexin V and PARP cleavage assays (Fig. 5).
Loss of cell-cycle control is important in carcinogenesis. Although genetic alterations of the cell-cycle regulator p27 have rarely been found in cancer, downregulation of p27 is frequently observed in human cancers and has been associated with increased cell proliferation and tumor aggressiveness [14,11,10,13]. In several studies, intratumoral injections of Ad-p27wt has been shown to partially suppress tumor formation in animal models [23,24,16,39]. In other experiments, Ad-p27 was the most potent of several cyclin kinase inhibitors in terms of inducing cell-cycle arrest and apoptosis and inhibiting tumorigenesis [21,20,22,23,16,39,17]. Our recombinant adenovirus expressing mutant p27 may be useful against breast carcinoma, although the possible antitumor effects of p27(T187A) must be confirmed by in vivo tumorigenesis assays in mice.
In line with these findings, other studies have shown that the p27(T187A) mutant is not ubiquitinated [28] and that transient expression of a p27(T187A) plasmid causes a G1 block that is resistant to cyclin E and is not modulated by cyclin E–CDK2 [26,27]. We found that stable expression and adenoviral transduction of either p27wt or p27(T187A) inhibited cell growth and induced G1 arrest in both 293T and breast cancer cells. However, overexpression of p27(T187A) induced growth inhibition and apoptosis to a greater extent than did overexpression of p27wt, regardless of the expression system used. These findings are consistent with those of another study in which Ad-p27(T187A) had a greater effect on cell-cycle arrest and apoptosis induction, because of its resistance to degradation, and suppressed the growth of established lung cancer xenografts [24]. These findings support our hypothesis that stabilizing p27 expression may be an effective strategy for inhibiting cancer cell growth.
The relationship between p27 and induction of apoptosis is still unclear. p27 may be indirectly associated with apoptosis (e.g., through CDK inhibition), and may be able to regulate the cell cycle or apoptosis to protect cells from over-growth and apoptosis-inducing stimulation. Naruse et al. [17] and Katayose et al. [22] have suggested that the growth-inhibitory effect of and apoptosis induction by overexpression of p27 requires expression of pRb. The pRb-regulated checkpoint in G1 is an important apoptotic checkpoint. Cyclin E-CDK2 is the primary complex that phosphorylates pRb, which prevents interactions of it with the E2F transcription factor.
We also found that levels of the cancer biomarkers cyclin D1, cyclin E, and Skp2 changed in response to overexpression of p27wt and p27(T187A). Specifically, we observed that ectopic p27wt and p27(T187A) led to accumulation or stabilization of cyclin D1 but that transduction of the mutant form of p27 led to more pronounced reduction of cyclin E levels. Skp2 levels were also reduced in p27wt- and p27(T187A)-overexpressing breast cancer cells. In some circumstances, p27 is thought to inhibit cyclin D-CDK4/6 complexes [4,5]. In other circumstances, p27 does not inhibit the activity of the complex but rather acts as an assembly factor [40]; in other words, p27 may not be inhibitory when bound to cyclin D1/CDK4, whereas p27 is inhibitory of cyclin E-CDK2 activity. All three oncoproteins are subject to cell-cycle regulation [41,42]. Cyclin D1 expression is induced primarily during G2 and remains constant through G1, where it is required for the cell to commit to another round of replication (reviewed in [43]). As cells enter S phase, cyclin D1 levels decline (reviewed in [43]). Our results indicate that expression of p27wt and p27(T187A) lead to G1 and sub-G1 arrest (data not shown), which would explain an increase in cyclin D1 levels (Fig. 6). Overexpression of p27 in either mutant or wt form reduced cyclin E levels. Cyclin E is an unstable protein degraded by two distinct pathways involving the ubiquitin–proteosome system (reviewed in [44]). During normal cell cycle, cyclin E levels are low during G1 and increase during the transition from G1 to S. The finding that p27 mutant was a more potent inducer of apoptosis than was p27wt in breast cancer cell lines MDA-MB-468, BT-549, and MDA-MB-231 may explain the dramatic reduction in cyclin E levels observed in cells transduced with the mutant p27. Skp2, also a cell cycle–regulated protein, is present at low levels in G1 and in late mitosis [42]. We showed that overexpression of p27wt or the p27(T187A) mutant in breast cancer cell lines greatly reduced Skp2 levels. Whether this observation is the cause or the consequence of overexpression of p27 is unclear. Skp2 degradation may result from accumulation of a CDK inhibitor that leads to G1 arrest. A recent report has shown that the degradation of the SCF component Skp2 during G1 is controlled by APC/Ccdh1 ubiquitin ligase [45]. The correlation between overexpression of p27 and APC/Ccdh1 ubiquitin pathway, if any, needs further investigation.
The control of p27 protein levels is affected by ubiquitin-dependent degradation [31,26,27,29,25], in a ubiquitin- and Skp2-independent manner at G1 [42,46], and by JAB1-dependent degradation [47]. The effect of p27 on the cell cycle is regulated mainly by its stability [48,49], but recent studies have shown that the function of p27 is also associated with its sub-cellular localization [50,51]. Besides Thr187, there are three phosphorylation sites Ser10, Thr157, and Thr198 that are involved in cellular localization [52,53]. Phosphorylation at Ser10 stabilizes p27 protein in G1 [53]. Phosphorylation at Thr157 by protein kinase B/Akt impairs the nuclear import of p27 but does not affect its stability in breast cancer and other cells [50,51]. Mutations in these phosphorylation sites may result in a more potent induction of apoptosis and inhibition of cell growth for breast cancer gene therapy.
Acknowledgments
We thank Drs. K. Keyomarsi of The University of Texas M. D. Anderson Cancer Center and M. Pagano of New York University for sharing reagents used in this study. We also thank P. Lo for editing this manuscript. A.M. is funded by the Association pour la Recherche sur le Cancer and the Fondation pour la Recherche Medicale. This study is supported by grant 1R01CA90853, core grants 5P50CA83639, P30CA16672 from the National Cancer Institute and the Susan G. Komen Breast Cancer Foundation.
References
- 1.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 2.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 3.Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polyak K, Lee MH, Erdjument-Bromage H, Ko3 A, Roberts JM, Tempst P, Massague J. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 5.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 6.Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, Kaushansky K, Roberts JM. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996;85:733–744. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- 7.Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Ko3 A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1) Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 8.Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 9.Porter PL, Malone KE, Heagerty PJ, Alexander GM, Gatti LA, Firpo EJ, Daling JR, Roberts JM. Expression of cell-cycle regulators p27Kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients. Nat Med. 1997;3:222–225. doi: 10.1038/nm0297-222. [DOI] [PubMed] [Google Scholar]
- 10.Catzavelos C, Bhattacharya N, Ung YC, Wilson JA, Roncari L, Sandhu C, Shaw P, Yeger H, Morava-Protzner I, Kapusta L, Franssen E, Pritchard KI, Slingerland JM. Decreased levels of the cell-cycle inhibitor p27Kip1 protein: prognostic implications in primary breast cancer. Nat Med. 1997;3:227–230. doi: 10.1038/nm0297-227. [DOI] [PubMed] [Google Scholar]
- 11.Mori M, Mimori K, Shiraishi T, Tanaka S, Ueo H, Sugimachi K, Akiyoshi T. p27 expression and gastric carcinoma. Nat Med. 1997;3:593. doi: 10.1038/nm0697-593. [DOI] [PubMed] [Google Scholar]
- 12.Cordon-Cardo C, Ko3 A, Drobnjak M, Capodieci P, Osman I, Millard SS, Gaudin PB, Fazzari M, Zhang ZF, Massague J, Scher HI. Distinct altered patterns of p27KIP1 gene expression in benign prostatic hyperplasia and prostatic carcinoma. J Natl Cancer Inst. 1998;90:1284–1291. doi: 10.1093/jnci/90.17.1284. [DOI] [PubMed] [Google Scholar]
- 13.Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, Jessup JM, Pagano M. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med. 1997;3:231–234. doi: 10.1038/nm0297-231. [DOI] [PubMed] [Google Scholar]
- 14.Esposito V, Baldi A, De Luca A, Groger AM, Loda M, Giordano GG, Caputi M, Baldi F, Pagano M, Giordano A. Prognostic role of the cyclin-dependent kinase inhibitor p27 in non-small cell lung cancer. Cancer Res. 1997;57:3381–3385. [PubMed] [Google Scholar]
- 15.Kawamata N, Morosetti R, Miller CW, Park D, Spirin KS, Nakamaki T, Takeuchi S, Hatta Y, Simpson J, Wilcyznski S, et al. Molecular analysis of the cyclin-dependent kinase inhibitor gene p27/Kip1 in human malignancies. Cancer Res. 1995;55:2266–2269. [PubMed] [Google Scholar]
- 16.Katner AL, Hoang QB, Gootam P, Jaruga E, Ma Q, Gnarra J, Rayford W. Induction of cell cycle arrest and apoptosis in human prostate carcinoma cells by a recombinant adenovirus expressing p27(Kip1) Prostate. 2002;53:77–87. doi: 10.1002/pros.10124. [DOI] [PubMed] [Google Scholar]
- 17.Naruse I, Hoshino H, Dobashi K, Minato K, Saito R, Mori M. Over-expression of p27kip1 induces growth arrest and apoptosis mediated by changes of pRb expression in lung cancer cell lines. Int J Cancer. 2000;88:377–383. [PubMed] [Google Scholar]
- 18.Ishii T, Fujishiro M, Masuda M, Goshima Y, Kitamura H, Teramoto S, Matsuse T. Effects of p27Kip1 on cell cycle status and viability in A549 lung adenocarcinoma cells. Eur Respir J. 2004;23:665–670. doi: 10.1183/09031936.04.00096204. [DOI] [PubMed] [Google Scholar]
- 19.Supriatno Harada K, Kawaguchi S, Onoue T, Yoshida H, Sato M. Characteristics of antitumor activity of mutant type p27Kip1 gene in an oral cancer cell line. Oral Oncol. 2004;40:679–687. doi: 10.1016/j.oraloncology.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Gorospe M, Huang Y, Holbrook NJ. p27Kip1 overexpression causes apoptotic death of mammalian cells. Oncogene. 1997;15:2991–2997. doi: 10.1038/sj.onc.1201450. [DOI] [PubMed] [Google Scholar]
- 21.Craig C, Wersto R, Kim M, Ohri E, Li Z, Katayose D, Lee SJ, Trepel J, Cowan K, Seth P. A recombinant adenovirus expressing p27Kip1 induces cell cycle arrest and loss of cyclin-Cdk activity in human breast cancer cells. Oncogene. 1997;14:2283–2289. doi: 10.1038/sj.onc.1201064. [DOI] [PubMed] [Google Scholar]
- 22.Katayose Y, Kim M, Rakkar AN, Li Z, Cowan KH, Seth P. Promoting apoptosis: a novel activity associated with the cyclin-dependent kinase inhibitor p27. Cancer Res. 1997;57:5441–5445. [PubMed] [Google Scholar]
- 23.Schreiber M, Muller WJ, Singh G, Graham FL. Comparison of the effectiveness of adenovirus vectors expressing cyclin kinase inhibitors p16INK4A, p18INK4C, p19INK4D, p21(WAF1/CIP1) and p27KIP1 in inducing cell cycle arrest, apoptosis and inhibition of tumorigenicity. Oncogene. 1999;18:1663–1676. doi: 10.1038/sj.onc.1202466. [DOI] [PubMed] [Google Scholar]
- 24.Park KH, Seol JY, Kim TY, Yoo CG, Kim YW, Han SK, Shim YS, Lee CT. An adenovirus expressing mutant p27 showed more potent antitumor effects than adenovirus-p27 wild type. Cancer Res. 2001;61:6163–6169. [PubMed] [Google Scholar]
- 25.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 26.Shea3 RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]
- 27.Vlach J, Hennecke S, Amati B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J. 1997;16:5334–5344. doi: 10.1093/emboj/16.17.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, Pagano M. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999;13:1181–1189. doi: 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U, Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999;1:207–214. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]
- 30.Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, Hershko A. The cell-cycle regulatory protein Cks1 is required for SCFSkp2-mediated ubiquitinylation of p27. Nat Cell Biol. 2001;3:321–324. doi: 10.1038/35060126. [DOI] [PubMed] [Google Scholar]
- 31.Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin–proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen H, Gitig DM, Ko3 A. Cell-free degradation of p27(kip1), a G1 cyclin-dependent kinase inhibitor, is dependent on CDK2 activity and the proteasome. Mol Cell Biol. 1999;19:1190–1201. doi: 10.1128/mcb.19.2.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conway KE, McConnell BB, Bowring CE, Donald CD, Warren ST, Vertino PM. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res. 2000;60:6236–6242. [PubMed] [Google Scholar]
- 34.Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, Siddik ZH, Mills GB, Claret FX. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. 2003;278:19245–19256. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- 35.Philipp-Staheli J, Payne SR, Kemp CJ. p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res. 2001;264:148–168. doi: 10.1006/excr.2000.5143. [DOI] [PubMed] [Google Scholar]
- 36.Fredersdorf S, Burns J, Milne AM, Packham G, Fallis L, Gillett CE, Royds JA, Peston D, Hall PA, Hanby AM, Barnes DM, Shousha S, O’Hare MJ, Lu X. High level expression of p27(kip1) and cyclin D1 in some human breast cancer cells: inverse correlation between the expression of p27(kip1) and degree of malignancy in human breast and colorectal cancers. Proc Natl Acad Sci USA. 1997;94:6380–6385. doi: 10.1073/pnas.94.12.6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Signoretti S, Di Marcotullio L, Richardson A, Ramaswamy S, Isaac B, Rue M, Monti F, Loda M, Pagano M. Oncogenic role of the ubiquitin ligase subunit Skp2 in human breast cancer. J Clin Invest. 2002;110:633–641. doi: 10.1172/JCI15795. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Keyomarsi K, Tucker SL, Buchholz TA, Callister M, Ding Y, Hortobagyi GN, Bedrosian I, Knickerbocker C, Toyofuku W, Lowe M, Herliczek TW, Bacus SS. Cyclin E and survival in patients with breast cancer. N Engl J Med. 2002;347:1566–1575. doi: 10.1056/NEJMoa021153. [DOI] [PubMed] [Google Scholar]
- 39.Rakkar AN, Li Z, Katayose Y, Kim M, Cowan KH, Seth P. Adenoviral expression of the cyclin-dependent kinase inhibitor p27Kip1: a strategy for breast cancer gene therapy. J Natl Cancer Inst. 1998;90:1836–1838. doi: 10.1093/jnci/90.23.1836. [DOI] [PubMed] [Google Scholar]
- 40.LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 41.Ekholm SV, Zickert P, Reed SI, Zetterberg A. Accumulation of cyclin E is not a prerequisite for passage through the restriction point. Mol Cell Biol. 2001;21:3256–3265. doi: 10.1128/MCB.21.9.3256-3265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hara T, Kamura T, Nakayama K, Oshikawa K, Hatakeyama S. Degradation of p27(Kip1) at the G(0)–G(1) transition mediated by a Skp2-independent ubiquitination pathway. J Biol Chem. 2001;276:48937–48943. doi: 10.1074/jbc.M107274200. [DOI] [PubMed] [Google Scholar]
- 43.Sa G, Guo Y, Stacey DW. The regulation of S phase initiation by p27kip1 in NIH3T3 cells. Cell Cycle. 2005;4:618–627. [PubMed] [Google Scholar]
- 44.Deshaies RJ. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435–467. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- 45.Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 2004;428:190–193. doi: 10.1038/nature02330. [DOI] [PubMed] [Google Scholar]
- 46.Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, Yoshida M, Nakayama K, Nakayama KI. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004;6:1229–1235. doi: 10.1038/ncb1194. [DOI] [PubMed] [Google Scholar]
- 47.Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature. 1999;398:160–165. doi: 10.1038/18230. [DOI] [PubMed] [Google Scholar]
- 48.Boehm M, Yoshimoto T, Crook MF, Nallamshetty S, True A, Nabel GJ, Nabel EG. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. EMBO J. 2002;21:3390–3401. doi: 10.1093/emboj/cdf343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem. 2002;277:14355–14358. doi: 10.1074/jbc.C100762200. [DOI] [PubMed] [Google Scholar]
- 50.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 51.Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 52.Fujita N, Sato S, Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2003;278:49254–49260. doi: 10.1074/jbc.M306614200. [DOI] [PubMed] [Google Scholar]
- 53.Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, Meloche S. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J. 2001;20:6672–6682. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]