

Abstract
Restrained molecular dynamics simulations were performed to study the interaction forces of a protein with the self-assembled monolayers (SAMs) of S(CH2)4(EG)4OH, S(CH2)11OH, and S(CH2)11CH3 in the presence of water molecules. The force-distance curves were calculated by fixing the center of mass of the protein at several separation distances from the SAM surface. Simulation results show that the relative strength of repulsive force acting on the protein is in the decreasing order of OEG-SAMs > OH-SAMs > CH3-SAMs. The force contributions from SAMs and water molecules, the structural and dynamic behavior of hydration water, and the flexibility and conformation state of SAMs were also examined to study how water structure at the interface and SAM flexibility affect the forces exerted on the protein. Results show that a tightly bound water layer adjacent to the OEG-SAMs is mainly responsible for the large repulsive hydration force.
INTRODUCTION
Surface resistance to protein adsorption is currently a subject of great interest and is critical to the performance of biosensors, implanted biomaterials, drug carriers, and coatings on ship hulls (1–3). A large number of experimental studies of protein adsorption to various artificial surfaces have been performed to understand the mechanism of surface resistance to protein adsorption (4–6). Poly (ethylene glycol) (PEG) polymers (7,8) or oligo (ethylene glycol) (OEG) self-assembled monolayers ([SAMs] 9–11) have been widely used to prevent protein adsorption from biological media. Steric repulsion or water barrier has often been proposed to explain protein resistance to surfaces. Steric repulsion, first proposed by De Gennes and co-workers (12) using free energy calculations and later improved by Szleifer et al. (13) using the single chain mean field theory, is mainly attributed to chain compression (conformational entropy loss) as the protein approaches the surface. This hypothesis is often used to explain PEG polymers with long chain length. The water barrier mechanism (14–16) arises due to tightly bound water at the interface, which forms a physical barrier to prevent direct contact between the protein and the surface. The water barrier hypothesis is often used to explain the OEG-SAMs (2) with short chain lengths. Unlike long polymer chains, those densely packed, shorter OEG-SAMs have less freedom for conformational change upon protein adsorption.
Protein adsorption is often studied by measuring the amounts of adsorbed protein on a surface using surface plasma resonance ([SPR] 9,10), Fourier transform infrared (15), and radiolabel techniques (17). An alternative way of investigating the protein-surface interactions is to directly evaluate the interaction forces between a specific protein and a chosen surface using scanning force microscopy (18), interfacial force microscopy ([IFM] 19), and surface force apparatus (20,21). These force measurements can distinguish between long-range attractive/repulsive interactions and short-range binding events (22). Many experimental efforts (18–24) have been dedicated to measuring the intermolecular forces as a function of separation distance between various proteins and a series of SAMs terminated with different functional groups using various force measurements.
These direct surface force measurements commonly encounter the following limitations. First, a difference between advancing and receding force profiles (i.e., force hysteresis) was often observed, indicating that the convective fluid flow induced by the movement of a cantilever tip has influence on the force-distance curves. Indeed, Kim et al. (19) confirmed that force-distance profiles depend on the tip-approach speeds due to the difficulty of draining a liquid from the region between the two approaching SAM surfaces. Second, it should be noted that the total interfacial force acting on a protein tip, measured by most surface force experiments, is composed of the contributions from solvent, surface, and the mechanical response of the tip and substrate. Kim et al. (19) pointed out that the force-distance profiles should be corrected by excluding the mechanical contributions from the tips and their corresponding supports to obtain protein-surface interactions. Third, the force-distance curves, even measured for the same protein-surface system, can be altered by changing the loading force on the tip. Sheth and Leckband (25) reported that the forces between streptavidin and PEG were repulsive at low compressive loads, but they became attractive at high compressive loads. All measurements were performed at forces that are much too low to denature the protein. Thus, force changes were due to the change of loading strength. Fourth, the variations of surface properties caused by surface defects or contaminations can also lead to nonreproducible or inconsistent results. Rixman and co-workers (21) and Kidoaki et al. (20) found that the adhesion forces between the protein tips and hydrophobic surfaces were much larger than those between protein tips and hydrophilic surfaces. However, Wang et al. (22) recently showed that the adhesion forces of protein-CH3-terminated SAMs and protein-OH-terminated SAMs were 0.87 nN and 2.54 nN, respectively. Their results are different from those reported by Kidoaki et al. (20) and Rixman et al. (21). In the work by Wang et al. (22), a large contact angle of 82° was reported for the OH-SAM surface, which could contribute to the difference.
Molecular simulations are well suited to the study of protein adsorption on surfaces and provide molecular-level information, which is complementary to experimental results. Our previous simulations (14) showed that the total number of hydrogen bonds between water molecules and OEG chains is higher and the flexibility of OEG-SAM chains is larger at intermediate OEG surface densities of 0.5 and 0.8 than those at higher or lower OEG surface density. Our recent SPR experimental results (26) showed that the OEG-SAMs adsorb proteins when their surface OEG densities are too high or too low, yet nonfouling at appropriate OEG densities. When comparing our simulation results (14) and our experimental observations (26), it appears that there is a correlation between protein resistance to OEG-SAMs and a large number of tightly bound water molecules around OEG chains and the rapid mobility of hydrated SAM chains. Thus, it is desirable to calculate the interaction forces between a protein and a given surface to directly correlate these forces with nonfouling properties of the surface. In this work, we performed molecular simulations to calculate the interaction forces between a protein and various CH3-, OH-, and OEG-terminated SAMs in the presence of water molecules as the protein approaches the SAM surface from a large separation down to intimate contact. The force-distance curves calculated from simulations not only directly evaluate repulsive interaction forces (a nonfouling surface) and attractive interaction forces (an adsorption surface) but also identify the molecular origin of the overall repulsive or attractive force from water and monolayers. It should be pointed out that the difference in force curves obtained from molecular simulations and force measurements is that molecular simulations provide purely the interaction forces between a protein and the surface, whereas force measurements contain both protein-surface interactions and hydrodynamic forces. These hydrodynamic forces could often dominate over protein-surface interaction forces. Thus, molecular simulations are better suited to accurately describing the interactions between protein, water, and SAM surfaces without interference from convective fluid flow or mechanical disruption. The tightly bound water layer and the flexibility of the SAM chains are also found to associate with interfacial forces between the protein and the SAM surfaces, thus the nonfouling behavior of a surface.
SIMULATION MODEL AND METHODOLOGY
Model systems
SAMs of alkanethiolates terminated with different functional groups (S(CH2)4(EG)4OH, S(CH2)11OH, and S(CH2)11CH3) on Au(111) were used to study protein adsorption. For each given SAM surface, a single protein (lysozyme) was manually placed at three distances (5, 10, and 20 Å) with respect to the SAM surface in the presence of explicit solvent water molecules and counterions (Fig. 1). OEG-, OH-, and CH3-SAMs have 132 chains each, which are densely packed in an 11 × 12 array, forming a  lattice structure with a sulfur-sulfur distance of 0.497 nm. The SAM surface has dimensions of 55 Å × 52 Å in the x, y plane. The alkanethiol chains were initially tilted by ∼30° from the surface normal (z axis) toward their next nearest neighbors. The sulfur atoms of the SAMs were assumed to be chemically bonded to the Au(111) substrate. The initial structures of the SAMs were built using the CHARMM program (27,28) and then energy was minimized in a vacuum. The OH- and CH3-SAMs have a zigzag configuration, whereas the OEG-SAMs have a helical configuration.
lattice structure with a sulfur-sulfur distance of 0.497 nm. The SAM surface has dimensions of 55 Å × 52 Å in the x, y plane. The alkanethiol chains were initially tilted by ∼30° from the surface normal (z axis) toward their next nearest neighbors. The sulfur atoms of the SAMs were assumed to be chemically bonded to the Au(111) substrate. The initial structures of the SAMs were built using the CHARMM program (27,28) and then energy was minimized in a vacuum. The OH- and CH3-SAMs have a zigzag configuration, whereas the OEG-SAMs have a helical configuration.
FIGURE 1.

Snapshots of protein (lysozyme) on OEG-terminated SAMs in the presence of explicit water molecules and counterions. The minimal separation distances between protein and OEG-SAM are (left) 5 Å, (middle) 10 Å, and (right) 20 Å.
Lysozyme is often used as a model protein to study adsorption behavior on surfaces since its structure, dynamics, and folding have been studied extensively by a wide range of experimental and theoretical techniques (29). The initial x-ray crystal structure of lysozyme (Protein Data Bank entry code 7LYZ), comprising 129 amino acids, served as the starting configuration of the simulations. Missing polar hydrogens were added to the crystal structure. The histidine (HIS), arginine (ARG), and lysine (LYS) residues were taken to be protonated, whereas glutamate (GLU) and aspartate (ASP) residues were taken to be deprotonated; four disulfide bonds were added; the N-terminus (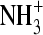 ) and the C-terminus (
) and the C-terminus ( ) were assigned a charge state of +1e and −1e, respectively. The protein has a total of 1960 atoms and a net charge of +8e.
) were assigned a charge state of +1e and −1e, respectively. The protein has a total of 1960 atoms and a net charge of +8e.
For the protein, CH3-, and OH-SAMs, an all-atom CHARMM22 force field (28), consisting of bond, Urey-Bradley, angle, dihedral, and improper terms, as well as nonbonded van der Waals (VDW) and Coulombic interactions, was used to describe the atomic interactions. For the water, the modified three-site point charge model (TIP3P) model was adopted where VDW interaction sites are on both hydrogen and oxygen atoms (28). For the OEG-SAMs, the Smith-Jaffe-Yoon potential model (30,31) was utilized because this empirical force field can reproduce very well the helical structure of OEG in solution. (Further details about the model of OEG and its force field parameters are given in references 30–33.)
Simulation methodology
We follow a two-step protocol from our previous simulations of protein interactions with OEG-SAMs (14). First, we carried out a series of Metropolis Monte Carlo (MC) simulations to determine the orientation of lysozyme at different SAM interfaces in continuous medium with a distance-dependent dielectric constant 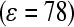 that mimics water. With preliminarily optimized orientation of the protein from MC simulations, the protein was positioned at different distances from the SAM surface and water molecules were added to a simulation box. Then, we performed molecular dynamics (MD) simulations of solvated systems by fixing the center of the mass of the protein.
that mimics water. With preliminarily optimized orientation of the protein from MC simulations, the protein was positioned at different distances from the SAM surface and water molecules were added to a simulation box. Then, we performed molecular dynamics (MD) simulations of solvated systems by fixing the center of the mass of the protein.
For MC simulations, lysozyme was initially placed at various separation distances (10–50 Å) above surfaces with a random orientation. The protein was either translated or rotated around its center of mass with the acceptance ratio of 0.5 using the Metropolis criteria. During simulations, the protein was modeled as a rigid molecule; the SAM surfaces were fixed in the x, y plane; and water was treated as an implicit solvent continuum model. With the model described above, only the nonbonded interactions, i.e., VDW and Coulombic, between the protein and the SAM surfaces were calculated at this stage. Each simulation was carried out for ∼106 MC steps.
After MC simulations, the optimal orientation of lysozyme at the SAM surface was obtained and lysozyme was placed above the SAM surface with the minimum separation distances of ∼5 Å, 10 Å, and 20 Å. Then, lysozyme and the SAM surfaces were immersed in a preequilibrated box of TIP3P water molecules with a density of 1 g/cm3. Counterions (one sodium and nine chlorines) were added to balance system charges. Water molecules that overlapped with the protein or the SAMs within 2.8 Å were removed. The whole system with the protein, water, SAMs, and counterions was initially minimized in energy for 4000 cycles using the conjugate gradient algorithm to remove any bad contacts between molecules. This minimized system was then gradually heated from 50 K to 300 K with 50 K increments in a short MD run of 20 ps while harmonically constraining the backbone atoms of the protein and the SAMs to their initial positions. This heating process allowed the initial relaxation of water molecules around the protein and the SAM surfaces.
For the equilibrium MD part, the starting configuration of the protein, water, SAMs, and counterions was taken from the final frame of the heating MD simulation, as shown in Fig. 1. The velocity Verlet method was used for the integration of the Newton's equation in the NVT ensemble with a time step of 1.0 fs. The Berendsen method was used to maintain the constant temperature of 300 K with a coupling constant of 0.1 ps. Initial velocities were assigned with a Maxwell-Bolzmann distribution at 300 K. Each simulation system was placed in a rectangular box. The periodic boundary condition and minimum image convention were applied to the x and y directions only. There is a hard wall on the top of the simulation box, and a reflective boundary condition is applied. The center of mass of the protein molecule was fixed so that the minimum separation distance between protein and SAMs was maintained constant during MD simulations. All sulfur atoms of SAMs were fixed, and all covalent bonds involving hydrogen atoms were constrained using the RATTLE algorithm with a geometric tolerance of 10−6. The short-range VDW interactions were calculated by the switch function at a twin range cutoff between 8 Å and 10 Å. The long-range electrostatic interactions were calculated by the force-shifting function at a cutoff distance of 12 Å. Early studies (34–36) showed that the atom-based force-shifting function can conserve energy, correctly predict the experimental data, and generate stable trajectories. The cell-linked neighbor list with a cutoff range of 1.32 nm was employed to accelerate the simulation. The total length of an MD simulation run is ∼1.5 ns. Configurations were saved every 1.0 ps after 1 ns for analysis. All simulations were performed on a 16-node Linux cluster Intel ×86 (CPU 2.2 GHz) using our BIOSURF program. We developed this generalized molecular simulation program for the study of a protein at biological interfaces. All initial structures were built using the CHARMM package (version c30b1).
RESULTS AND DISCUSSION
MC simulations were first performed to determine protein orientation. During MC simulations, the energetically favorable orientation of the protein on each SAM surface was obtained by calculating the nonbonded interactions of protein with the SAM surface. MD simulations were then carried out to calculate the interaction forces between the protein and different OEG-, OH-, and CH3-SAMs. During MD simulations, the force-distance profiles, protein conformation, dynamical water structure, and SAM conformation were examined. In all MD simulations, temperature was maintained at 300 K and total potential energy approached a plateau, indicating that the equilibrium state was reached. Average properties of lysozyme, SAMs, and water molecules are summarized in Table 1.
TABLE 1.
Simulation results for protein, the SAMs, and water
| Model system Separation distance (Å) | 5 | CH3-SAMs 10 | 20 | 5 | OH-SAMs 10 | 20 | 5 | OEG-SAMs 10 | 20 |
|---|---|---|---|---|---|---|---|---|---|
| Protein | |||||||||
| Cα RMSD (Å) | 1.42 | 1.36 | 1.35 | 1.34 | 1.30 | 1.32 | 1.27 | 1.22 | 1.29 |
| Radius of gyration (Å) | 14.07 | 14.20 | 14.10 | 14.15 | 14.12 | 14.13 | 14.09 | 14.01 | 14.04 |
| SAM surfaces | |||||||||
| RMSD (Å) | 0.77 | 0.62 | 0.46 | 0.93 | 0.65 | 0.62 | 1.00 | 0.82 | 0.77 |
 (degree) (degree) |
33.4 | 33.3 | 33.1 | 32.7 | 32.2 | 31.0 | 7.6 | 8.9 | 6.8 |
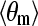 (degree) (degree) |
33.7 | 33.7 | 33.3 | 33.5 | 33.8 | 32.5 | 29.5 | 31.1 | 29.1 |
| Water | |||||||||
| Hydrogen bonds | 0.0 | 0.0 | 0.0 | 216.2 | 226.5 | 221.8 | 272.9 | 294.5 | 295.0 |
| Diffusion coefficients (10−5 cm2/s) | 1.93 | 2.04 | 2.88 | 1.80 | 2.02 | 2.32 | 0.26 | 0.63 | 0.20 |
Force-distance profiles
Fig. 2 shows the net intermolecular forces on the protein from the SAMs and water molecules as a function of separation distances between the protein and different CH3-, OH-, and OEG-terminated SAMs. The separation distance is defined by the nearest distance between the protein VDW surface and the end groups of the last heavy atoms in the SAM chains. For the hydrophilic OH- and OEG-SAMs, repulsive forces are observed for the whole range studied, as expected. Repulsive forces increase monotonically as the separation distance is decreased. For the OEG-SAMs, a nearly exponential repulsive force acting on the protein was observed, in agreement with experimental results (21). The OEG-SAMs provide much stronger repulsive force than OH-SAMs. For the hydrophobic CH3-SAMs, force profiles show that weak attractive interactions occur at a larger separation distance (20 Å), whereas weak repulsive interactions occur at a smaller separation distance (5 or 10 Å). It should be noted that, contrary to experimental results, the weak repulsive force was observed for the CH3-SAM surface as the protein was closer to the surface from simulations. This is because within the simulation timescale the protein does not denature, as it should be on the hydrophobic surface observed in experiments.
FIGURE 2.
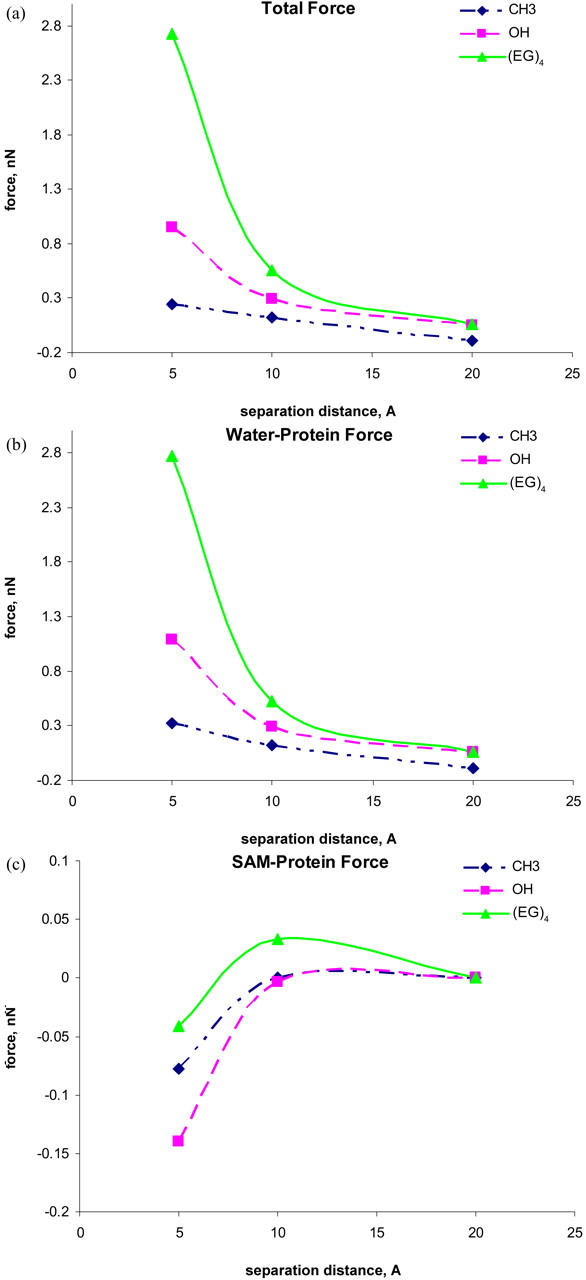
(a) Force-distance curves between the protein and three SAM surfaces in the presence of water molecules, (b) forces on the protein from water, and (c) forces on the protein from SAM surfaces. The lines are drawn to guide eyes.
The root mean-square derivations (RMSD) can be used to characterize the conformational changes of the protein. As listed in Table 1, the RMSD values of the protein are ∼1.38 Å, 1.32 Å, and 1.26 Å on the CH3-SAMs, the OH-SAMs, and the OEG-SAMs, respectively. These RMSD values also confirm that the protein does not undergo large local deformation or denaturation during the simulations. For the CH3-SAM, the weak repulsive force is due to the fact that as a “hydrophilic” (undenatured) protein approaches the hydrophobic CH3-SAM surface, the hydration layer around the approaching protein is compressed by close contact with the CH3-SAM, leading to repulsive steric hydration force. It is interesting to note that this repulsive force is small, i.e., protein adsorption on the CH3-SAM has a low energy barrier. Thus, the protein will cross the barrier and adsorb on the CH3-SAM.
Molecular simulations can describe the behavior of protein adsorption at the very early stage but are not able to describe protein denature on the CH3-SAM because of a short timescale in simulation studies. Kim and co-workers (19) reported measurements of the interaction forces between functionalized probe tips and different CH3-, OH-, and OEG-terminated SAMs using IFM. Corrected force-distance profiles showed that the OEG-SAMs generate a stronger repulsive force than the OH-SAMs. In addition, it is well known that the hydrophobic SAMs enhance protein adsorption, whereas neutral hydrophilic SAMs reduce protein adsorption (16,20,37,38). Thus, simulation results are in good agreement with those of experiments. To better understand the origin of the intermolecular forces between the protein and SAMs in the presence of water molecules, the contributions to the total interfacial force from water molecules and the SAMs are shown in Fig. 2, b and c, respectively.
It can be seen that the water-contribution curves are almost identical to the total interfacial force curves in all cases. The hydration force becomes more repulsive as the separation distance decreases. This finding implies that the interactions between the SAM surface and water molecules via hydrogen bonding create an energy barrier for protein adsorption. In the work by Kim et al. (19), a thick, interphase water layer with high viscosity (six orders of magnitude larger than that of bulk water) at the OEG-SAMs was observed, which may account for the protein resistance of OEG-SAMs. For the hydrophilic SAMs, the repulsive force arises at a relatively large separation distance due to the compression between two hydration layers around the protein and hydrophilic SAM surfaces. For the hydrophobic SAMs, the nonpolar nature of the CH3-SAMs is unable to accommodate water molecules to form the hydration layer around CH3-SAM chains. Thus, when a protein is brought closer to the hydrophobic CH3-SAMs without the hydration layer, the strong, attractive hydrophobic interactions between the protein and the CH3-SAMs may easily cause the protein to expose its internal hydrophobic residues to the surface, leading to protein adsorption.
The SAM-contribution curves (excluding hydration water around the SAM) present very small forces (∼0.05 nN) on the protein. The forces were found to become slightly attractive with decreasing separation distance, indicating that all SAMs themselves are not intrinsically protein resistant. Instead, the nanoscale structures of SAMs (i.e., surface hydrophilicity, packing density, and conformation structure) strongly affect the hydration of the SAM surface, which has a determinable influence on protein resistance. To understand the impact of hydration water and SAM structure on the interaction forces between the protein and the SAM surface, water behavior and SAM flexibility were studied in detail as described below.
Water behavior
Our previous work (14) showed that the tightly bound water layer at protein-SAM interfaces plays an important role in protein adsorption. We analyzed the total number of hydrogen bonds between the SAMs and water molecules, as shown in Table 1. Hydrogen bonds can be defined on the basis of either energetic or geometric criteria. In this study, we used the geometric criterion to determine hydrogen bonds. A hydrogen bond exists if the donor-acceptor distance is <0.35 nm and the hydrogen donor-acceptor angle is smaller than 60°. For the CH3-SAMs, no hydrogen bonds form because hydrogen-bonding acceptors are not available in the CH3-SAM chains. The number of hydrogen bonds between hydrophilic SAMs and water molecules is ∼17% larger in the OEG-SAMs than in the OH-SAMs for all values of separation distances. This is consistent with the hypothesis of protein resistance we supported in previous work, i.e., the surface containing a large number of hydrogen bonds with water molecules has better protein-resistant properties. The comparison of the force-distance curves with hydrogen bonds between water molecules and SAMs reveals that the surfaces involving a large number of hydrogen bonds with water molecules produce large repulsive forces on the protein, leading to better protein resistance. It is more difficult for the protein to squeeze out tightly bound water molecules from the interfacial layer to the bulk. Thus, those water molecules form a barrier to prevent direct contact between protein and surface.
To examine the dynamic behavior of hydration water at the protein-SAM interfaces, we calculated the self-diffusion coefficient (SDC) from the mean square displacement as a function of time. The SDC is defined as
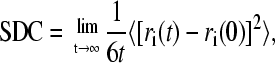 |
where 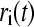 is the coordinates of atom i at time t and
is the coordinates of atom i at time t and 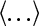 indicates ensemble average. The mobility of water molecules can be nicely quantified in terms of diffusivity. Simulation results show that the SDC of water molecules at the hydrophobic SAMs is larger than those of water molecules at the hydrophilic SAMs. The average SDC at the CH3-SAMs is 2.28 × 10−5 cm2/s, which is close to bulk water of 2.26 × 10−5 cm2/s. But, the average SDCs at both OH- and OEG-SAMs are smaller than bulk water. Water diffusivity at the OEG-SAMs (0.36 × 10−5 cm2/s) is reduced by an order of magnitude as compared to the one at the OH-SAMs (2.05 × 10−5 cm2/s), implying that the decreased diffusion coefficient is a consequence of increased hydrogen bonds between SAMs and water molecules. These hydration water molecules involving a large number of hydrogen bonds with SAMs prefer to stay longer and are more strongly bound at the interface.
indicates ensemble average. The mobility of water molecules can be nicely quantified in terms of diffusivity. Simulation results show that the SDC of water molecules at the hydrophobic SAMs is larger than those of water molecules at the hydrophilic SAMs. The average SDC at the CH3-SAMs is 2.28 × 10−5 cm2/s, which is close to bulk water of 2.26 × 10−5 cm2/s. But, the average SDCs at both OH- and OEG-SAMs are smaller than bulk water. Water diffusivity at the OEG-SAMs (0.36 × 10−5 cm2/s) is reduced by an order of magnitude as compared to the one at the OH-SAMs (2.05 × 10−5 cm2/s), implying that the decreased diffusion coefficient is a consequence of increased hydrogen bonds between SAMs and water molecules. These hydration water molecules involving a large number of hydrogen bonds with SAMs prefer to stay longer and are more strongly bound at the interface.
SAM flexibility
In addition, the flexibility of chains in various SAMs also plays an important role in protein adsorption. The flexibility of the SAMs is characterized by the RMSD values. As shown in Table 1, for a given separation distance, the RMSD increases in the order of OEG-SAMs > OH-SAMs > CH3-SAMs, indicating that the molecular chain of OEG is more flexible than those in the OH-SAMs, which is more flexible than CH3-SAMs. Simulation results also show that, for given SAMs, RMSD increases with decreasing separation distance. The flexibility of chains can be affected by surface packing density and conformational structure of SAMs. In this work, all SAM surfaces have the same packing densities because a 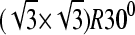 lattice structure was employed. Thus, the flexibility of chain is mainly determined by the conformational structure of SAMs. As shown in Table 1, the mean system tilt
lattice structure was employed. Thus, the flexibility of chain is mainly determined by the conformational structure of SAMs. As shown in Table 1, the mean system tilt 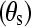 and the molecular tilt
and the molecular tilt 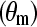 were used to characterize the conformational structure of SAMs. The mean system tilt
were used to characterize the conformational structure of SAMs. The mean system tilt  is the angle between the z axis and the vector connecting from the first atom to the last atom of the same chain. The molecular tilt
is the angle between the z axis and the vector connecting from the first atom to the last atom of the same chain. The molecular tilt  is the average angle between the z axis and the average vector passing through the adjacent atoms in a given molecular chain. The difference between
is the average angle between the z axis and the average vector passing through the adjacent atoms in a given molecular chain. The difference between 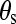 and
and  reflects the extent of an order state for a given molecular chain. It can be seen that the difference between
reflects the extent of an order state for a given molecular chain. It can be seen that the difference between  and
and 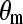 in the CH3-SAMs and OH-SAMs was close to 1°. This small difference indicates an ordered conformation. In contrast, the OEG-SAMs showed larger conformational disorder, in which the difference between
in the CH3-SAMs and OH-SAMs was close to 1°. This small difference indicates an ordered conformation. In contrast, the OEG-SAMs showed larger conformational disorder, in which the difference between  and
and 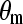 is 23°. To further examine the conformation of molecular chains in the SAM, Fig. 3 shows the distribution of dihedral angles involving only heavy atoms for all SAMs. We define a trans dihedral angle corresponding to
is 23°. To further examine the conformation of molecular chains in the SAM, Fig. 3 shows the distribution of dihedral angles involving only heavy atoms for all SAMs. We define a trans dihedral angle corresponding to  and a gauche dihedral angle corresponding to
and a gauche dihedral angle corresponding to  . As shown in Fig. 3, the CH3-SAMs and OH-SAMs have trans conformation because two sharp peaks of the CC-CC angles occur at ±180°C, whereas the OEG-SAMs have a helical conformation because the OC-CO angles exhibit a large population of the gauche conformation at ±66°C and the CC-OC angles have both gauche and trans states, in agreement with our previous simulation study and experimental results obtained from the wide angle x-ray diffraction (41). Thus, the flexibility of chains is related to the conformation structure of SAMs. The CH3- and OH-SAMs with an ordered, all-trans conformation have small flexibility, whereas OEG-SAMs with a disordered, helical conformation have large flexibility.
. As shown in Fig. 3, the CH3-SAMs and OH-SAMs have trans conformation because two sharp peaks of the CC-CC angles occur at ±180°C, whereas the OEG-SAMs have a helical conformation because the OC-CO angles exhibit a large population of the gauche conformation at ±66°C and the CC-OC angles have both gauche and trans states, in agreement with our previous simulation study and experimental results obtained from the wide angle x-ray diffraction (41). Thus, the flexibility of chains is related to the conformation structure of SAMs. The CH3- and OH-SAMs with an ordered, all-trans conformation have small flexibility, whereas OEG-SAMs with a disordered, helical conformation have large flexibility.
FIGURE 3.

Probability distribution of the dihedral torsions for (a) the CC-CC torsion in the methyl-terminated SAMs, (b) the CC-CC torsion in the hydroxyl-terminated SAMs, (c) the OC-CO torsion in the OEG-terminated SAMs, and (d) the CC-OC torsion in the OEG-terminated SAMs.
Hypothesis of protein resistance
It is well known from experimental observations that the OEG-SAMs are highly protein resistant, the OH-SAMs have low protein adsorption, and the CH3-SAMs have high protein adsorption. The force-distance curves calculated from our simulations are quantitatively consistent with experimental observations, i.e., the relative repulsive force acting on the protein from various SAMs increases in an order of CH3-SAMs < OH-SAMs < OEG-SAMs. Simulation results show that the total interfacial force mainly comes from hydration water, not from the SAM surface. This indicates that the tightly bound water layer between the protein and the SAMs via hydrogen bonds is associated directly with the total force acting on the protein. This force becomes more repulsive as the number of hydrogen bonds between water molecules and SAMs increases. This is because when water molecules form hydrogen bonds with the hydrophilic sites, strong water-SAMs interactions will prevent the protein from approaching the surface closely. Thus, the formation of the tightly bound hydration layer with low diffusivity leads to large repulsive hydration forces, which can prevent the protein from direct contact with the surface.
To generate the tightly bound water layer, both terminal hydrophilicity and interior hydrophilicity (39) in the SAM chains are required because the SAMs with hydrophilic terminal and interior groups are able to strongly interact with water molecules through hydrogen bonds. Herrwerth and co-workers (39) showed that OH-SAMs do not exhibit protein resistance as effective as the OEG-SAMs because no hydrophilic interior is present. In addition to hydration water, the conformational flexibility of chains also plays a role in protein adsorption to some extent. Simulation results show the correlation between the flexibility of chains and the interaction forces on the protein. The more flexible SAM chain has a larger repulsive force. The flexibility of chains is determined by the conformational structure of SAMs when the surface density of SAMs is fixed. The CH3- and OH-terminated SAMs with a highly ordered, all-trans conformation exhibit small chain flexibility and have small repulsive forces on the protein, whereas the OEG-SAMs with disordered, helical conformation show large chain flexibility and have large repulsive force on the protein. This is because the helical, disordered OEG-SAMs allow more water to penetrate into the surface to form tightly bound hydrogen bond networks as compared with all-trans, ordered OH- and CH3-SAMs, leading to the better nonfouling behavior of OEG-SAMs.
Our SPR experiments (26) show that both fibrinogen and lysozyme do not adsorb onto the disordered OH-terminated OEG-SAMs of intermediate OEG surface densities but adsorb to a certain degree onto the ordered OH-terminated OEG-SAMs of higher OEG surface densities. Vanderah and co-workers (40) observe a similar trend of protein adsorption as a function of CH3-terminated OEG surface densities. In our simulation work, we used OH-terminated OEG-SAMs as in our previous experiments (26) instead of CH3-terminated SAMs to avoid any possible hydrophobic effects at higher packing densities. It should be pointed out that even though 100% OEG-SAMs have higher protein adsorption than 80% OEG-SAMs, 100% OEG-SAMs have still four times lower protein adsorption than OH-terminated SAMs as showed in Fig. 7 of our previous experimental work (14). In summary, our simulation results suggest that the hydrogen bonds, chain flexibility, and conformational structure are highly correlated and have an interplay influence on the interaction forces between the protein and the surface. The SAMs with tightly bound water, high chain flexibility, and disordered conformational structure will generate a large repulsive force on the protein, leading to resistance to protein adsorption.
CONCLUSIONS
In this work, we performed restrained molecular simulations to directly calculate the interaction forces exerted on a protein from the CH3-, OH-, and OEG-terminated SAMs in the presence of water molecules as the protein approaches the SAMs from a large separation down to intimate contact. The force-distance curves show that the repulsive forces acting on the protein decrease in the following order: OEG-SAMs > OH-SAMs > CH3-SAMs. The total interfacial forces mainly come from hydration water, not from the SAM surface itself in all cases studied. This indicates that the tightly bound water layer adjacent to the SAMs is mainly responsible for the large repulsive force. Based on our simulation results, it can be seen that the SAMs having a large number of hydrogen bonds with water molecules exhibit a larger repulsive force on the protein. In addition, the large chain flexibility and disordered conformation structure of the SAMs also play a role to some extent in producing the repulsive force on the protein. Thus, a large number of tightly bound water molecules around OEG chains and the high flexibility of OEG chains are the key factors to determine the nonfouling properties of a surface. For the CH3-SAM, it is interesting to note the small repulsive force between the protein and the surface as the protein approaches the surface, i.e., protein adsorption on the CH3-SAM has a low energy barrier, which allows the protein to cross the barrier and adsorb on the CH3-SAM. The force-distance curve obtained from molecular simulations presented in this work is a very useful tool to quickly evaluate the nonfouling behavior of a surface.
Acknowledgments
We gratefully acknowledge the National Science Foundation for financial support under CMS-0225622 and CTS-0433753. This work is also supported by a graduate research award to J.Z. from the Center for Nanotechnology at the University of Washington.
References
- 1.Horbett, T. A., and J. L. Brash. 1995. Proteins at interfaces. II. Fundamentals and Applications. In ACS Symposium Series 602. American Chemical Society, Washington, D.C.
- 2.Ostuni, E., R. G. Chapman, R. E. Holmlin, S. Takayama, and G. M. Whitesides. 2001. A survey of structure-property relationships of surfaces that resist the adsorption of proteins. Langmuir. 17:5605–5620. [Google Scholar]
- 3.Kane, R. S., P. Deschatelets, and G. M. Whitesides. 2003. Kosmotropes form the basis of protein-resistant surfaces. Langmuir. 19:2388–2391. [Google Scholar]
- 4.Chapman, R. G., E. Ostuni, M. N. Liang, G. Meluleni, E. Kim, L. Yan, G. Pier, H. S. Warren, and G. M. Whitesides. 2001. Polymeric thin films that resist the adsorption of proteins and the adhesion of bacteria. Langmuir. 17:1225–1233. [Google Scholar]
- 5.Tegoulia, V. A., W. Rao, A. T. Kalambur, J. F. Rabolt, and S. L. Cooper. 2001. Surface properties, fibrinogen adsorption, and cellular interactions of a novel phosphorylcholine-containing self-assembled monolayer on gold. Langmuir. 17:4396–4404. [Google Scholar]
- 6.Murphy, E. F., J. R. Lu, J. Brewer, J. Russell, and J. Penfold. 1999. The reduced adsorption of proteins at the phosphoryl choline incorporated polymer-water interface. Langmuir. 15:1313–1322. [Google Scholar]
- 7.Harris, J. M., editor. 1992. Poly(ethylene glycol) Chemistry: Biotechnical and Biomedical Applications. Plenum Press, New York.
- 8.Bailey, F. E., Jr., and J. Y. Koleske. 1984. Poly(ethylene Oxide). Academic Press, New York.
- 9.Pale-Grosdemange, C., E. S. Simon, K. L. Prime, and G. M. Whitesides. 1991. Formation of self-assembled monolayers by chemisorption of derivatives of oligo(ethylene glycol) of structure HS(CH2)11(OCH2CH2)mOH on gold. J. Am. Chem. Soc. 113:12–20. [Google Scholar]
- 10.Prime, K. L., and G. M. Whitesides. 1991. Self-assembled organic monolayers: model systems for studying adsorption of proteins at surfaces. Science. 252:1164–1167. [DOI] [PubMed] [Google Scholar]
- 11.Prime, K. L., and G. M. Whitesides. 1993. Adsorption of proteins onto surfaces containing end-attached oligo(ethylene oxide): a model system using self-assembled monolayers. J. Am. Chem. Soc. 115:10714–10721. [Google Scholar]
- 12.Jeon, S. I., J. H. Lee, J. D. Andrade, and P. G. De Gennes. 1991. Protein—surface interactions in the presence of polyethylene oxide: I. Simplified theory. J. Colloid Interface Sci. 142:149–158. [Google Scholar]
- 13.McPherson, T., A. Kidane, I. Szleifer, and K. Park. 1998. Prevention of protein adsorption by tethered poly(ethylene oxide) layers: experiments and single-chain mean-field analysis. Langmuir. 14:176–186. [Google Scholar]
- 14.Zheng, J., L. Li, S. Chen, and S. Jiang. 2004. Molecular simulation study of water interactions with oligo (ethylene glycol)-terminated alkanethiol self-assembled monolayers. Langmuir. 20:8931–8938. [DOI] [PubMed] [Google Scholar]
- 15.Pertsin, A. J., and M. Grunze. 2000. Computer simulation of water near the surface of oligo(ethylene glycol)-terminated alkanethiol self-assembled monolayers. Langmuir. 16:8829–8841. [Google Scholar]
- 16.Archambault, J. G., and J. L. Brash. 2004. Protein repellent polyurethane-urea surfaces by chemical grafting of hydroxyl-terminated poly(ethylene oxide): effects of protein size and charge. Colloids Surf. B. Biointerfaces. 33:111–120. [DOI] [PubMed] [Google Scholar]
- 17.Dupont-Gillain, C. C., C. M. J. Fauroux, D. C. J. Gardner, and G. J. Leggett. 2003. Use of AFM to probe the adsorption strength and time-dependent changes of albumin on self-assembled monolayers. J. Biomed. Mater. Res. 67A:548–558. [DOI] [PubMed] [Google Scholar]
- 18.Sethuraman, A., M. Han, R. S. Kane, and G. Belfort. 2004. Effect of surface wettability on the adhesion of proteins. Langmuir. 20:7779–7788. [DOI] [PubMed] [Google Scholar]
- 19.Kim, H. I., J. G. Kushmerick, J. E. Houston, and B. C. Bunker. 2003. Viscous “interphase” water adjacent to oligo(ethylene glycol)-terminated monolayers. Langmuir. 19:9271–9275. [Google Scholar]
- 20.Kidoaki, S., and T. Matsuda. 1999. Adhesion forces of the blood plasma proteins on self-assembled monolayer surfaces of alkanethiolates with different functional groups measured by an atomic force microscope. Langmuir. 15:7639–7646. [Google Scholar]
- 21.Rixman, M. A., D. Dean, C. E. Macias, and C. Ortiz. 2003. Nanoscale intermolecular interactions between human serum albumin and alkanethiol self-assembled monolayers. Langmuir. 19:6202–6218. [Google Scholar]
- 22.Wang, M. S., L. B. Palmer, J. D. Schwartz, and A. Razatos. 2004. Evaluating protein attraction and adhesion to biomaterials with the atomic force microscope. Langmuir. 20:7753–7759. [DOI] [PubMed] [Google Scholar]
- 23.Feldman, K., G. Hähner, N. D. Spencer, P. Harder, and M. Grunze. 1999. Probing resistance to protein adsorption of oligo(ethylene glycol)-terminated self-assembled monolayers by scanning force microscopy. J. Am. Chem. Soc. 121:10134–10141. [Google Scholar]
- 24.Efremova, N. V., S. R. Sheth, and D. E. Leckband. 2001. Protein-induced changes in poly(ethylene glycol) brushes: molecular weight and temperature dependence. Langmuir. 17:7628–7636. [Google Scholar]
- 25.Sheth, S. R., and D. E. Leckband. 1997. Measurements of attractive forces between proteins and end-grafted poly(ethylene glycol) chains. Proc. Natl. Acad. Sci. USA. 94:8399–8404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li, L., S. Chen, J. Zheng, B. D. Ratner, and S. Jiang. 2005. Protein adsorption on oligo(ethylene glycol)-terminated alkanethiolate self-assembled monolayers: the molecular basis for nonfouling behavior. J. Phys. Chem. B. 109:2934–2941. [DOI] [PubMed] [Google Scholar]
- 27.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 28.MacKerell, A. D., D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher III, B. Rourx, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 29.Smith, L. J., A. E. Mark, C. M. Dobson, and W. F. van Gunsteren. 1995. Comparison of MD simulations and NMR experiments for hen lysozyme. Analysis of local fluctuations, cooperative motions, and global changes. Biochemistry. 34:10918–10931. [DOI] [PubMed] [Google Scholar]
- 30.Smith, G. D., R. L. Jaffe, and D. Y. Yoon. 1993. Force field for simulations of 1,2-dimethoxyethane and poly(oxyethylene) based upon ab initio electronic structure calculations on model molecules. J. Phys. Chem. 97:12752–12759. [Google Scholar]
- 31.Smith, G. D., O. Borodin, and D. Bedrov. 2002. A revised quantum chemistry-based potential for poly(ethylene oxide) and its oligomers in aqueous solution. J. Comput. Chem. 23:1480–1488. [DOI] [PubMed] [Google Scholar]
- 32.Tasaki, K. 1996. Poly(oxyethylene)-water interactions: a molecular dynamics study. J. Am. Chem. Soc. 118:8459–8469. [Google Scholar]
- 33.Tasaki, K. 1996. Conformation and dynamics of poly(oxyethylene) in benzene solution: solvent effect from molecular dynamics simulation. Macromolecules. 29:8922–8933. [Google Scholar]
- 34.Steinbach, P. J., and B. R. Brooks. 1994. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 15:667–683. [Google Scholar]
- 35.Norberg, J., and L. Nilsson. 2003. Advances in biomolecular simulations: methodology and recent applications. Q. Rev. Biophys. 36:257–306. [DOI] [PubMed] [Google Scholar]
- 36.Beck, D. A. C., R. S. Armen, and V. Daggett. 2005. Cutoff size need not strongly influence molecular dynamics results for solvated polypeptides. Biochemistry. 44:609–616. [DOI] [PubMed] [Google Scholar]
- 37.Norde, W. 1986. Adsorption of proteins from solution at the solid-liquid interface. Adv. Colloid Interface Sci. 25:267–340. [DOI] [PubMed] [Google Scholar]
- 38.Jiang, X., D. A. Bruzewicz, M. M. Thant, and G. M. Whitesides. 2004. Palladium as a substrate for self-assembled monolayers used in biotechnology. Anal. Chem. 76:6116–6121. [DOI] [PubMed] [Google Scholar]
- 39.Herrwerth, S., W. Eck, S. Reinhardt, and M. Grunze. 2003. Factors that determine the protein resistance of oligoether self-assembled monolayers—internal hydrophilicity, terminal hydrophilicity, and lateral packing density. J. Am. Chem. Soc. 125:9359–9366. [DOI] [PubMed] [Google Scholar]
- 40.Vanderah, D. J., H. La, J. Naff, V. Silin, and K. A. Rubinson. 2004. Control of protein adsorption: molecular level structural and spatial variables. J. Am. Chem. Soc. 126:13639–13641. [DOI] [PubMed] [Google Scholar]
- 41.Harris, D. J., T. J. Bonagamba, M. Hong, and K. Schmidt-Rohr. 2000. Conformation of poly(ethylene oxide)-hydroxybenzene molecular complexes studied by solid-state NMR. Macromolecules. 33:3375–3381. [Google Scholar]