

Abstract
Uptake and consecutive phosphorylation of mannitol in Escherichia coli is catalyzed by the mannitol permease EnzymeIImtl. The substrate is bound at an extracellular-oriented binding site, translocated to an inward-facing site, from where it is phosphorylated, and subsequently released into the cell. Previous studies have shown the presence of both a high- and a low-affinity binding site with KD-values in the nano- and micromolar range, respectively. However, reported KD-values in literature are highly variable, which casts doubts about the reliability of the measurements and data analysis. Using an optimized binding measurement system, we investigated the discrepancies reported in literature, regarding both the variability in KD-values and the binding stoichiometry. By comparing the binding capacity obtained with flow dialysis with different methods to determine the protein concentration (UV-protein absorption, Bradford protein detection, and a LDH-linked protein assay to quantify the number of phosphorylation sites), we proved the existence of only one mannitol binding site per dimeric species of unphosphorylated EnzymeIImtl. Furthermore, the affinity of EnzymeIImtl for mannitol appeared to be dependent on the protein concentration and seemed to reflect the presence of an endogenous ligand. The dependency could be simulated assuming that >50% of the binding sites were occupied with a ligand that shows an affinity for EnzymeIImtl in the same range as mannitol.
INTRODUCTION
The mannitol permease EnzymeIImtl (EIImtl) from Escherichia coli binds extracellular mannitol, transports it across the membrane, and releases it inside the cell as mannitol-1-phosphate. The phosphoryl moiety originates from the high-energy donor PEP and is transferred by two cytosolic kinases, Enzyme I (EI) and HPr. EIImtl is phosphorylated by HPr at His-554 in its IIA-domain, from where the phosphoryl group is transferred to Cys-384 in the IIB-domain. It was proposed that in the unphosphorylated carrier mannitol resides at an outward-facing binding site, whereas upon phosphorylation of both the A- and B-domains, mannitol is translocated to an inward-facing binding site, either by movement of the mannitol molecule itself or complete binding site reorientation (1,2). The existence of both an outward- and inward-facing binding site was proposed on the basis of association and dissociation kinetics from both sides of the membrane (3).
Published results on mannitol binding experiments with wild-type EIImtl reveal large variations in the dissociation constants (KD), ranging from 35 nM up to several micromolars (1,4–14). The fact that some authors reported both a high and a low mannitol binding affinity suggests two possible mannitol binding sites. It has been shown that the enzyme is dimeric both in the membrane and in the detergent-solubilized state (7,15–19). Thus, both subunits may have a different conformation and harbor a mannitol-binding site with unequal KD. A second mannitol-binding site would be consistent with translocation of mannitol from an outward- to an inward-facing binding site. Also, it has been proposed that EIImtl exposes a single binding site to either side of the membrane in an alternating fashion (1,3,20). Lolkema included a second mannitol binding site into computational models and performed simulations that showed that a second site could explain some of the complex kinetic behavior of EIImtl, both in the translocation pathway and phosphorylation reactions. Finally, it was proposed that EIImtl contains one mannitol binding site in its native membrane and two in a purified form (21).
The reported high-affinity dissociation constants vary ∼30-fold, from 35 nM up to 1 μM. Because substrate affinity and binding stoichiometry are such crucial parameters for modeling the working mechanism of an enzyme, this is an unacceptable situation. Moreover, in our own studies we were confronted with a strong dependency of the apparent KD on the concentration of the enzyme. In this article, we clarify the contradiction existing in the binding data regarding mannitol binding stoichiometry and affinity, using flow dialysis in combination with computer simulations.
MATERIALS AND METHODS
Chemicals and reagents
D-[1-3H(N)]mannitol (17.0 Ci/mmol, batch no. 3499-326) was purchased from NEN Research Products (Boston, MA). D-[1-14C]mannitol (59.0 mCi/mmol, batch no. 78) was purchased from Amersham Biosciences (Uppsala, Sweden). Radioactivity measurements were performed using Emulsifier Scintillator Plus obtained from Packard (Groningen, The Netherlands). Q-Sepharose Fast Flow was from Amersham Biosciences. Ni-NTA resin was from Qiagen (Valencia, CA); l-histidine and N-ethylmaleimide (NEM) were from Fluka (Milwaukee, WI). BSA, LDH and β-NADH were from Sigma (St. Louis, MO). Decylpoly(ethylene glycol) 300 (decylPEG) was obtained from Kwant High Vacuum Oil Recycling and Synthesis (Bedum, The Netherlands). The detergent C10E5 (decyl pentaethylene glycol ether) was synthesized and purified as described (22). His-tagged versions of EI and HPr were created, using standard genetic tools as will be described elsewhere. All other chemicals used were analytical grade.
Cell growth, isolation of ISO membrane vesicles, and purification
The plasmid harboring the wild-type mtlA gene with a thrombin-cleavable C-terminal His-tag (pMamtlaPrEII6his) was transformed and subsequently grown in E. coli LGS322 (F− thi-1, hisG1, argG6, metB1, tonA2, supE44, rspL104, lacY1, galT6, gatR49, gatA50, Δ(mtlA′p), mtlDc, Δ(gutR′MDBA-recA)) as described (6,19). LGS322, not transformed, and thus not expressing the mtlA gene was used in control experiments (LGSminus).
ISO membrane vesicles were prepared by passage of cells though a French press at 10,000 PSI, essentially as described (23). The membrane vesicles were washed once in 25 mM Tris-HCl, pH 7.6, 5 mM DTT, and 1 mM NaN3, and quickly frozen in small aliquots in liquid nitrogen before storage at −80°C. For mannitol binding experiments or purification of EIImtl, membrane vesicles were placed at 37°C for quick thawing and thereafter directly placed on ice until further use.
Wild-type EIImtl was purified by Ni-NTA affinity chromatography. Briefly, membrane vesicles at ∼2 mg protein/mL were solubilized in 25 mM Tris-HCl, pH 8.5, 50 mM NaCl, 10 mM 2-mercaptoethanol, 2% (v/v) decylPEG, and 25 mM imidazole for 10 min at room temperature. After spinning down the nonsolubilized fraction (10 min, 250,000× g, 4°C), the supernatant was mixed with washed Ni-NTA resin by stirring for 1 h at 4°C. After draining the flow-through, the column was subsequently washed with 10 column volumes buffer A (25 mM Tris-HCl, pH 8.5, 300 mM NaCl, 0.3% decylPEG, 2 mM GSH, plus 10 mM imidazole), and 10 column volumes buffer B (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.3% decylPEG, plus 2 mM GSH). After batchwise elution from Ni-NTA with 80 mM l-histidine (in 25 mM Tris-HCl, pH 7.6, 0.25% decylPEG, plus 2 mM GSH), 100 mM NaCl was added to the samples. Protein purity of the samples was confirmed with SDS-PAGE analysis.
If required, further purification of the protein samples was performed on a MonoQ HR 5/5 column (Pharmacia Biotech, Piscataway, NJ) as described (24). The Ni-NTA purified protein samples were ∼4–5 times diluted in 25 mM Tris-HCl, pH 7.6, 0.25% decylPEG, plus 2 mM GSH before loading. The column was washed for 1 h at 0.25 mL min−1 with the same buffer supplemented with 100 mM NaCl. Elution of the proteins was started by applying a linear NaCl gradient from 100 to 400 mM in the same buffer.
Activity measurements
The nonvectorial PEP-dependent phosphorylation activity, catalyzed by EIImtl, was measured as described (25). Briefly, the assay mixture contained 25 mM Tris-HCl, pH 7.6, 5 mM DTT, 5 mM MgCl2, 5 mM PEP, 350 nM EI, 17 μM HPr, with or without 0.25% decylPEG, and rate-limiting amounts of EIImtl (in nanomolar range). After incubation of the mixture for 5 min at 30°C, the reaction was started by adding 1 mM 14C-mannitol. The reaction was quenched at given time intervals by loading the samples on Dowex AG1-X2 columns. After washing the column with four column volumes of H2O, formed 14C-mannitol-1-P was eluted using two column volumes of 0.2 N HCl and quantified by liquid scintillation counting.
The number of phosphorylation sites of Ni-NTA purified EIImtl was determined by measuring the conversion of PEP into pyruvate, using a lactate dehydrogenase linked assay (26). Mixtures of 52.8 units LDH, 2 μM EI, 4 μM HPr, 100 μM NADH, 20 mM Tris-HCl, pH 7.6, 5 mM MgCl2, 0.25% decylPEG, plus 1 mM DTT were prepared, with or without EIImtl. The mixtures were incubated for 2 min at 30°C, after which the reaction was started by the addition of 1 mM PEP. The oxidation of NADH was monitored by following the absorbance at 340 nm (ɛNADH = 6220 L mol−1 cm−1). After 10 min, when the reaction was complete, 10 μM mannitol was added. The difference in the extent of NADH oxidation between the control and the reaction with EIImtl, after extrapolation to the point of PEP addition, was used to estimate the number of phosphorylation sites, and hence the concentration of EIImtl, assuming two phosphorylation sites per monomer (9). To test if all phosphate groups were quantitatively donated to EIImtl, half of the phosphate-accepting groups on EIImtl were made unavailable by quenching Cys-384 with NEM. Protein samples were desalted using a Micro Bio-Spin 6 column (Bio-Rad, Hercules, CA), after which 1 mM NEM was added. After incubation for 10 min at room temperature, the reaction was stopped by adding 5 mM DTT. The LDH assay was carried out as described above.
Mannitol binding experiments
Mannitol binding was quantified with the flow dialysis technique (13). In short, membrane vesicles (or purified protein samples) were suspended in 25 mM Tris-HCl, pH 7.6, 5 mM DTT, 5 mM MgCl2, either with or without detergent. The mixture was placed at 25°C for 5 min, after which the sample was loaded into the upper chamber of the flow dialysis cell. Titration was performed by several small additions from a concentrated radiolabeled mannitol stock. All stock solutions (from 25 μM to 1.5 mM) contained 10 μM 3H-mannitol from the radiolabeled stock solution, supplemented with 1H-mannitol up to the desired concentration. Each stock solution thus contained identical amounts of radioactivity. The data analysis is described in the next section.
Theoretical considerations and data analysis
Equilibrium between a ligand (S) and a binding site on a protein (E) is described by
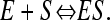 |
(1) |
The concentration of unbound, free ligand (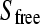 ) is obtained from the general binding equation
) is obtained from the general binding equation
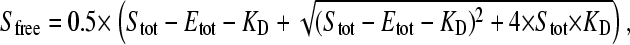 |
(2) |
where 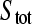 and
and 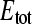 represent the total ligand concentration and the concentration of the binding sites, respectively. The equilibrium constant KD is defined as:
represent the total ligand concentration and the concentration of the binding sites, respectively. The equilibrium constant KD is defined as:
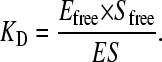 |
(3) |
With our flow dialysis setup free, radioactive substrate, added at each titration point n, leaks through a semipermeable membrane to a fraction collector. The signal Cn after addition n (in CPM values) is proportional to 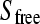 at the specific titration points and is defined by:
at the specific titration points and is defined by:
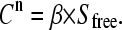 |
(4) |
The parameter β relates the measured radioactivity (Cn) and the concentration of free ligand (Sfree) in the upper compartment of the flow dialysis system and is measured in an experiment with no binding sites added (13). The data (Cn-values) collected with the mannitol binding experiment, together with the values for Stot, can now be analyzed using Eqs. 2 and 4 to calculate the total concentration of binding sites (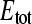 ) and the affinity for the substrate (KD), on the assumption that the simple binding model (Eq. 1) holds. In case of a three-parameter fitting routine, the data were fitted for
) and the affinity for the substrate (KD), on the assumption that the simple binding model (Eq. 1) holds. In case of a three-parameter fitting routine, the data were fitted for 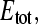 KD, and β. The routines were written in the software package Mathematica 5.0, using the Levenberg-Marquardt nonlinear least-squares minimization algorithm, for fitting of the data (available on request). Details for the treatment of systematic and nonsystematic errors are described in detail (13). Estimates of errors in the binding parameters are based on Monte Carlo simulations of 200 data sets, using a value of 0.08 μl as the uncertainty in each addition (Σ-value, see Veldhuis et al. (13)), unless stated otherwise.
KD, and β. The routines were written in the software package Mathematica 5.0, using the Levenberg-Marquardt nonlinear least-squares minimization algorithm, for fitting of the data (available on request). Details for the treatment of systematic and nonsystematic errors are described in detail (13). Estimates of errors in the binding parameters are based on Monte Carlo simulations of 200 data sets, using a value of 0.08 μl as the uncertainty in each addition (Σ-value, see Veldhuis et al. (13)), unless stated otherwise.
We used computer simulations to investigate the impact of impurities in the radioactive stock or the effect of occupied binding sites (endogenous ligand) on the observed values for 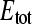 and KD. For this purpose, the binding equations were adapted to a one-site (E) two-ligand (S1 and S2) model. With this model virtual data sets (mimicking titration experiments) were generated, and subsequently analyzed with the simple one-site one-ligand model (Eq. 2) to solve for
and KD. For this purpose, the binding equations were adapted to a one-site (E) two-ligand (S1 and S2) model. With this model virtual data sets (mimicking titration experiments) were generated, and subsequently analyzed with the simple one-site one-ligand model (Eq. 2) to solve for 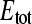 and KD.
and KD.
The following equations (5–7) were solved numerically for S1free, which is proportional to the measured radioactivity. Several situations were mimicked, whereby the second ligand concentration (S2tot) was kept proportional to either S1tot (the second ligand as a contamination of the ligand stock solution) or to Etot (the second ligand as a contamination of the EIImtl stock solution):
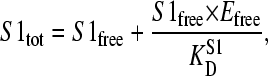 |
(5) |
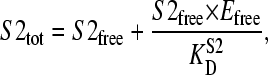 |
(6) |
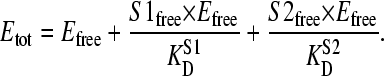 |
(7) |
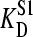 and
and 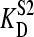 represent the affinities of E for S1, and S2, respectively.
represent the affinities of E for S1, and S2, respectively.
RESULTS
The affinity of EIImtl for mannitol in detergent-solubilized ISO membrane vesicles
To investigate the substrate binding properties of EIImtl, the flow dialysis technique was used. The detergent decylPEG was added to solubilize the ISO membrane vesicles, thereby making all binding sites accessible to mannitol. To obtain reliable Etot- and KD-values, the Etot/KD-ratio was kept between 3 and 30 at all membrane vesicle concentrations and the total amount of substrate at the end of the titrations was ∼5 times the amount of binding sites (13). Fig. 1 shows some typical titration experiments at Etot-concentrations ranging from ∼500 nM to 40 μM (corresponding with ∼0.2–∼20 mg/mL of total membrane protein). The solid lines represent the fitted curves. The best-fit values of the parameters Etot and KD for all the titration experiments are summarized in Fig. 2. Fig. 2 A shows that, as anticipated, the apparent concentration of binding sites (Etot) increased linearly with the concentration of detergent-solubilized membrane vesicles used (open circles). Surprisingly, we found that the apparent affinity constant for mannitol varied strongly with the amount of detergent-solubilized membrane vesicles, as shown in Fig. 2 C (open circles): the affinity decreased from ∼120 nM at the lowest (∼0.5 μM sites) to ∼1 μM at the highest vesicle concentrations that could be used for this batch (∼15 μM sites). When an even higher enzyme concentration was used (from a vesicle batch with a higher expression level, Etot = 40.4 μM), fitting of the binding data resulted in a KD-value of 2.3 μM (Fig. 1 D).
FIGURE 1.

Titration curves for different amounts of detergent-solubilized membrane vesicles. The solid lines represent nonlinear least-squares curve fits for KD and Etot. The slope of the dashed line represents the β-value, relating measured radioactivity with the free concentration of mannitol in the upper chamber of the flow dialysis system (see Methods and Materials). The dotted line is a computer simulated curve obtained with a KD of 100 nM and the corresponding value of Etot from the actual experiment. (A) Membrane vesicles (9 μL) with 25 μM 3H-mtl stock. (B) Membrane vesicles (64.5 μL) with 300 μM 3H-mtl stock. (C) Membrane vesicles (200 μL) with 800 μM 3H-mtl stock. (D) Membrane vesicles (409.5 μL; different batch) with 2 mM 3H-mtl stock. Mixture volumes were 450 μL, of which 380 μL was pipetted into the upper chamber of the flow dialysis system.
FIGURE 2.

Binding parameters plotted against vesicle or protein amount. (A) Observed Etot concentration against the amount of vesicles used for intact (•) and detergent solubilized (○) ISO membrane vesicles. The solid lines represent linear fits. (B) Observed KD against the amount of intact ISO membrane vesicles (•). (C) Observed KD against the amount of solubilized ISO membrane vesicles (○). (D) Observed KD against the observed Etot for different Ni-NTA purified protein preparations. Error bars were determined using Monte Carlo simulations with Σ = 0.08 and 500 data sets (13).
The decrease in affinity for mannitol at higher membrane vesicle concentrations was not caused by nonspecific binding, because addition of membrane vesicles from an EIImtl-deficient E. coli LGS strain (LGSminus) at ∼8 mg/mL to an Etot of ∼500 nM, did not affect the apparent binding parameters (results not shown). Accordingly, titration experiments with only LGSminus vesicles did not result in any mannitol binding, as was shown previously (13).
Effect of decylPEG on the binding parameters
In the titration experiments with detergent solubilized-membrane vesicles, the ratio decylPEG/EIImtl was not constant: the concentration of decylPEG varied from 0.25% at the lowest to 1.0% at the highest membrane vesicle concentration. To check for an effect of the amount of decylPEG on the binding parameters, titrations were performed at a fixed vesicle concentration (corresponding to Etot of ∼1 μM), over a 20-fold range of decylPEG (from 0.25 to 5.0%, final concentration). At 0.25% decylPEG, fitting of the binding data resulted in a KD of 89 ± 9 nM and an Etot of 1043 ± 40 nM. Using 1.0% decylPEG, we obtained KD- and Etot-values of 132 ± 13 and 1164 ± 41 nM, respectively. Finally, at 5.0% decylPEG, the KD- and Etot-values were 101 ± 11 and 913 ± 41 nM, respectively. These results show that the KD was not affected by increasing amounts of decylPEG, i.e., increasing decylPEG/EIImtl ratios. We conclude that variations in the decylPEG/EIImtl ratios cannot explain the observed variation of the KD with Etot.
The affinity of EIImtl for mannitol in intact ISO membrane vesicles
The association state of EIImtl has been studied extensively both in the purified and membrane-solubilized form (17,25,27,28). From this work it is most probable that with the concentrations used in the titrations, solubilized EIImtl is predominantly dimeric. To test whether a monomer/dimer oligomerization process could explain the observed variations in KD, the titrations were repeated with intact ISO membrane vesicles, where the amount of vesicles is not expected to influence the association equilibrium. Fig. 2 shows that also for intact membrane vesicles the affinity for mannitol varied strongly with the concentration of membrane vesicles used. We conclude that variations in the oligomerization state can be excluded as a cause of the dependence of the affinity for mannitol on the concentration of EIImtl.
The affinity of purified EIImtl for mannitol
EIImtl was purified using Ni-NTA chromatography, and the protein samples were >95% pure, as judged from SDS-PAGE analysis (data not shown). Similar titrations as described above were performed with these purified protein samples. In all cases the observed Etot increased linearly with the amount of purified protein used. The KD showed a similar, albeit less steep, dependence on the protein concentration as observed for the (detergent-solubilized) membrane vesicles (Fig. 2 D). However, at an Etot of ∼1 μM the observed KD was two times higher (∼250 nM) than in the detergent-solubilized membrane vesicles. Further purification of the Ni-NTA purified protein, using MonoQ ion-exchange chromatography, did not yield significantly different results: at Etot-values of 1.4 and 6.4 μM, the KD-values were 303 and 594 nM, respectively.
Binding stoichiometry
To determine the stoichiometry of substrate binding, it is important to know the concentration of functional protein with high accuracy. The concentration of Ni-NTA purified EIImtl was estimated with a number of techniques: protein UV-absorption spectroscopy, Bradford protein detection, and a LDH-coupled spectroscopic assay to determine the number of phosphorylation sites. Determination of the protein concentration by UV absorption, using an extinction coefficient of 31,190 L mol−1 cm−1 at 280 nm (calculated according to Pace et al. (29)), resulted in 21.0 μM of total protein. The Bradford protein detection method, using BSA as a standard, resulted in 18.8 ± 0.6 μM total protein (n = 3). The result of the LDH-coupled assay is shown in Fig. 3 and shows that the presence of EIImtl resulted in an additional decrease in absorption (Δ, dotted lines), indicative of PEP consumption, specifically by EIImtl. Extrapolation to the point of PEP addition (see Materials and Methods for details) indicated an EIImtl concentration of the purified protein preparation of 20.7 μM. Subsequent addition of 10 μM mannitol (arrow) resulted in an equivalent PEP consumption. The correspondence between the numbers from each of the measurements shows that all EIImtl units are functional in phosphorylation. Similar results were obtained for different preparations of purified EIImtl.
FIGURE 3.

Quantitative phosphorylation of EIImtl. The control mixture contained 52.8 units LDH, 2 μM EI, 4 μM HPr, 100 μM NADH, 20 mM Tris-HCl, pH 7.6, 5 mM MgCl2, 0.25% decylPEG, plus 1 mM DTT. EIImtl was added to a concentration of ∼4 μM. The reaction was started by addition of 1 mM PEP, and monitored by following the change in absorbance at 340 nm (ɛNADH = 6220 L mol−1 cm−1). After 10 min, when the reaction was complete, 10 μM of mannitol was added. The dotted lines represent linear fits through the data points after the reaction was complete. Extrapolation to the point where PEP was added yielded the amount of phosphate groups donated. The difference between the control (no EIImtl) and the sample with EIImtl yielded the amount of phosphorylation sites associated with EIImtl (Δ).
Using a purified protein preparation containing 20.2 ± 1.2 μM (average from the results of the methods mentioned above) of EIImtl monomeric subunits, we observed 10.3 μM of mannitol binding sites. This indicates that one molecule of mannitol is bound per dimeric EIImtl. From this it must be concluded that the putative second mannitol binding site observed earlier does not exist, at least in the purified enzyme. The specific mannitol phosphorylation activity of EIImtl in intact and detergent-solubilized membrane vesicles was 1.8 and 2.0 μM mtl-1-P/min/nM EIImtl, respectively. For Ni-NTA purified EIImtl the specific activity was slightly lower (1.6 μM mtl-1-P/min/nM EIImtl). Because EIImtl behaves similarly in purified form and in its native environment regarding binding and phosphorylation (see also Lolkema et al. (30)), it is fair to conclude that also embedded in the membrane the transporter has only one binding site. Furthermore, for the experiments performed with membrane vesicles, a linear relation was observed between the amount of membrane vesicles and Etot.
The dependence of the apparent affinity constant for mannitol on the concentration of EIImtl explained by a second ligand
The affinity for mannitol appeared to be highly dependent on the amount of protein material used (intact or detergent-solubilized membrane vesicles, and purified protein). To explain this dependency we considered several possibilities: incorrect radioligand concentration, impurities in the 3H-mannitol stock, or nonempty binding sites in EIImtl. We used computer simulations to investigate the effect of an unidentified second ligand, competing with radioactive mannitol for the same binding site. It could be present as: i), a contamination of the radioactive mannitol stock solution, or ii), a contamination of the protein stock solution, implying that the mannitol binding sites may not be empty at the start of our titrations, as is usually assumed.
Impurities in the 3H-mannitol stock
The most straightforward way to check for impurities or an incorrect radioligand stock concentration is to perform titration experiments at a fixed protein concentration, using fixed substrate stock concentrations, and different ratios between unlabeled and radiolabeled species. We performed two titration experiments at an Etot of ∼1 μM, both with a mannitol stock solution of 20 μM. A radioactive mannitol stock solution was prepared with uncut radioactive mannitol, which was adjusted to 20 μM with water and used for the first titration. For the second titration a cold mannitol stock solution of the same concentration (20 μM) was prepared and mixed in a 1:1 ratio with the radioactive stock solution from the first titration. Both titration experiments are shown in Fig. 4. Fitting of the data of the uncut 3H-mannitol experiment using three-parameter fitting (KD, Etot, and β; (13)) resulted in values for the KD, Etot, and β-value of 117 ± 11 nM, 277 ± 17 nM, and 14.72 ± 0.24 CPM nM−1, respectively. For the cut-up data set, values for the KD, Etot, and β-value of 109 ± 10 nM, 284 ± 18 nM, and 7.59 ± 0.14 CPM nM−1, respectively, were obtained. The β-value for the cut-up experiment was found to be two times lower than the β-value for the uncut experiment, because half of the radioactivity was used. The similarity between the two binding isotherms and the fitting results, demonstrated that the concentration of 3H-radiolabeled mannitol, as given by the manufacturer, was correct. We estimated that not >5% impurity could be present in our 3H-mannitol stock solution.
FIGURE 4.

Substrate cut-up experiment to check for impurities or an incorrect radioligand stock concentration. Membrane vesicles (3.75 μL, corresponding to ∼0.4 mg/ml of total membrane protein) were added to 450 μL mixture, supplemented with 0.25% decylPEG. The mixture (380 μL) was pipetted into the upper chamber of the flow dialysis system. The curved lines represent nonlinear least-squares curve fits for KD, Etot, and β, for the titration experiment with uncut (solid), and 2× cut-up (dashed) 20 μM 3H-mannitol. The straight lines represents the β-values for uncut (solid), and 2× cut-up (dashed) 20 μM 3H-mannitol stocks.
Nonempty binding sites
Computer simulations were used to examine the possible effect of “nonempty” binding sites (occupied with endogenous ligand) on our titrations. Our model assumes that a nonradioactive second ligand present in the protein sample competes with the same site as mannitol and that equilibrium is established fast on the timescale of our titrations. In one set of simulations we assumed the endogenous ligand to be present at a concentration equal to 75% of the concentration of binding sites employed. Etot was varied from 1 to 10 μM and the dissociation constant of the second ligand (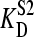 ) was varied from 1 to 10 times that of mannitol (
) was varied from 1 to 10 times that of mannitol (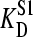 = 100 nM). Fig. 5 shows the effect of such an endogenous ligand on the apparent binding parameters
= 100 nM). Fig. 5 shows the effect of such an endogenous ligand on the apparent binding parameters 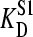 (Fig. 5 A), and Etot (Fig. 5 B), as a function of the protein concentration. In all cases there was a clear variation of the apparent KD (
(Fig. 5 A), and Etot (Fig. 5 B), as a function of the protein concentration. In all cases there was a clear variation of the apparent KD (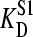 ) with the concentration of protein binding sites (Etot): higher protein concentrations resulted in higher KD-values, which is similar to the effect observed experimentally (Fig. 2 B). Of course the effect of a second (endogenous) ligand becomes weaker when its affinity for the protein decreases. The apparent value for Etot (observed Etot) was slightly lower than the value used for the simulations (added Etot), but this effect was more or less the same at all protein concentrations used, resulting in linear relationships between the observed and “actual” concentration of binding sites, again precisely as was found experimentally (Fig. 2 A). Simulations of this type showed that values of
) with the concentration of protein binding sites (Etot): higher protein concentrations resulted in higher KD-values, which is similar to the effect observed experimentally (Fig. 2 B). Of course the effect of a second (endogenous) ligand becomes weaker when its affinity for the protein decreases. The apparent value for Etot (observed Etot) was slightly lower than the value used for the simulations (added Etot), but this effect was more or less the same at all protein concentrations used, resulting in linear relationships between the observed and “actual” concentration of binding sites, again precisely as was found experimentally (Fig. 2 A). Simulations of this type showed that values of 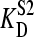 in the range of 100–500 nM may well explain our experimental results. This leaves mannitol itself as a possible candidate for the unidentified second ligand, apparently copurified with EIImtl. However, it is not very likely that mannitol would be present in the binding sites, because it is not produced by E. coli. One could argue that mannitol is present in the rich Luria Broth media, but growth of the same E. coli strain LGS322 (transformed with wild-type EIImtl, pMamtlaPrEII6his) on M9 minimal medium (31) with glucose as sole carbon source, showed a similar dependency of the KD on the protein concentration (data not shown). Furthermore, the LDH-linked assay should have resulted in a higher number of phosphoryl groups accepted from PEP, because endogenous mannitol (or any ligand that can be phosphorylated) would also act as an acceptor of phosphoryl groups from PEP (see also Fig. 3). To ensure that all phosphoryl groups were donated specifically to EIImtl, phosphorylation of Cys-384 was prevented by quenching with NEM. Again, the LDH-linked assay was performed and the data are shown in Fig. 6. Extrapolation of the absorbance change (after 4 min) to the point when PEP was added resulted in 4.6 μM of phosphate donated to reduced EIImtl (Δ1), and 2.5 μM of phosphate donated to NEM-quenched EIImtl (Δ2). From this it could be concluded that either ∼9% (0.2 μM) of EIImtl was not labeled by NEM, or that there is ligand present that could be phosphorylated, causing the extra 9% of donated phosphate groups. To discriminate between these two possibilities, we determined the phosphorylation activity of both preparations (Fig. 6, inset). The NEM-quenched sample (circles) retained ∼15% phosphorylation activity compared with the untreated sample (squares), indicating that not all Cys-384 residues were alkylated and explaining the 9% extra phosphorylation groups. We therefore conclude that >95% of the phosphate groups were specifically donated to EIImtl, and that the proposed endogenous ligand cannot be phosphorylated by EIImtl.
in the range of 100–500 nM may well explain our experimental results. This leaves mannitol itself as a possible candidate for the unidentified second ligand, apparently copurified with EIImtl. However, it is not very likely that mannitol would be present in the binding sites, because it is not produced by E. coli. One could argue that mannitol is present in the rich Luria Broth media, but growth of the same E. coli strain LGS322 (transformed with wild-type EIImtl, pMamtlaPrEII6his) on M9 minimal medium (31) with glucose as sole carbon source, showed a similar dependency of the KD on the protein concentration (data not shown). Furthermore, the LDH-linked assay should have resulted in a higher number of phosphoryl groups accepted from PEP, because endogenous mannitol (or any ligand that can be phosphorylated) would also act as an acceptor of phosphoryl groups from PEP (see also Fig. 3). To ensure that all phosphoryl groups were donated specifically to EIImtl, phosphorylation of Cys-384 was prevented by quenching with NEM. Again, the LDH-linked assay was performed and the data are shown in Fig. 6. Extrapolation of the absorbance change (after 4 min) to the point when PEP was added resulted in 4.6 μM of phosphate donated to reduced EIImtl (Δ1), and 2.5 μM of phosphate donated to NEM-quenched EIImtl (Δ2). From this it could be concluded that either ∼9% (0.2 μM) of EIImtl was not labeled by NEM, or that there is ligand present that could be phosphorylated, causing the extra 9% of donated phosphate groups. To discriminate between these two possibilities, we determined the phosphorylation activity of both preparations (Fig. 6, inset). The NEM-quenched sample (circles) retained ∼15% phosphorylation activity compared with the untreated sample (squares), indicating that not all Cys-384 residues were alkylated and explaining the 9% extra phosphorylation groups. We therefore conclude that >95% of the phosphate groups were specifically donated to EIImtl, and that the proposed endogenous ligand cannot be phosphorylated by EIImtl.
FIGURE 5.

Computer simulations to examine the effect of an endogenous ligand. Each data point (either KD or Etot) was simulated using 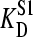 = 100 nM, stock solution concentration S1 = 80 × Etot, with Etot varying from 1 to 10 μM. The amount of endogenous ligand was set at 75% on a mol/mol ratio compared with the added amount of sites. In the simulations the value for
= 100 nM, stock solution concentration S1 = 80 × Etot, with Etot varying from 1 to 10 μM. The amount of endogenous ligand was set at 75% on a mol/mol ratio compared with the added amount of sites. In the simulations the value for 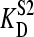 was varied from 100 to 1000 nM in several steps. (A) The observed
was varied from 100 to 1000 nM in several steps. (A) The observed 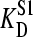 plotted against Etot. (B) The observed value for Etot plotted against the added amount of Etot.
plotted against Etot. (B) The observed value for Etot plotted against the added amount of Etot.
FIGURE 6.

Quantitative phosphorylation of Cys-384-quenched EIImtl. For experimental conditions see legend of Fig. 3. The dotted lines represent linear fits through the data points from 4 to 10 min. The difference between the control (EI, HPr) and EIImtl (with (Δ2), or without (Δ1) NEM) is the amount of phosphorylation sites associated with EIImtl. The inset represents the phosphorylation activity of the EIImtl samples, i.e., the amount of mannitol-1-P formed in time; (▪) untreated sample; (•) NEM-quenched sample; (♦) blank sample with no EIImtl.
Dissociation of mannitol from EIImtl
The simulations presented in Fig. 5, indicated that an endogenous ligand could explain the experimental results, but only if present in considerable amounts with an affinity that is comparable to the affinity of EIImtl for mannitol itself. This implies that much of the endogenous ligand must remain bound during purification of the protein. To test whether this is a plausible possibility, we incubated 2.5 nmol of EIImtl (corresponding with 1.25 nmol of binding sites) in 500 μL buffer A (25 mM Tris-HCl, pH 7.6, 0.25% decylPEG, 2 mM GSH) with a small excess of 3H-mannitol for 3 min at room temperature. Subsequently, the protein was bound to Q-Sepharose (250 μL), the column was washed with 19 column volumes of buffer A, and the protein was eluted with nine column volumes of buffer B (buffer A, supplemented with 300 mM NaCl). Fig. 7 shows the time course of the radioactivity appearing in the eluate during the washing and elution steps, with (solid symbols) and without (open symbols) EIImtl. Assuming all binding sites were occupied with 3H-mannitol, the difference in the amount of 3H-mannitol that was bound and the amount that eluted with the protein showed that even after this extensive washing procedure ∼39% of the available binding sites were still occupied by 3H-mannitol.
FIGURE 7.

Dissociation of mannitol from EIImtl. A mixture (500 μL), with (solid symbols) or without (open symbols) 5 μM EIImtl (1.25 nmol binding sites in buffer A, 25 mM Tris-HCl, pH 7.6, 0.25% decylPEG, plus 2 mM GSH), was incubated with 6.67 μM 3H-mannitol (3.34 nmol) for 3 min at room temperature and applied onto 250 μL Q-Sepharose. The column was subsequently batchwise washed with 19 column volumes of buffer A, and the protein was batchwise eluted with 9 column volumes of buffer B (buffer A, supplemented with 300 mM NaCl). Fraction 1 is the flow through, after which washing was started. The arrow indicates the start of elution. All fractions were tested for radioactivity by liquid scintillation counting. The shaded triangles with numbers point the fractions for which samples were prepared for SDS-PAGE analysis (inset). Sample 0 represents the starting material, before loaded to the Q-Sepharose column.
DISCUSSION
We have characterized the substrate binding properties of EIImtl, using flow dialysis, and conclude that the unphosphorylated protein has a single high-affinity mannitol binding site per dimeric EIImtl. Binding isotherms generated at different protein concentrations did not reveal a low-affinity binding site even at the highest protein concentrations.
In our binding experiments we observed a significant dependence of the dissociation constant for mannitol on the protein concentration: an increase in protein concentration resulted in a significant increase in the apparent dissociation constant. As shown in Fig. 1 the binding isotherms could be fitted appropriately with a simple one-site one-ligand model: the χ2-values did not indicate that this model was inadequate.
Computer simulations showed that such binding behavior could be expected when competing ligands are present in either the substrate stock or in the protein preparation. Several authors have reported either chemical and/or radiochemical impurities in their radiolabeled substrate stocks (32–37; and the references cited in Merrill and Lewis (37)). On the basis of the manufacturers quality sheets, the compounds were >95% pure. However, analysis of the radiolabeled compounds revealed in some cases purity as low as 30%. Transfer constants for permeation of 14C-sucrose across the blood-brain barrier in rats were overestimated up to 8 times for certain batches of 14C-sucrose (38). The effects of impurities in radiolabeled stocks on the binding parameters can be dramatic (39–41). We excluded the possibility of impurities in the radiolabeled stock solutions with the substrate cut-up control experiment (Fig. 4).
The presence of endogenous ligand associated with EIImtl seems a more plausible explanation, as shown by the simulations in Fig. 5. The simulations further show that, to explain the observed effects, a considerable fraction of the sites should be occupied with an endogenous ligand that has an affinity similar to that of mannitol itself. In agreement with this model is the observation that the dependence is less strong in the purified protein samples (Fig. 2 D, diamonds), where more of the endogenous ligand appears to have been removed during purification. Indeed, we showed that only very extensive washing will remove endogenous mannitol from the binding sites (Fig. 7). If this explanation is correct, our best estimate for the KD would be that observed at the lowest protein concentrations, where the effect of an endogenous ligand is smallest. Reliable binding measurements at even lower protein concentrations are not possible with the flow dialysis technique (13).
For some oligopeptide binding proteins (OppA from Lactococcis lactis and Salmonella typhimurium, AppA from Bacillus subtilis) and for the histidine-binding protein (HisJ) from S. typhimurium endogenous ligands were observed after purification (42–45). Apparently, some oligopeptides bind with such a strong affinity that purification does not remove the bound ligands. In the crystal structure of a mutant of the lactose permease (LacY) from E. coli showed an unidentified disaccharide bound at the sugar-binding site (46).
With flow dialysis a radioactive tracer is used to measure the binding parameters. In these experiments any unlabeled endogenous ligand will be exchanged with the tracer in the course a binding experiment. Therefore, the concentration of binding sites appears unaffected but their affinity appears to decrease at increasing concentrations. When techniques like fluorescence are used to measure ligand binding, endogenous ligand will affect most the apparent value for Etot, because endogenous ligand will typically diminish the total change in fluorescence observable during a titration. This is indeed what we observed for a single-tryptophan mutant (EIImtl-W30, data not shown): different purified preparations showed different degrees of fluorescence increase upon addition of mannitol.
The next logical step would be to remove the endogenous ligand or discover its nature. For the OppA oligopeptide binding protein (43), endogenous ligands could be removed by unfolding, washing, and subsequent refolding. However, this is not possible for membrane proteins like EIImtl. Despite several attempts, we were unable to identify the proposed competing ligand. A colorimetric assay to determine the content of alditols was hampered by a high background (47). An enzymatic method to follow the oxidation of mannitol, using mannitol dehydrogenase, was not sensitive enough at the expected concentrations of endogenous ligand (48). Electron spray mass spectrometry suffered from the high background of detergent, even after removal of >99% of the detergent by water/diethylether liquid phase separation (data not shown). Also, extensive dialysis of membrane vesicles did not (completely) abolish the KD dependence on the enzyme concentration. Further research is necessary to elucidate the nature of the proposed endogenous ligand.
In conclusion, we demonstrated that dimeric, unphosphorylated EIImtl has only a single high-affinity binding site. This new insight into the substrate-binding mechanism will have consequences for models of the phosphotransferase system in general. The “restricted access model”, describing the mannitol-concentration-dependent translocation of binding sites, explains most of the kinetic properties of EIImtl (21). Numerical analysis of the kinetic scheme, however, provided no clues regarding coupling of binding sites within each monomer and our conclusion that each dimer has only one high-affinity biding site is compatible with this kinetic scheme. Also, for the IICBglc dimer of the glucose permease (EIIglc), a model has been proposed with both a high- and a low-affinity binding site (49), but direct evidence for this is lacking. Although EIImtl and EIIglc are not homologous, they behave catalytically in a similar manner and we feel that a reevaluation of the number of substrate binding sites in EIIglc may be necessary.
Acknowledgments
We thank Joost H. A. Folgering for critical reading of the manuscript.
This work was supported by the Netherlands Foundation for Chemical Research (C.W.) with financial aid from the Netherlands Organization for the Advancement of Scientific Research.
Abbreviations used: EIImtl, EnzymeIImtl from Escherichia coli; CPM, counts per minute; decylPEG, decylpoly(ethyleneglycol)300; DTT, dithiothreitol; Etot, total amount of binding sites; EI, Enzyme I from Escherichia coli; GSH, reduced glutathion; HPr, heat-stable protein from Escherichia coli; ISO, inside out; KD, dissociation constant; LDH, lactate dehydrogenase; Mtl, mannitol; NEM, N-ethylmaleimide; PEP, phosphoenolpyruvate; UV, ultraviolet.
References
- 1.Lolkema, J. S., D. Swaving-Dijkstra, R. H. Ten Hoeve-Duurkens, and G. T. Robillard. 1990. The membrane-bound domain of the phosphotransferase Enzyme IImtl of Escherichia coli constitutes a mannitol translocation unit. Biochemistry. 29:10659–10663. [DOI] [PubMed] [Google Scholar]
- 2.Lolkema, J. S., and G. T. Robillard. 1992. The enzymes II of the phosphoenolpyruvate-dependent carbohydrate transport system. New Compr. Biochem. 21:135–168. [Google Scholar]
- 3.Lolkema, J. S., D. Swaving-Dijkstra, and G. T. Robillard. 1992. Mechanics of solute translocation catalyzed by Enzyme IImtl of the phosphoenolpyruvate-dependent phosphotransferase system of Escherichia coli. Biochemistry. 31:5514–5521. [DOI] [PubMed] [Google Scholar]
- 4.Boer, H., R. H. Ten Hoeve-Duurkens, J. S. Lolkema, and G. T. Robillard. 1995. Phosphorylation site mutants of the mannitol transport protein EnzymeIImtl of Escherichia coli: studies on the interaction between the mannitol translocating C-domain and the phosphorylation site on the energy coupling B-domain. Biochemistry. 34:3239–3247. [DOI] [PubMed] [Google Scholar]
- 5.Briggs, C. E., S. S. Khandekar, and G. R. Jacobson. 1992. Structure/function relationships in the Escherichia coli mannitol permease: identification of regions important for membrane insertion, substrate binding and oligomerization. Res. Microbiol. 143:139–149. [DOI] [PubMed] [Google Scholar]
- 6.Grisafi, P. L., A. Scholle, J. Sugiyama, C. Briggs, G. R. Jacobson, and J. W. Lengeler. 1989. Deletion mutants of the Escherichia coli K-12 mannitol permease: dissection of transport-phosphorylation, phospho-exchange, and mannitol-binding activities. J. Bacteriol. 171:2719–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lolkema, J. S., H. Kuiper, R. H. Ten Hoeve-Duurkens, and G. T. Robillard. 1993a. Mannitol-specific Enzyme II of the phosphoenolpyruvate-dependent phosphotransferase system of Escherichia coli: physical size of Enzyme IImtl and its domain IIBA and IIC in the active state. Biochemistry. 32:1396–1400. [DOI] [PubMed] [Google Scholar]
- 8.Meijberg, W., G. K. Schuurman-Wolters, and G. T. Robillard. 1998. Thermodynamic evidence for conformational coupling between the B and C domains of the mannitol transporter of Escherichia coli, Enzyme IImtl. J. Biol. Chem. 273:7949–7956. [DOI] [PubMed] [Google Scholar]
- 9.Pas, H. H., R. H. Ten Hoeve-Duurkens, and G. T. Robillard. 1988. Bacterial phosphoenolpyruvate-dependent phosphotransferase system: mannitol-specific EII contains two phosphoryl binding sites per monomer and one high-affinity mannitol binding site per dimer. Biochemistry. 27:5520–5525. [DOI] [PubMed] [Google Scholar]
- 10.Saraceni-Richards, C. A., and G. R. Jacobson. 1997. A conserved glutamate residue, Glu-257, is important for substrate binding and transport by the Escherichia coli mannitol permease. J. Bacteriol. 179:1135–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saraceni-Richards, C. A., and G. R. Jacobson. 1997. Subunit and amino acid interactions in the Escherichia coli mannitol permease: a functional complementation study of coexpressed mutant permease proteins. J. Bacteriol. 179:5171–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swaving-Dijkstra, D., J. Broos, J. S. Lolkema, H. Enequist, W. Minke, and G. T. Robillad. 1996. A fluorescence study of single tryptophan-containing mutants of EnzymeIImtl of the Escherichia coli phosphoenolpyruvate-dependent mannitol transport system. Biochemistry. 35:6628–6634. [DOI] [PubMed] [Google Scholar]
- 13.Veldhuis, G., E. P. P. Vos, J. Broos, B. Poolman, and R. M. Scheek. 2004. Evaluation of the flow-dialysis technique for analysis of protein-ligand interactions: an experimental and a Monte Carlo study. Biophys. J. 86:1959–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weng, Q.-P., and G. R. Jacobson. 1993. Role of a conserved histidine residue, His-195, in the activities of the Escherichia coli mannitol permease. Biochemistry. 32:11211–11216. [DOI] [PubMed] [Google Scholar]
- 15.Roossien, F. F., and G. T. Robillard. 1984. Mannitol-specific carrier protein from the Escherichia coli phosphoenolpyruvate-dependent phosphotransferase system can be extracted as a dimer from the membrane. Biochemistry. 23:5682–5685. [DOI] [PubMed] [Google Scholar]
- 16.Stephan, M. M., and G. R. Jacobson. 1986. Membrane disposition of the Escherichia coli mannitol:permease: identification of membrane-bound and cytoplasmic domains. Biochemistry. 25:4046–4051. [DOI] [PubMed] [Google Scholar]
- 17.Pas, H. H., J. C. Ellory, and G. T. Robillard. 1987. Bacterial phosphoenolpyruvate-dependent phosphotransferase system: association state of membrane-bound mannitol-specific enzyme II demonstrated by inactivation. Biochemistry. 26:6689–6696. [DOI] [PubMed] [Google Scholar]
- 18.Khandekar, S. S., and G. R. Jacobson. 1989. Evidence for two distinct conformations of the Escherichia coli mannitol permease that are important for its transport and phosphorylation functions. J. Cell. Biochem. 39:207–216. [DOI] [PubMed] [Google Scholar]
- 19.Boer, H., R. H. Ten Hoeve-Duurkens, G. K. Schuurman-Wolters, A. Dijkstra, and G. T. Robillard. 1994. Expression, purification and kinetic characterization of the mannitol transport domain of the phosphoenolpyruvate-dependent mannitol phosphotransferase system of Escherichia coli. J. Biol. Chem. 269:17863–17871. [PubMed] [Google Scholar]
- 20.Elferink, M. G. L., A. J. M. Driessen, and G. T. Robillard. 1990. Functional reconstitution of the purified phosphoenolpyruvate-dependent mannitol-specific transport system of Escherichia coli in phospholipid vesicles: coupling between transport and phosphorylation. J. Bacteriol. 172:7119–7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lolkema, J. S. 1993. A method to study complex enzyme kinetics involving numerical analysis of enzymatic schemes. J. Biol. Chem. 268:17850–17860. [PubMed] [Google Scholar]
- 22.Swaving-Dijkstra, D., J. Broos, and G. T. Robillard. 1996. Membrane proteins and impure detergents: procedures to purify membrane proteins to a degree suitable for tryptophan fluorescence spectroscopy. Anal. Biochem. 240:142–147. [DOI] [PubMed] [Google Scholar]
- 23.Broos, J., F. Ter Veld, and G. T. Robillard. 1999. Membrane protein-ligand interactions in Escherichia coli vesicles and living cells monitored via a biosynthetically incorporated tryptophan analogue. Biochemistry. 38:9798–9803. [DOI] [PubMed] [Google Scholar]
- 24.Broos, J., G. B. Strambini, M. Gonnelli, E. P. P. Vos, M. Koolhof, and G. T. Robillard. 2000. Sensitive monitoring of the dynamics of a membrane-bound transport protein by tryptophan phosphorescence spectroscopy. Biochemistry. 39:10877–10883. [DOI] [PubMed] [Google Scholar]
- 25.Robillard, G. T., and M. Blaauw. 1987. Enzyme II of the Escherichia coli phosphoenolpyruvate-dependent phosphotransferase system: protein-protein and protein-phospholipid interactions. Biochemistry. 26:5796–5803. [DOI] [PubMed] [Google Scholar]
- 26.Gunnewijk, M. G. W., and B. Poolman. 2000. Phosphorylation state of HPr determines the level of expression and the extent of phosphorylation of the lactose transport protein of Streptococcus thermophilus. J. Biol. Chem. 275:34073–34079. [DOI] [PubMed] [Google Scholar]
- 27.Lolkema, J. S., and G. T. Robillard. 1990. Subunit structure and activity of the mannitol-specific enzyme II of the Escherichia coli phosphoenolpyruvate-dependent phosphotransferase system solubilized in detergent. Biochemistry. 29:10120–10125. [DOI] [PubMed] [Google Scholar]
- 28.Roossien, F. F., W. Van Es-Spiekman, and G. T. Robbilard. 1986. Dimeric Enzyme IImtl of the E. coli phosphoenolpyruvate-dependent phosphotransferase system. Cross-linking studies with bifunctional sulfhydryl reagents. FEBS Lett. 196:284–290. [DOI] [PubMed] [Google Scholar]
- 29.Pace, C. N., F. Vajdos, L. Fee, G. Grimsley, and T. Gray. 1995. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4:2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lolkema, J. S., R. H. Ten Hoeve-Duurkens, and G. T. Robillard. 1993. Steady state kinetics of mannitol phosphorylation catalysed by Enzyme IImtl of the Escherichia coli phosphoenolpyruvate-dependent phosphotransferase system. J. Biol. Chem. 268:17844–17849. [PubMed] [Google Scholar]
- 31.Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 32.Ball, W. P., and P. V. Roberts. 1991. Long-term sorption of halogenated organic chemicals by aquifer material. 1. Equilibrium. Environ. Sci. Technol. 25:1223–1237. [Google Scholar]
- 33.Bartlett, S. E., and M. T. Smith. 1995. The apparent affinity of morphine-3-glucuronide at μ1-opoid receptors results from morphine contamination: demonstration using HPLC and radioligand binding. Life Sci. 57:609–615. [DOI] [PubMed] [Google Scholar]
- 34.Drenth, E. H., and R. A. de Zeeuw. 1982. Radiochemical purity control of radiolabeled drugs. Int. J. Appl. Radiat. Isot. 33:681–683. [DOI] [PubMed] [Google Scholar]
- 35.Gu, B., O. R. West, and R. L. Siegrist. 1995. Using 14C-labeled radiochemicals can cause experimental error in studies of the behavior of volatile organic compounds. Environ. Sci. Technol. 29:1210–1214. [DOI] [PubMed] [Google Scholar]
- 36.Honoré, B. 1987. Protein binding studies with radiolabeled compounds containing radiochemical impurities. Anal. Biochem. 162:80–88. [DOI] [PubMed] [Google Scholar]
- 37.Merrill, E. J., and A. D. Lewis. 1974. Purity of radiolabeled chemicals. Anal. Chem. 46:1114–1116. [Google Scholar]
- 38.Preston, E., D. O. Foster, and P. A. Mills. 1998. Effects of radiochemical impurities on measurements of transfer constants for [14C]sucrose permeation of normal and injured blood-brain barrier of rats. Brain Res. Bull. 45:111–116. [DOI] [PubMed] [Google Scholar]
- 39.Builder, S. E., and I. H. Segel. 1978. Equilibrium ligand binding assays using labeled substrates: nature of the errors introduced by radiochemical impurities. Anal. Biochem. 85:413–424. [DOI] [PubMed] [Google Scholar]
- 40.Lazareno, S., and N. J. M. Birdsall. 2000. Effects of contamination on radioligand binding parameters. Trends Pharmacol. Sci. 21:57–60. [DOI] [PubMed] [Google Scholar]
- 41.Segel, I. H. 1994. The effects of labeled and unlabeled impurities on the analysis of equilibrium binding and initial velocity data by means of Scatchard plots. J. Theor. Biol. 171:267–280. [Google Scholar]
- 42.Ames, G. F. L., C. E. Liu, A. K. Joshi, and K. Nikaido. 1996. Liganded and unliganded receptors interact with equal affinity with the membrane complex of periplasmic permeases, a subfamily of traffic ATPases. J. Biol. Chem. 271:14264–14270. [DOI] [PubMed] [Google Scholar]
- 43.Lanfermeijer, F. C., A. Picon, W. N. Konings, and B. Poolman. 1999. Kinetics and consequences of binding of nona- and dodecapeptides to the oligopeptide binding protein (OppA) from Lactococcus lactis. Biochemistry. 38:14440–14450. [DOI] [PubMed] [Google Scholar]
- 44.Levdikov, V. M., E. V. Blagova, J. A. Brannigan, L. Wright, A. A. Vagin, and A. J. Wilkinson. 2005. The structure of the oligopeptide-binding protein AppA, from Bacillus subtilis in complex with a nanopeptide. J. Mol. Biol. 345:879–892. [DOI] [PubMed] [Google Scholar]
- 45.Tame, J. R. H., G. N. Murshudov, E. J. Dodson, T. K. Neil, G. G. Dodson, C. F. Higgins, and A. J. Wilkinson. 1994. The structural basis of sequence-independent peptide binding by OppA protein. Science. 264:1578–1581. [DOI] [PubMed] [Google Scholar]
- 46.Abramson, J., I. Smirnova, V. Kasho, G. Verner, H. R. Kaback, and S. Iwata. 2003. Structure and mechanism of the lactose permease of Escherichia coli. Science. 301:610–615. [DOI] [PubMed] [Google Scholar]
- 47.Sanchez, J. 1998. Colorimetric assay of alditols in complex biological samples. J. Agric. Food Chem. 46:157–160. [DOI] [PubMed] [Google Scholar]
- 48.Graefe, H., B. Gütschow, H. Gehring, and L. Dibbelt. 2003. Sensitive and specific photometric determination of mannitol in human serum. Clin. Chem. Lab. Med. 41:1049–1055. [DOI] [PubMed] [Google Scholar]
- 49.Garcia-Alles, L. F., V. Navdaeva, S. Haenni, and B. Erni. 2002. The glucose-specific carrier of the Escherichia coli phosphotransferase system. Eur. J. Biochem. 269:4969–4980. [DOI] [PubMed] [Google Scholar]