

Abstract
Lung surfactant causes the surface tension, γ, in the alveoli to drop to nearly zero on exhalation; in the upper airways γ is ∼30 mN/m and constant. Hence, a surface tension gradient exists between alveoli and airways that should lead to surfactant flow out of the alveoli and elimination of the surface tension gradient. However, the lung surfactant specific protein SP-C enhances the resistance to surfactant flow by regulating the ratio of solid to fluid phase in the monolayer, leading to a jamming transition at which the monolayer transforms from fluidlike to solidlike. The accompanying three orders of magnitude increase in surface viscosity helps minimize surfactant flow to the airways and likely stabilizes the alveoli against collapse.
INTRODUCTION
Lung surfactant is a complex mixture of lipids and proteins originating from the type II cells that line the alveolar epithelial surfaces of humans and other mammals (1,2). Lung surfactant reduces the surface tension in the alveolar spaces, which minimizes the work of breathing, promotes uniform lung inflation, and prevents alveolar collapse during the cycles of lung expansion and compression. A lack of functional surfactant due to prematurity leads to neonatal respiratory distress syndrome (NRDS; 1,3). Surfactant inhibition is implicated in the development of acute RDS in infants and adults (2). Surfactant replacement therapy, typically with extracts of bovine or porcine lung surfactants, has proven to be of great clinical value in reducing the effects of NRDS (2,3).
To maintain proper lung function, the surface tension, γ, in the alveoli must drop to nearly zero on exhalation (2,4,5). In the trachea, γ is ∼30 mN/m (6) and in the connecting airways, γ is ∼15 mN/m (7). As a result, a surface tension gradient exists among the trachea, airways, and the alveoli during exhalation. Surface tension gradients induce flow and the transport of surfactant and other interfacial materials in the direction of the higher surface tension (8,9), which is likely necessary to promote removal of particulates and other debris from the deep lung (2).
For surface tension driven flows in the lungs, the rate of surfactant transport is proportional to 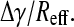 Δγ is the difference in surface tension between the alveoli (where surfactant is generated and expressed) and the airways (where no surfactant is expressed), and Reff is an effective flow resistance that depends on the surface viscosity, ηs, of the surfactant film (9), the bulk viscosity of the epithelial lining fluid, η (10–12), the thickness of the epithelial lining fluid (9,11,12), and the branching and geometry of the airways, surface interactions, particulates, etc. (2,13–15). Instillation of replacement surfactants in premature infants (in which the higher surface tension is in the alveolus) occurs by surface tension driven flows in the smaller airways and alveoli and takes only minutes to occur in practice (10,11). Reversing the direction of the surface tension gradient in normal lungs would suggest that the flow of surfactant from the lungs is just as fast in the absence of a sufficiently high flow resistance. Moreover, the total internal area of the terminal bronchioles is ∼3000 cm2, and the total internal area of the tracheobronchial tree from larynx to terminal bronchioles is ∼4000 cm2 (16). By comparison, alveolar surface area is 80–150 m2 or ∼200–300 times larger. Hence, only a fraction of the alveolar surfactant could line the whole airway. For surface tension gradients to be maintained, there must be a strong resistance to surfactant flow at low surface tension. One way to enhance the flow resistance would be for the surface viscosity to increase substantially with decreasing surface tension (increasing Reff) to offset the increasing surface tension gradient, Δγ, during exhalation (15), thereby keeping
Δγ is the difference in surface tension between the alveoli (where surfactant is generated and expressed) and the airways (where no surfactant is expressed), and Reff is an effective flow resistance that depends on the surface viscosity, ηs, of the surfactant film (9), the bulk viscosity of the epithelial lining fluid, η (10–12), the thickness of the epithelial lining fluid (9,11,12), and the branching and geometry of the airways, surface interactions, particulates, etc. (2,13–15). Instillation of replacement surfactants in premature infants (in which the higher surface tension is in the alveolus) occurs by surface tension driven flows in the smaller airways and alveoli and takes only minutes to occur in practice (10,11). Reversing the direction of the surface tension gradient in normal lungs would suggest that the flow of surfactant from the lungs is just as fast in the absence of a sufficiently high flow resistance. Moreover, the total internal area of the terminal bronchioles is ∼3000 cm2, and the total internal area of the tracheobronchial tree from larynx to terminal bronchioles is ∼4000 cm2 (16). By comparison, alveolar surface area is 80–150 m2 or ∼200–300 times larger. Hence, only a fraction of the alveolar surfactant could line the whole airway. For surface tension gradients to be maintained, there must be a strong resistance to surfactant flow at low surface tension. One way to enhance the flow resistance would be for the surface viscosity to increase substantially with decreasing surface tension (increasing Reff) to offset the increasing surface tension gradient, Δγ, during exhalation (15), thereby keeping 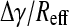 small.
small.
How does lung surfactant make the necessary adjustments in the two-dimensional surface viscosity as a function of surface tension? Many of the common mechanisms for enhancing solution viscosity in three dimensions, for example, polymer entanglements, have no analogy in two dimensions. The changes in surface viscosity must accompany the changes in monolayer phase behavior that occur with decreasing surface tension (increasing surface pressure). At low surface pressures, there are typically fluid phases, with liquidlike molecular correlations and low surface viscosity (14,15). As the surface pressure increases to levels expected in the lungs (>40 mN/m), solid phases, with long-range molecular correlations, form. Over a wide range of surface pressure in multicomponent lung surfactant monolayers, these solid and fluid phases can coexist (13,15,17). We have found that the surface viscosity at phase coexistence can undergo a dramatic increase due to the jamming of solid phase domains at a critical surface area fraction (13). Rather small changes in the solid/fluid phase ratio can lead to large changes in surface viscosity (13,15). Here we show that the lung surfactant specific protein SP-C (18), the function of which has not yet been fully established, interacts with the lipids of the monolayer to regulate the surface viscosity. The SP-C protein manages this by selectively removing fluid phase lipids from the monolayer at low surface tensions to form multilayer patches associated with the monolayer (19,20). The solid domains jam together, and the surface viscosity increases by orders of magnitude, increasing the resistance to surface tension gradient induced flow (13). SP-C increases the surface viscosity by almost an order of magnitude as compared to lipid monolayers of the same lipid composition. In addition to enhancing jamming, the hydrophobic helical part of SP-C stitches adjacent monolayers together, leading to a higher surface viscosity (19,20). At the higher surface tensions that accompany inhalation, the fluid phase lipids are reincorporated into the monolayer, and the monolayer viscosity decreases, allowing the film to be adsorbed or respread and cover the increased alveolar surface (2).
The lung surfactant film mechanical properties at coexistence are the two-dimensional analog of a three-dimensional suspension of solids in a liquid (13,21). In three-dimensional suspensions, as the solid volume fraction, φ, approaches the volume fraction for random close packing, φc, the viscosity diverges as 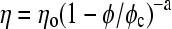 ; where η is the steady-shear viscosity of the dispersion, ηo is the viscosity of the suspending liquid, and the exponent, a, varies from 1 to 2 depending on the interactions between the solids (22). The surface viscosity of lipid monolayers at coexistence for a variety of compositions, surface pressures, and temperatures diverges in a similar fashion:
; where η is the steady-shear viscosity of the dispersion, ηo is the viscosity of the suspending liquid, and the exponent, a, varies from 1 to 2 depending on the interactions between the solids (22). The surface viscosity of lipid monolayers at coexistence for a variety of compositions, surface pressures, and temperatures diverges in a similar fashion: 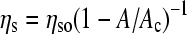 ; where ηso is the surface viscosity of the continuous, fluid phase, and ηs is the effective surface viscosity of the monolayer (13). A is the area fraction of the solid phase, and Ac is a critical solid phase fraction at which the solid domains jam into each other and the viscosity diverges. Relatively small changes in the fluid/solid ratio can lead to order of magnitude changes in the surface viscosity near Ac (13). By adding or subtracting fluid phase lipids from the monolayer in a reversible fashion, the lung surfactant protein SP-C leads to the divergence of the surface viscosity of the monolayers at the appropriate surface tension to dramatically increase the flow resistance of surfactant from the alveoli.
; where ηso is the surface viscosity of the continuous, fluid phase, and ηs is the effective surface viscosity of the monolayer (13). A is the area fraction of the solid phase, and Ac is a critical solid phase fraction at which the solid domains jam into each other and the viscosity diverges. Relatively small changes in the fluid/solid ratio can lead to order of magnitude changes in the surface viscosity near Ac (13). By adding or subtracting fluid phase lipids from the monolayer in a reversible fashion, the lung surfactant protein SP-C leads to the divergence of the surface viscosity of the monolayers at the appropriate surface tension to dramatically increase the flow resistance of surfactant from the alveoli.
This molecular picture may explain other macroscopic aspects of lung mechanics. SP-C knockout mice show reduced lung viscoelasticity in comparison to normal mice; the higher monolayer viscosity in the presence of SP-C may stabilize the alveoli against collapse at end expiration (23,24). The viscosity-regulating function of SP-C can be partially replaced by SP-B, but SP-B does not pull as much fluid phase from the monolayer as SP-C, nor does it have the ability to bind monolayers together (20,25). This may explain why SP-C knockout mice survive (23,24) but are more susceptible to disease (24) or lung injury (26), especially when SP-B is depleted. Birds, whose lungs consist of rigid tubular air capillaries that do not change volume or surface area, have no SP-C (27).
EXPERIMENTAL
Materials
Dipalmitoylphosphatidylcholine (DPPC) and palmitoyloleoylphosphatidyl-glycerol (POPG) were purchased from Avanti Polar Lipids (Alabaster, AL; purity >99%) and palmitic acid (PA) from Sigma Chemical (St Louis, MO; purity >99%). The lipids were mixed in weight ratios of 68:22:8 in 3:1 chloroform/methanol. This lipid mixture is chosen to mimic the functional properties of natural lung surfactants both in vitro and in vivo (28) and is similar to the clinically used replacement lung surfactant, Survanta. However, the composition of native surfactant varies significantly from species to species, and the compositions of replacement surfactants vary even more due to difficulties in extraction and in the number and type of additives used, so there is no universally accepted surfactant composition (29). Peptide mimics of the lung surfactant proteins SP-B and SP-C were synthesized as described elsewhere (20) and added to the lipid mixture in 3:1 chloroform/methanol. Water was prepared using a Millipore Milli-Q system and had a resistivity >18 MΩ-cm−1.
Survanta is an organic solvent extract of minced bovine lungs produced for clinical use by Ross Laboratories (Columbus, OH). After extraction, Survanta contains mainly phospholipids, neutral lipids, fatty acids, and two surfactant specific proteins, SP-B and SP-C (29). The extract is then supplemented with synthetic DPPC, PA, and tripalmitin to standardize the Survanta composition and optimize its in vitro performance. The resulting formulation contains ∼70% DPPC and other saturated phospholipids and 10% fatty acids, mainly PA. There is <1% (by weight) of surfactant protein, which is primarily SP-C. Survanta is sold and used as an aqueous suspension in physiological saline with a concentration of 25 mg/ml phospholipids. For our experiments, Survanta was diluted in buffer (NaCl 150 mM, CaCl2 2 mM, NaHCO3 0.2 mM, and pH = 7) to a lipid concentration of 1 or 2 mg/ml and was spread onto the same buffer solution in the Langmuir trough. Typically, sufficient Survanta was added to raise the surface pressure to ∼10 mN/m before compression was initiated.
Curosurf (Chiesi Farmaceutici, Parma, Italy) is an organic extract of porcine lungs and contains 50%–65% saturated phosphatidylcholines, mainly DPPC (29). Curosurf contains both SP-B and SP-C but has minimal free fatty acids. Curosurf was also diluted in buffer (NaCl 150 mM, CaCl2 2 mM, NaHCO3 0.2 mM, and pH = 7) to a lipid concentration of 1 or 2 mg/ml and was spread onto the same buffer solution in the Langmuir trough. Sufficient Curosurf was added to raise the surface pressure to ∼10 mN/m before compression was initiated.
Methods
Pressure-area isotherms were collected using a custom built Langmuir trough with a Wilhelmy-type pressure-measuring device and a computer-controlled barrier. The model monolayers were spread dropwise from the 3:1 chloroform/methanol solvent with Hamilton syringes (∼0.5 mg/ml) onto ultra pure water for protein free mixtures (MilliQ, 18.3mΩ.cm) or onto saline buffer (150 mM NaCl, 2 mM CaCl2, 0.2 mM NaHCO3) at pH = 7.0 ± 0.1 for samples with proteins. The initial compressions were begun after ∼10 min to allow for complete evaporation of the solvent. Survanta and Curosurf were spread as aqueous suspensions by syringe; sufficient surfactant was added to the trough to obtain a surface pressure of ∼10 mN/m at full trough expansion. For fluorescence imaging, the lipid mixtures were doped with 0.5–1 mol % of the fluorescent lipid Texas Red-DHPE (Molecular Probes, Eugene, OR). The fluorescent probe segregates to disordered or fluid phases, which then appear bright in images. The probe is absent from the solid or liquid condensed (LC) phases, which appear black or dark (17). The fluorescence imaging system is described elsewhere (20).
Magnetic needle viscometer
The steady-shear surface viscosity was measured in a custom built magnetic needle viscometer (21). To achieve a constant force on the magnetic needle (Fm), the trough was positioned off-center between two Helmholtz coils (21). A 14 mm wide channel formed by two glass plates was made along the axis of the magnetic gradient within which a magnetic needle (a Teflon rod 3 cm long and 2 mm diameter) was floated. The amplitude of the magnetic force, Fm, was adjusted by varying the current in the coils via two power supplies. Above the channel, a video camera recorded the needle motion. The video signal was then digitized and the needle speed derived from these images (13,14,21). The absolute drag on the needle was determined from the initial and terminal velocity of the needle and calibrated to known DPPC surface shear viscosity (13,14,21). The density of the needle is greater than that of water, so the needle typically sinks at ∼50 mN/m; the surface tension of the interface is not high enough to keep the needle floating. Hence, we cannot measure the surface viscosity over the entire physiologically relevant range of 40–72 mN/m. Each viscosity measurement was performed at a fixed surface pressure and temperature and was repeated a minimum of three times.
Atomic force microscopy
Selected films were transferred to mica substrates for atomic force microscopy (AFM) imaging using a custom built transfer system (30). A modified Nanoscope III FM (Digital Instruments, Santa Barbara, CA) was used for imaging. A low-resolution fluorescence optical microscope was used to position the AFM tip onto specific regions of the sample. Once the desired regions were located, AFM imaging was done with a 150 μm × 150 μm (J) scanner in contact mode. Silicon nitride tips with a spring constant of 0.12 N/m were used. Exerting large forces on the sample was a concern during imaging, so samples were checked often for deformation. This was done by imaging for a few minutes on a smaller region (∼20 μm), then zooming out to check if damage had been done to the scanned region.
RESULTS AND DISCUSSION
The surface viscosities of individual lipids and mixtures common to lung surfactants (29) measured with a custom designed magnetic needle viscometer (13,14,21) are shown in Fig. 1. POPG is always in the liquid expanded (LE) phase at physiological conditions and is representative of the unsaturated lipids in lung surfactant (31). The surface viscosity of POPG remains <0.001 mN-s/m over the entire range of surface pressure. In contrast, the viscosity of PA increases suddenly at π = 22 mN/m. PA monolayers have a crystalline packing with the molecules in a tilted herringbone motif (L2′′ phase) below 22 mN/m. Above 22 mN/m, the lattice is untilted (32,33) and ηs jumps by almost an order of magnitude. The viscosity is correlated with the longer correlation lengths and tighter grain boundaries in the untilted phase (32,33), even though the probe used to measure viscosity is macroscopic (14).
FIGURE 1.

Surface shear viscosity, ηs, as a function of surface pressure, π, for Langmuir monolayers of POPG, PA, DPPC, DPPC/POPG 4:1, DPPC/POPG/PA 68:22:8 (wt/wt) at 25°C. The POPG monolayer remains in the fluid state at all π, thus ηs remains low. The PA monolayer undergoes a transition from a tilted to a nontilted phase at 22 mN/m, which triggers a dramatic increase of ηs. DPPC surface viscosity in the LE phase (below ∼15 mN/m) is similar to POPG. In the LC phase, ηs increases exponentially. Both DPPC/POPG (bottom inset image) and DPPC/POPG/PA (top inset image) films show coexistence between solid phase islands (black in inset images) and a continuous fluid phase (bright in inset images) for surface pressures >10 mN/m. Both images shown are taken at 30 mN/m; PA increases the fraction of solid phase at a given π, which also increases ηs.
DPPC, the dominant lipid in native lung surfactant, undergoes a LE to LC first order transition at 12 mN/m at 25°C; the coexistence pressure increases with increasing temperature (34,35). In the LE phase, the viscosity is ∼.001 mN-s/m, similar to POPG. In the LC phase, the viscosity increases exponentially with surface pressure (8,14). DPPC/POPG (68:22 wt/wt) mixtures exhibit coexistence between a DPPC-rich LC phase and a POPG-rich LE phase (35) at surface pressures above ∼10 mN/m (fluorescence micrographs inset to Fig.1). POPG inhibits the growth of the LC domains without affecting the molecular packing (35), causing the surface viscosity to decrease relative to pure DPPC. Adding PA to the DPPC/POPG mixture (68:22:8 wt/wt) leads to a significant increase in both the fraction of solid phase at a given surface pressure and the surface viscosity (inset to Fig. 1). PA causes a decrease in the molecular tilt and an increase in the molecular cohesion of DPPC crystals (13,34). LC phase islands of cocrystallized DPPC and PA (dark in images) coexist with a continuous DPPC/POPG LE phase (bright; 13,35). ηs increases with the area fraction of solid, as can be seen by the comparison of the fluorescence images with and without PA (13). The size and polydispersity of the crystalline domains influences the critical solid phase fraction at which the viscosity diverges, Ac (13). The surface viscosity at coexistence is well described by 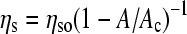 , and changes from the low values of the continuous fluid phase to that of the solid phase as the solid domains jam into each other as the solid phase fraction approaches Ac (13). At higher temperatures, the transitions are shifted to higher surface pressures; the transition depends only on the solid phase area fraction and Ac (13).
, and changes from the low values of the continuous fluid phase to that of the solid phase as the solid domains jam into each other as the solid phase fraction approaches Ac (13). At higher temperatures, the transitions are shifted to higher surface pressures; the transition depends only on the solid phase area fraction and Ac (13).
However, the maximum viscosity of the pure lipid films is ∼0.05 mN-s/m, which may be insufficient to arrest surface tension driven flow or to help keep alveoli from collapsing at the end of exhalation. Figs. 2 and 3 show that adding 5 wt % of a synthetic version of lung surfactant specific protein SP-C (SP-Cff; 18,20) increases the DPPC/POPG/PA surface viscosity from ∼.03 to .43 mN-s/m at π = 45 mN/m, at which the isotherm shows a plateau. This lipid mixture also shows a strong hysteresis; on compression, the viscosity stays low up to ∼40 mN/m then increases by ∼3 orders of magnitude as the surface pressure is increased slightly. On expansion, the viscosity decreases gradually with decreasing surface pressure. Further increasing the SP-C concentration to 15% does not change the surface viscosity significantly. Adding SP-C to pure POPG monolayers or other LE phase monolayers (data not shown) does not increase the surface viscosity; SP-C accentuates the percolation transition in those films that show a LE-LC coexistence (13).
FIGURE 2.

Surface-pressure-area isotherm of DPPC/POPG/PA (68:22:8 wt %) monolayers with varying weight fractions of SP-Cff. Adding SP-Cff shifts the isotherms to larger area/molecule, showing that the protein has incorporated into the interface. The pronounced plateau at ∼40 mN/m shows that the monolayer loses material or is strongly condensed. This is consistent with the multilayer formation shown in Fig. 4.
FIGURE 3.

Surface shear viscosity, ηs, as a function of surface pressure, π, for DPPC/POPG/PA (68:22:8 wt %) monolayers with varying weight fractions of SP-Cff. The surface viscosity on compression remains low up to ∼40 mN/m, at which it increases by more than three orders of magnitude. On expansion, the surface viscosity decreases more slowly with decreasing surface pressure, showing a marked hysteresis. Five percent SP-Cff increases the surface viscosity by a factor of 5 over the lipid alone. Increasing the SP-Cff to 15% provides only a marginal increase in the surface viscosity.
The origin of the viscosity increase is made obvious by AFM images of DPPC/POPG/PA films with 2 wt % SP-Cff transferred to mica substrates below (Fig. 4 A) and above (Fig. 4 B) the plateau (19,20). The AFM images show that below the plateau, the fluid phase regions of the monolayer are ∼1 nm lower (darker in image, see height trace below) than the solid phase regions. However, for the monolayers transferred just above the plateau, where the dramatic change in the viscosity is observed, the fluid regions have thickened to be ∼5 nm higher than the solid phase domains. This is consistent with the formation of three-layer thick areas in the fluid phase. The plateau in the isotherms of these films (Fig. 2) is consistent with a loss of interfacial area corresponding to the removal of fluid phase lipids from the interface to form these multilayer patches. The clinical surfactants Curosurf and Survanta, which both contain native SP-C, also show multilayer patches in the fluid phases (15).
FIGURE 4.

AFM images of DPPC/POPG/PA + SP-Cff films. (A) Below the plateau in the isotherm, the fluid phase regions are 1 nm lower (see height trace) than the condensed regions. (B) Above the plateau, the fluid regions have thickened to be ∼5 nm higher than the condensed domains. This is consistent with the formation of three-layer thick areas in the fluid phase. The plateau in the isotherms corresponds to the removal of fluid phase lipids from the interface to form these multilayer patches. SP-C acts to regulate the solid/fluid phase ratio causing the surface viscosity to diverge.
Native SP-C is a 4.2 kDa, dipalmitoylated, 35-residue peptide, of which 23 residues are hydrophobic; both SP-C (19,20) and SP-B (25,36,37) are primarily located in the fluid phase domains of the monolayer. SP-C has a transbilayer orientation similar to that of integral membrane proteins and adopts an α-helical conformation between residues 9 and 34 (18). The N-terminal segment includes two palmitoylcysteinyls. The length of the α-helix is ∼3.7 nm and orients along the acyl chains of lipids in a monolayer or bilayer environment (18,38). Hence, it is likely that the multilayers seen in Fig. 4 B are held together by the transmembrane SP-C (19). Native SP-C, palmitoylated human recombinant SP-C, and synthetic SP-Cff have similar effects in promoting the monolayer to multilayer transition (19,20).
The π-A isotherms for DPPC/POPG/PA with various concentrations of dSP-B1-25, a peptide mimic of native SP-B, are shown in Fig. 5. At a given surface pressure, the average area per molecule increases with the peptide concentration, showing the insertion of the protein within the monolayer. On all the isotherms, there is a smooth kink at low pressure and a shallow plateau at higher pressures, which are displaced toward lower pressures as the peptide concentration increases. The plateau goes from ∼40 mN/m for the pure lipid film to ∼32 mN/m with 10% dSP-B1-25. The monolayers collapse at the same pressure, ∼66 mN/m.
FIGURE 5.

Pressure-area isotherms of 68:22:8 DPPC/POPG/PA lipid mixture with various amounts of dSP-B1-25, a peptide mimic of native SP-B (25). Increasing the dSP-B1-25 fraction moves the isotherms to the right, indicating that the dSP-B1-25 is retained in the film. The collapse pressure stays the same.
The corresponding surface viscosity measurements (Fig. 6) also show two regimes. For surface pressures below the plateau, ηs is low and almost constant, as is the case for films containing SP-C (Fig. 3). At the plateau surface pressure, the surface viscosity increases; however, there is only about a 50% increase in the maximum ηs value of the film with 5% dSP-B1-25 compared to the lipid-only mixture. This is compared to the factor of 5 with the same amount of SP-C added (Fig. 3). dSP-B1-25 also is primarily located in the fluid phase domains and also removes fluid phase lipids from the monolayer on compression, but not nearly as efficiently as SP-C (25). As can be seen in the AFM images in Fig. 7, dSP-B1-25 leads to the formation of isolated protrusions from the fluid phase at high surface pressure (25). With no dSP-B1-25 in the films (Fig. 7 A), the solid phases are smooth and slightly higher (∼0.5 nm) than the fluid phase, which is mottled light and dark gray. With 5 wt % dSP-B1-25 in the film (Fig. 7 B), small white protrusions emerge from the fluid phase domains, with height ranging from ∼5 to 10 nm. Increasing the dSP-B1-25 to 10 wt % increases the lateral dimensions of the protrusions but not their density (Fig. 7 C). dSP-B1-25 removes material from the fluid phase, as does SP-C, but the location and distribution of the material is quite different. The effect on the surface viscosity is, as expected, not as great. SP-B also does not have the ability to link monolayers together as does the transmembrane SP-C (15,19,20,31).
FIGURE 6.

Surface viscosity as a function of surface pressure for the dSP-B1-25 containing films. Increasing the dSP-B1-25 content to 5% only increases the surface viscosity by ∼50%, compared to more than a factor of 5 increase caused by an equal amount of SP-C. Increasing the dSP-B1-25 fraction increases the surface viscosity, suggesting that more fluid phase lipids are removed from the monolayer, consistent with the images in Fig. 7.
FIGURE 7.

AFM images of lipid films transferred to mica substrates at π = 40 mN/m. (A) 0 wt % dSP-B1-25. With no dSP-B1-25, the solid phase is smooth and slightly higher (∼0.5 nm) than the fluid phase, which is mottled light and dark gray. (B) 5 wt % dSP-B1-25. As is the case for SP-C, SP-B is located in the fluid phase domains. The solid phase domains remain smooth and are free of protrusion. dSP-B1-25 leads to the formation of isolated small protrusions from the fluid phase domains, with heights ranging from ∼5−10 nm. SP-B removes fluid phase lipids from the monolayer on compression, but not nearly as efficiently as SP-C. (C) 10 wt % dSP-B1-25. Increasing the dSP-B1-25 to 10 wt % increases the lateral dimensions of the protrusions but not their density. dSP-B1-25 removes material from the fluid phase, as does SP-C, but the location and distribution of the material is quite different, leading to a smaller effect on the surface viscosity.
Fig. 8 shows that the clinically used replacement surfactants, Survanta and Curosurf, have very similar surface viscosity as does the lipid and SP-C mixture. Survanta is an extract of bovine lung surfactant supplemented with DPPC and PA, whereas Curosurf is an extract of porcine lungs (29). Both clinical surfactants show a dramatic increase in surface viscosity at ∼40–45 mN/m, consistent with the SP-C induced percolation of the solid phase fraction. As expected, Survanta has a higher viscosity due to the increased DPPC and PA content, which increases the solid phase fraction at a given surface pressure (see Fig. 1). Increasing the temperature of the Survanta films just moves the transition to a higher surface pressure, consistent with the temperature dependence of the solid phase area fraction. (The Curosurf transition at 37° is at too high a surface pressure for our instrument to measure.) AFM images show that both Survanta and Curosurf form multilayer patches in the fluid phase domains at high surface pressures (15).
FIGURE 8.

Surface shear viscosity of Survanta (bovine lung extract supplemented with DPPC and PA) and Curosurf (porcine lung extract) replacement lung surfactants. Both show similar behavior to the lipid mixtures in that the surface viscosity strongly increases at a surface pressure (our magnetic needle surface viscometer can only measure up to ∼50 mN/m) corresponding to an SP-C induced percolation of the solid phase domains in the monolayer. Survanta has a lower transition surface pressure than Curosurf at 25°C, as the added PA and DPPC in Survanta increase the solid phase fraction as in Fig. 1 (inset). Raising the temperature of the Survanta film to 37° moves the transition to higher surface pressure, consistent with the observation that the solid phase fraction is smaller at higher temperatures (13,34). Raising the temperature of the Curosurf film to 37° presumably moves the transition to a surface pressure higher than could be measured using the magnetic needle viscometer (data not shown).
CONCLUSIONS
Relating these dramatic changes in viscosity to their physiological implications is not obvious, as the optimal surface viscosity of a lung surfactant film has not been established. The in vivo function of SP-C is also open to question. A further complication is that the surface viscosity can only be measured over a part of the physiologically relevant surface pressure range of 35–70 mN/m due to limitations of the magnetic needle surface viscometer (21). However, from our previous work and theoretical analyses of the origin of the high surface viscosity (13,15), it is likely that the surface viscosity increases further as the solid phase fraction increases with surface pressure over the physiological range up to monolayer collapse (15). Hence, the resistance to surface tension driven flow should continue to remain high over the entire physiological range of surface pressures.
That regulation of surface viscosity is an important role for SP-C is consistent with recent observations of SP-C deficient animals. One strain of SP-C deficient mice is viable at birth and grows normally without apparent pulmonary abnormalities (23) but develops pulmonary problems after 1 year of age (24). The absence of SP-C decreases the hysteresivity, which describes the mechanical coupling between energy dissipative forces and tissue-elastic properties, consistent with a decrease in the surface viscosity. Lower than normal levels of SP-B in SP-C deficient mice led to lung dysfunction (24). This suggests that the function of SP-C can be carried out to some extent by SP-B in otherwise normal lungs, consistent with our observations that SP-B increases the surface viscosity of monolayers, but not as much as SP-C. In a different strain of SP-C deficient mice, however, there were more severe abnormalities in airway resistance, tissue damping, and hysteresivity, suggesting significant changes in the mechanics of the lung surfactant system. It was suggested that differences in shear forces over time contributed to the disruption of lung structure and function that were observed in the SP-C deficient mice (24). The control over surfactant viscosity demonstrated by SP-C appears to be important to the mechanics of normal lung function; a lack of SP-C leads to lung damage over time (24).
The ability of SP-C to regulate surface viscosity may be illustrative of a more generic biological control over the mechanical properties of two-dimensional systems including cell membranes (39,40). Recent two-photon and fluorescence micrographs show that phase coexistence occurs in lipid bilayers as well as monolayers (39,40). This has led to speculation that lipid phase separation is responsible for “raft” formation with its implications for protein localization and function in cell membranes. The shear viscosity of fluid phases in two dimensions is quite low; by comparison the viscosity of condensed phases is very large. However, as shown here, it is not necessary to undergo a complete phase transition to switch from low to high viscosity due to the jamming of the solid phase domains. The mechanical properties of bilayer membranes might be controlled by subtle adjustments in the area ratios of a more viscous and a less viscous phase. Large changes in the membrane properties could thus be induced by relatively small changes in the membrane composition or local environment. Proteins similar to the lung surfactant specific SP-B or SP-C, which have the ability to condense or remove unsaturated lipids, may act to tune the solid/liquid phase ratio. The ability of cell membranes to control local membrane phase separation and, hence, the membrane mechanical properties may be necessary for a variety of cell recognition and binding events (39–41).
Acknowledgments
The authors thank A. Barba for her active participation in the project. We also thank H. W. Taeusch for supplying Survanta and Curosurf, and J. Clements for enlightening us about lung physiology.
Financial support was provided from National Institutes of Health grant HL-51177 and the University of California Tobacco Related Disease Research Program, grant 11RT-0222.
References
- 1.Clements, J. A., and M. E. Avery. 1998. Lung surfactant and neonatal respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 157:S59–S66. [DOI] [PubMed] [Google Scholar]
- 2.Notter, R. H. 2000. Lung surfactant: basic science and clinical applications. In Lung Biology in Health and Disease. Claude Lenfant, editor. Marcel Dekker, New York, Basel.
- 3.Goerke, J. 1998. Pulmonary surfactant: functions and molecular composition. Biochim Biophys Acta. 1408:79–89. [DOI] [PubMed] [Google Scholar]
- 4.Schürch, S., J. Goerke, and J. A. Clements. 1978. Direct determination of volume and time-dependence of alveolar surface tension in excised lungs. Proc. Natl. Acad. Sci. USA. 75:3417–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schürch, S., J. Goerke, and J. A. Clements. 1976. Direct determination of surface tension in the lung. Proc. Natl. Acad. Sci. USA. 73:4698–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Im Hof, V., P. Gerh, V. Gerber, M. M. Lee, and S. Schürch. 1997. In vivo determination of surface tension in the horse and in vitro model studies. Respir. Physiol. 109:81–93. [DOI] [PubMed] [Google Scholar]
- 7.Geiser, M., S. Schürch, and P. Gehr. 2003. Influence of surface chemistry and topography of particles on their immersion into the lung's surface-lining layer. J. Appl. Physiol. 94:1793–1801. [DOI] [PubMed] [Google Scholar]
- 8.Sacchetti, M., H. Yu, and G. Zografi. 1993. In-plane steady shear viscosity of monolayers at the air/water interface and its dependence on free area. Langmuir. 9:2168–2171. [Google Scholar]
- 9.Stone, H. A. 1995. Fluid motion of monomolecular films in a channel flow geometry. Phys. Fluids. 7:2931–2937. [Google Scholar]
- 10.Bull, J. L., L. K. Nelson, J. T. J. Walsh, M. R. Glucksberg, S. Schurch, and J. B. Grotberg. 1999. Surfactant-spreading and surface-compression disturbance on a thin viscous film. J. Biomed. Eng. 121:89–98. [DOI] [PubMed] [Google Scholar]
- 11.Espinosa, F. F., and R. D. Kamm. 1999. Bolus dispersal through the lungs in surfactant replacement therapy. J. Appl. Physiol. 86:391–410. [DOI] [PubMed] [Google Scholar]
- 12.Espinosa, F. F., A. H. Shapiro, J. J. Fredberg, and R. D. Kamm. 1993. Spreading of exogenous surfactant in an airway. J. Appl. Physiol. 75:2028–2039. [DOI] [PubMed] [Google Scholar]
- 13.Ding, J., H. E. Warriner, and J. A. Zasadzinski. 2002. Viscosity of two-dimensional suspensions. Phys. Rev. Lett. 88:168102. [DOI] [PubMed] [Google Scholar]
- 14.Alonso, C., and J. Zasadzinski. 2004. Linear dependence of surface drag on surface viscosity. Phys. Rev. E. 69:021602. [DOI] [PubMed] [Google Scholar]
- 15.Alonso, C., T. Alig, J. Yoon, H. E. Warriner, F. Bringezu, and J. A. Zasadzinski. 2004. More than a monolayer: relating lung surfactant structure and mechanics to composition. Biophys. J. 87:4188–4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weibel, E. R. 1963. Morphometry of the Human Lung. Academic Press, New York.
- 17.McConnell, H. 1991. Structures and transitions in lipid monolayers at the air-water interface. Annu. Rev. Phys. Chem. 42:171–195. [Google Scholar]
- 18.Johansson, J. 1998. Structure and properties of surfactant protein C. Biochim. Biophys. Acta. 1408:161–172. [DOI] [PubMed] [Google Scholar]
- 19.von Nahmen, A., M. Schenk, M. Sieber, and M. Amrein. 1997. The structure of a model pulmonary surfactant as revealed by scanning force microscopy. Biophys. J. 72:463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding, J., D. Y. Takamoto, A. Von Nahmen, M. M. Lipp, K. Y. C. Lee, A. J. Waring, and J. A. Zasadzinski. 2001. Effects of lung surfactant proteins, SP-B and SP-C, and palmitic acid on monolayer stability. Biophys. J. 80:2262–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding, J. Q., H. E. Warriner, J. A. Zasadzinski, and D. K. Schwartz. 2002. Magnetic needle viscometer for Langmuir monolayers. Langmuir. 18:2800–2806. [Google Scholar]
- 22.Brady, J. F. 1993. The rheological behavior of concentrated colloidal dispersions. J. Chem. Phys. 99:567–581. [Google Scholar]
- 23.Glasser, S. W., M. S. Burhans, T. R. Korfhagen, C. L. Na, P. D. Sly, G. F. Ross, M. Ikegami, and J. A. Whitsett. 2001. Altered stability of pulmonary surfactant in SP-C-deficient mice. Proc. Natl. Acad. Sci. USA. 98:6366–6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glasser, S. W., E. A. Detmer, M. Ikegami, C. L. Na, M. T. Stahlman, and J. A. Whitsett. 2003. Pneumonitis and emphysema in SP-C gene targeted mice. J. Biol. Chem. 278:14291–14298. [DOI] [PubMed] [Google Scholar]
- 25.Ding, J., I. Doudevski, H. Warriner, A. J. Waring, and J. A. Zasadzinski. 2003. Nanostructural changes in lung surfactant monolayers induced by interactions between POPG and surfactant protein SP-B. Langmuir. 19:1539–1550. [Google Scholar]
- 26.Ikegami, M., T. E. Weaver, J. J. Conkright, P. D. Sly, G. F. Ross, J. A. Whitsett, and S. W. Glasser. 2002. Deficiency of SP-B reveals protective role of SP-C during oxygen lung injury. J. Appl. Physiol. 92:519–526. [DOI] [PubMed] [Google Scholar]
- 27.Bernhard, W., P. L. Haslam, and J. Floros. 2004. From birds to humans: new concepts on airways relative to alveolar surfactant. Am. J. Respir. Cell Mol. Biol. 30:6–11. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka, Y., T. Takei, T. Aiba, K. Masuda, A. Kiuchi, and T. Fujiwara. 1986. Development of synthetic lung surfactants. J. Lipid Res. 27:475–485. [PubMed] [Google Scholar]
- 29.Bernhard, W., J. Mottaghian, A. Gebert, G. A. Rau, H. von der Hardt, and C. F. Poets. 2000. Commercial versus native surfactants. Am J. Respir Crit Care Med. 162:1524–1533. [DOI] [PubMed] [Google Scholar]
- 30.Lee, K. Y. C., M. M. Lipp, D. Y. Takamoto, E. Ter-Ovanesyan, J. A. Zasadzinski, and A. J. Waring. 1998. Apparatus for the continuous monitoring of surface morphology via fluorescence microscopy during monolayer transfer to substrates. Langmuir. 14:2567–2572. [Google Scholar]
- 31.Takamoto, D. Y., M. M. Lipp, A. Von Nahmen, K. Y. C. Lee, A. J. Waring, and J. A. Zasadzinski. 2001. Interaction of lung surfactant proteins with anionic phospholipids. Biophys. J. 81:153–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riviere, S., S. Henon, J. Meunier, D. K. Schwartz, M.-W. Tsao, and C. M. Knobler. 1994. Textures and phase transitions in Langmuir monolayers of fatty acids. A comparative Brewster angle microscope and polarized fluorescence microscope study. J. Chem. Phys. 101:10045–10051. [Google Scholar]
- 33.Kaganer, V. M., H. Mohwald, and P. Dutta. 1999. Structure and phase transitions in Langmuir monolayers. Rev. Mod. Phys. 71:779–819. [Google Scholar]
- 34.Lee, K. Y. C., A. Gopal, A. Von Nahmen, J. A. Zasadzinski, J. Majewski, G. S. Smith, P. B. Howes, and K. Kjaer. 2002. Influence of palmitic acid and hexadecanol on the phase transition temperature and molecular packing of dipalmitoylphophatidyl-choline monolayers at the air-water interface. J. Chem. Phys. 116:774–783. [Google Scholar]
- 35.Bringezu, F., J. Q. Ding, G. Brezesinski, and J. A. Zasadzinski. 2001. Changes in model lung surfactant monolayers induced by palmitic acid. Langmuir. 17:4641–4648. [Google Scholar]
- 36.Lipp, M. M., K. Y. C. Lee, D. Y. Takamoto, J. A. Zasadzinski, and A. J. Waring. 1998. Coexistence of buckled and flat monolayers. Phys. Rev. Lett. 81:1650–1653. [Google Scholar]
- 37.Lipp, M. M., K. Y. C. Lee, J. A. Zasadzinski, and A. J. Waring. 1996. Phase and morphology changes in lipid monolayers induced by SP-B protein and its amino-terminal peptide. Science. 273:1196–1199. [DOI] [PubMed] [Google Scholar]
- 38.Gericke, A., C. R. Flach, and R. Mendelsohn. 1997. Structure and orientation of lung surfactant SP-C and L-α-dipalmitoylphosphatidylcholine in aqueous monolayers. Biophys. J. 73:492–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baumgart, T., S. T. Hess, and W. W. Webb. 2003. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature. 425:821–824. [DOI] [PubMed] [Google Scholar]
- 40.Keller, S. L. 2003. Miscibility transitions and lateral compressibility in liquid phases of lipid monolayers. Langmuir. 19:1451–1456. [Google Scholar]
- 41.Korlach, J., P. Schwille, W. W. Webb, and G. W. Feigenson. 1999. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 96:8461–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]