

Abstract
To elucidate the mechanisms of cooperativity of cytochrome P450eryF an SH-reactive fluorescent probe was introduced close to the substrate-binding site. Cys-154, the only accessible cysteine, was eliminated by site-directed mutagenesis, and a novel cysteine was substituted for Ser-93 in the B′/C loop. S93C, C154A, C154S, S93C/C154A, and S93C/S154C were characterized in terms of affinity for 1-pyrenebutanol (1-PB), cooperativity, and ionic-strength dependence of the 1-PB-induced spin shift. S93C/C154S retains the key functional properties of the wild-type, and modification by three different SH-reactive probes had little effect on the characteristics of the enzyme. The labeled proteins exhibited fluorescence resonance energy transfer from 1-PB to the label, which allowed us to resolve two substrate-binding events, and to determine the corresponding KD values (KD1 = 1.2 ± 0.2 μM, KD2 = 9.4 ± 0.8 μM). Using these values for analysis of the substrate-induced spin transition, we demonstrate that the interactions of P450eryF with 1-PB are consistent with a sequential binding mechanism, where substrate interactions at a higher-affinity site cause a conformational transition crucial for the binding of the second substrate molecule and subsequent spin shift. This transition is apparently associated with an important rearrangement of the system of salt links in the proximity of Cys-154.
INTRODUCTION
In the last decade homo- and heterotropic cooperativity have emerged as important properties of a number of cytochromes P450, including human 3A4, 2C9, 1A2, 2D6, and 2B6 (1–4). The most prominent examples of cooperativity are provided by P450 3A4, the most abundant P450 in human liver (2). Despite extensive studies, a generalized mechanism of this apparent allostery is still missing. Starting from the work of Korzekwa and co-authors (5,6) and Guengerich and co-authors (7) the prevailing hypothesis is that cytochromes P450 exhibiting cooperativity accommodate multiple substrate molecules in one large binding pocket. A loose fit of a single substrate molecule requires the binding of a second ligand for efficient binding and/or catalysis (6,8). However, the possibility that P450 cooperativity reflects a true case of allostery, involving an effector-induced conformational transition in the enzyme has also been discussed extensively (7,9–12). In this case, an effector-binding site might be remote from the active site (7). Observation of distal binding sites for palmitic acid in P450 2C8 (13) and for progesterone in P450 3A4 (14) in recently resolved x-ray structures provides important support for this possibility.
Studies of mutual effects of different substrates and effectors of P450 3A4 have compelled some authors to suggest three or more ligand binding sites in this enzyme (15–18), although direct physical evidence is lacking. The situation becomes even more complex when various indications of conformational heterogeneity of P450 3A4 are incorporated into mechanisms of cooperativity (11,19–21), leading to a nested allostery model for this enzyme (10). This model, which is based on the presumption of two stable conformers, each possessing the properties of an allosteric enzyme with two binding sites, may provide a viable alternative to a scheme with three or more binding sites. Formation of P450 oligomers in the microsomal membrane and in reconstituted systems has been proposed as one explanation for such persistent conformational heterogeneity (11). Therefore, understanding of the mechanisms of cooperativity in P450 has exhibited a stepwise transition from simple “space-filling” models (6,8) to more dynamic and complex schemes. Accordingly, the current concept of P450 cooperativity tends to approach classical models of allosteric enzymes involving effector-induced conformational transitions in the enzyme and, possibly, modulation of its oligomerization state.
Awareness of the increasing complexity of P450 cooperativity necessitates novel approaches and experimental techniques in the field. Unfortunately, the analysis of P450 cooperativity from steady-state kinetics of substrate oxidation cannot provide any information on the connection between the binding of substrates and effectors and subsequent conformational transitions in the enzyme. Of particular importance is the relationship between the binding of each substrate and/or effector molecule and the spin equilibrium of the heme iron, which is thought to be an important determinant of catalytic efficiency and coupling (22–27). Thus, to distinguish between different models of cooperativity and to understand molecular mechanisms of modulation of P450 activity by allosteric ligands it is critical to be able to monitor enzyme-ligand interactions directly.
To establish such an approach we made use of cytochrome P450eryF (P450 107A1) from Saccharopolyspora erythraea as a model. P450eryF exhibits homotropic (28,29) and heterotropic (30) cooperativity with such substrates as androstenedione, testosterone, 7-benzoxyquinoline, α-naphthoflavone, and pyrene derivatives (12,29–32). This soluble bacterial enzyme lacking the complexity of membranous monooxygenase systems provides a valuable reference point for further studies of eukaryotic cytochromes P450. In our initial experiments 1-pyrenebutanol (1-PB) was introduced as a fluorescent substrate of P450eryF (29). Interactions with 1-PB were monitored by the substrate-induced spin shift as well as by fluorescence resonance energy transfer (FRET) from the substrate to the heme of the enzyme. The results suggest that the binding of 1-PB to the higher-affinity binding site alone does not modulate the spin equilibrium, which is observed only in the complex of P450 with two molecules of 1-PB (29). This conclusion also appears to be valid for P450 3A4, based on recent studies of the testosterone-induced spin shift in monomeric P450 3A4 incorporated into a nanoscale lipid bilayer (Nanodisks) (33), as well as studies of the effect of interactions with testosterone and α-naphthoflavone on the tryptophan fluorescence of the enzyme (34). Further elaboration of the mechanism of P450 cooperativity clearly requires a similar direct approach to assess KD at the second (lower-affinity) binding site, which is essential to assess the relevancy of any particular model of enzyme-substrate interactions.
Furthermore, our recent studies of the effect of ionic strength on P450eryF revealed a rearrangement of intramolecular electrostatic interactions in the proximity of Cys-154 as an important element of the mechanisms of cooperativity (12). The results indicate that the modulation of the degree of hydration of the P450 heme pocket plays an important role in allostery. Further exploration of this hypothesis would be greatly facilitated by knowledge of the dynamics of protein-bound water in the interactions of P450eryF with allosteric ligands. Perturbation of protein-ligand interactions by hydrostatic pressure provides a unique approach to unravel the mechanisms of protein transitions involving changes in interactions with the solvent (35–37). Unfortunately, the quantum yield of the fluorescence of pyrenes and their spectral properties are known to be affected by the formation of excimers, which is dependent on pyrene concentration, viscosity of solution, polarity of the media, hydrostatic and osmotic pressure, and temperature. Therefore, techniques based on FRET from 1-PB to the heme of P450 are not well suited for pressure perturbation experiments.
The objective of this study was to resolve the binding of the first and the second 1-PB molecules to P450eryF with a more selective FRET-based assay involving a covalently bound fluorescent probe as an acceptor for FRET from the substrate molecule. Of particular interest are SH-reactive fluorescent probes, since wild-type P450eryF contains only two cysteine residues, namely Cys-154 and Cys-342. Only Cys-154 is accessible to modification, as Cys-342 serves as a ligand to the heme iron. However, modification of Cys-154 with 7-ethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin (CPM) had a crucial effect on the cooperativity and spin equilibrium in P450eryF (12). Based on analysis of 3D crystal structures of P450eryF we therefore introduced a new cysteine residue in place of Ser-93 in the solvent-exposed part of the B′/C loop. This residue is in the proximity of the substrate-binding pocket, and the surface location was expected to allow replacement with cysteine and modification by an appropriate SH-reactive probe without perturbing the enzyme conformation. Incorporation of the label at Cys-154 was prevented by replacement with alanine or serine.
Here we describe studies with P450eryF mutants bearing various substitutions at position 154, together with a detailed characterization of a double (S93C/C154S) mutant and its derivatives labeled with SH-reactive probes, such as CPM, N-[2-(1-maleimidyl)ethyl]-7-(diethylamino)coumarin-3-carboxamide (MDCC), and monobromobimane (mBBr). This mutant retains the major properties of the wild-type enzyme, and its modification with CPM, MDCC, or mBBr has no significant effect on interactions with 1-PB, substrate-induced spin shift, or cooperativity. The binding of 1-PB to the labeled mutant results in FRET to the fluorescent probe, offering an excellent tool for the studies of P450-ligand interactions.
This newly elaborated technique allowed us to resolve two substrate-binding events and to determine the dissociation constant for 1-PB bound at each of two sites in P450eryF. This finding represents an advance in unraveling the relationship between each of the substrate-binding events and the conformational transitions in the enzyme and on developing a reliable model of cooperativity in cytochromes P450. In addition to the results on enzyme-substrate interactions in the labeled S93C/C154S mutant, we also provide a comparative analysis of the effect of ionic strength on cooperativity and spin transition in a series of P450eryF mutants at position 154, which indicates that the mechanism of allostery involves a ligand-induced conformational rearrangement in the vicinity of Cys-154 that is associated with changes in the hydration of this residue.
MATERIALS AND METHODS
Chemicals
CPM, MDCC, and mBBr were the products of Molecular Probes (Eugene, OR). Oligonucleotide primers for PCR were obtained from Sigma Genosys (Woodlands, TX). The QuikChange site-directed mutagenesis kit was from Stratagene (La Jolla, CA). All other chemicals were of analytical grade and used without further purification.
Site-directed mutagenesis
All single mutants were generated using overlap extension PCR, and His-tagged P450eryF (wild-type) as the template (31) and primers:
Table 2.
| Mutation | DNA positions | Primer |
|---|---|---|
| C154A | 447-474 | 5′-CATCAAGGTCATCGCAGAGCTGCTCGGC-3′ |
| 5′-GCCGAGCAGCTCTGCGATGACCTTGATG-3′ | ||
| C154S | 447-474 | 5′-CATCAAGGTCATCTCTGAGCTGCTCGGC-3′ |
| 5′-GCCGAGCAGCTCAGAGATGACCTTGATG-3′ | ||
| S93C | 263-292 | 5′-CCAACATGGGTACCTGTGACCCGCCGACCC-3′ |
| 5′-GGGTCGGCGGGTCACAGGTACCCATGTTGG-3′ |
In the latter primer, in addition to the main TGC→TGT replacement, we made a silent GGC→GGT mutation to introduce an additional restriction site. S93C/C154S and S93C/C154A double mutants were obtained using S93C as the template and the same primers specified above for C154S and C154A single mutants. All constructs were sequenced to ensure that only the desired mutations were present (Protein Chemistry Core Laboratory, The University of Texas Medical Branch, Galveston, TX).
Protein expression and purification
P450eryF was expressed in Escherichia coli, and purified on a metal-affinity column, as described (31). All mutants produced nearly the same expression levels as the wild-type P450eryF. Protein yields upon purification varied from 24 to 57%. Protein was stored at −80°C in 0.1 M Na-Hepes buffer pH 7.4, 1 mM EDTA, and 1 mM dithiotreitol (DTT) containing 10% glycerol (buffer A).
Preparation and characterization of P450eryF labeled with fluorescent probes
Before labeling of S93C/C154S P450eryF with SH-reactive fluorescent probes it was necessary to remove DTT present in the storage buffer (buffer A, see above). The procedure of DTT removal was designed to prevent oxidation of SH groups with possible formation of disulfide-linked protein dimers. DTT was first replaced by 2 mM Tris(2-carboxyethyl)phosphine (TCEP), an SH-protecting agent. For this purpose, 250 μl of a concentrated (400–600 μM) solution of protein was passed through Bio-Spin®-6 column (Bio-Rad Laboratories, Hercules, CA) equilibrated with 0.1 M Na-Hepes buffer (Hepes(−)-buffer) containing 2 mM TCEP. Protein solution was then diluted to 2.5 ml by argon-saturated Hepes(−) buffer and concentrated to 250 μl in an argon-flushed Centrisart-I (cutoff 20,000) concentrator (Sartorius AG, Goettingen, Germany). This dilution-concentration cycle was then repeated. To modify this DTT-free preparation with CPM, MDCC, or mBBr the sample containing 60–80 nmol of P450 was diluted to 20 μM with argon-saturated Hepes(−)-buffer and incubated under argon gas at constant stirring with a 1.2-fold molar excess of CPM, MDCC, or mBBr added as a 2–3 mM solution in acetone-methanol (1:1, v/v). The reaction of CPM, MDCC, and mBBr with the protein was followed by an increase in the fluorescence of the probes. The intensity of fluorescence at 450–490 nm was continuously monitored using an MC2000-2 rapid scanning spectrometer (Ocean Optics, Dunedin, FL) with LED-380 light-emitting diode (Ocean Optics) as a source of excitation light. Incubation for 5–6 h at 25°C was required for modification of 95% of P450eryF S93C/C154S, as determined by the fitting of the kinetics of fluorescence increase to the equation of second-order reaction kinetics. When this level of modification was reached, the sample was concentrated to 250–300 μl on a Viva-Spin 4-ml 30,000 MWCO concentrator (Vivascience AG, Hannover, Germany) and transferred into buffer A by gel-exclusion chromatography on Bio-Spin-6 spin column. Excess of the DTT-adduct of the label was then removed using Cabiochem Detergent-Out hydrophobic absorber spin columns (EMD Biosciences, La Jolla, CA).
Because the bands of absorbance of CPM (λmax = 385 nm), MDCC (λmax = 419 nm) and mBBr (λmax = 396 nm) overlap considerably with the Soret bands of both low- and high-spin cytochrome P450, we used a special technique to determine the exact degree of labeling. For this purpose the sample of labeled P450eryF was treated with 60 mM hydrogen peroxide, which causes destruction of the heme and complete disappearance of the heme absorbance bands. This process was followed by repetitive measurement of absorbance spectra in the 340–700 nm region. Complete disappearance of the heme bands was achieved in 50–70 min of treatment. The difference between the initial spectrum and the spectrum obtained after complete bleaching was used to determine the exact concentration of P450 using known standard spectra of the low-spin, high-spin, and P420 states of P450eryF (29). The amount of label was determined from the absorbance spectrum of the bleached (heme-free) protein using the spectrum of a water solution of glutathione adduct of the appropriate label as a standard. In these determinations, we used our SpectraLab program (38) for linear least-square fitting of the spectra by a combination of spectral standards and a 1-to 2-order polynomial, which was used to compensate for possible turbidity of the media.
When used with MDCC and mBBr the above procedure of chemical modification of P450eryF S93C/C154S results in preparations with a molar ratio of label to P450 of 0.8–1.0 and protein recovery as high as 80–90%. In the case of modification with CPM, the recovery of P450eryF after modification was ∼50% due to a significant content of labeled P450 apo-protein (see Results), which was removed by ion-exchange chromatography on D15X weakly cationic ion-exchange membrane adsorber (Sartorius AG, Germany) as follows. The solution containing 40–50 nmol of P450eryF-CPM obtained by the above procedure was diluted with nine volumes of water and passed through a D15 membrane equilibrated with 10 mM Na-Hepes buffer, pH 7.4, 1 mM DTT, and 1 mM EDTA. Passage of the sample through the membrane was repeated until complete absorption of the protein was achieved. The membrane was then washed with 20 ml of 0.1 M Na-Hepes buffer, pH 7.4, 1 mM DTT, and 1 mM EDTA (Hepes(+)-buffer). Further washing with 0.1 M KCl in the same buffer resulted in elution of some amount (≤3% of the initial amount of P450) of unmodified P450eryF. P450eryF-CPM was then eluted with 0.2 M KCl in Hepes(+) buffer. Further washing of the membrane with 0.5 M KCl in the same buffer resulted in elution of heme-free protein also bearing the CPM label. The CPM/P450 molar ratio in the sample eluted in 0.2 M KCl was determined to be 0.95–1.0. This preparation was then concentrated to 150–200 μM P450 with a Viva-Spin concentrator and transferred into buffer A using a Bio-Spin 6 column.
1-PB binding and FRET assays
The absorbance and fluorescence spectra were measured with an MC2000-2 multichannel CCD rapid-scanning spectrometer (Ocean Optics) equipped with one absorbance and one fluorescence channel and a custom-made thermostated cell chamber with a magnetic stirrer. L7893 UV–Vis fiber optics light source (Hamamatsu Photonics, Tokyo, Japan). When the fluorimetric titrations were done with an MC2000-2 multichannel CCD rapid-scanning spectrometer, a pulsed PX-2 Xe-lamp (Ocean Optics) with an interference filter (53385, Oriel Instruments, Stratford, CT) having a transmittance band with 10-nm bandwidth centered at 330 nm was used as a source of the excitation light. The setup used in these FRET experiments permitted simultaneous registration of the transmittance and fluorescence spectra of the same sample, which allowed instant correction of the spectra of fluorescence for the changes in the intensity of the excitation light during the experiment. Alternatively, a computerized Hitachi F-2000 fluorescence spectrometer equipped with a custom-made thermostated cell holder and a magnetic stirrer was used. In this case, the excitation wavelength was set to 331 nm with a 10-nm bandwidth, and the fluorescence emission spectra were monitored in the region of 360–570 nm with a 10-nm bandwidth. In this case the spectra of absorbance of the sample taken before and after the experiment were used to calculate apparent absorbance spectra at each particular point of the titration, which were then used to correct the spectra of fluorescence for the changes in the intensity of the excitation light. All experiments were done at 25°C with continuous stirring in Hepes(+) buffer. The desired ionic strength of the medium was achieved by addition of an appropriate amount of KCl (as a 3-M stock solution in the Hepes buffer). An 8–15 mM stock solution of 1-PB in acetone was used in our titration experiments. The concentration of 1-PB in acetone solutions was determined by the absorbance at 343 nm using the extinction coefficient of 41,000 mM−1 cm−1 (39).
Data processing
The analysis of the series of spectra obtained in our spectrophotometric and fluorimetric titration/dilution experiments was done using a principal component analysis method, which is also known as singular value decomposition technique, as described earlier (38,40). To interpret the spectral transitions in terms of the changes in the concentration of P450 species, we used a least-squares fitting of the spectra of principal components by the set of the spectral standards of pure high-spin, low-spin, and P420 species of P450eryF (11). All data treatment and fitting, as well as the data acquisition were performed using our SpectraLab software package (38).
RESULTS
Effect of mutations at positions 93 and 154 on the interactions of P450eryF with 1-PB and substrate-induced spin shift
To introduce a unique accessible cysteine residue in close proximity of the substrate-binding pocket of P450eryF, we substituted Ser-93 with cysteine (S93C) and eliminated Cys-154 by substitution with alanine or serine (C154A and C154S, respectively). The newly obtained mutants were characterized in terms of the interactions of the enzyme with 1-PB, cooperativity, and the amplitude of the substrate-induced spin shift, ΔFh, as well as the effect of ionic strength on the 1-PB-induced spin shift and cooperativity. According to our recent results (12), an increase in ionic strength augments considerably the amplitude of the substrate-induced spin shift in P450eryF. In the 0.01–0.6 M range of ionic strength this effect is correlated with a prominent decrease in the cooperativity of the enzyme. The ionic strength dependence was characterized quantitatively with a semiempirical relationship similar to that used by Peyser and co-authors to analyze the effect of ionic strength on the conformation of myosin (41). Following this approach, the fraction of the high-spin heme protein in the enzyme-substrate complex (Fh) is represented as a function of the degree of occupation of a putative ion-binding site in the protein (41):
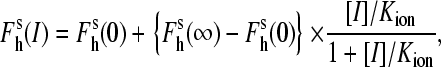 |
(1) |
where [I] is the ion concentration (which is formally equivalent to the ionic strength in this context), 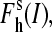
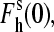 and
and 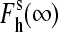 are the high-spin fraction of P450 at ion concentration [I], and at zero and infinite ionic strength, respectively, and Kion is the equilibrium constant of ion binding to a putative ion binding site in the enzyme. Despite the evident oversimplifications in this model, Eq. 1 fits the experimental ionic strength dependencies of
are the high-spin fraction of P450 at ion concentration [I], and at zero and infinite ionic strength, respectively, and Kion is the equilibrium constant of ion binding to a putative ion binding site in the enzyme. Despite the evident oversimplifications in this model, Eq. 1 fits the experimental ionic strength dependencies of 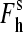 fairly well (see Fig. 2), providing a necessary starting point for a comparative analysis of P450eryF mutants in terms of responsiveness to ionic strength changes.
fairly well (see Fig. 2), providing a necessary starting point for a comparative analysis of P450eryF mutants in terms of responsiveness to ionic strength changes.
FIGURE 2.

Effect of ionic strength on the high-spin content in the 1-PB-bound P450eryF wild-type, its mutants, and labeled derivatives. (a) Data obtained with the wild-type enzyme (○), S93C (•), C154S (▵), C154A (▴), S93C/C154S (□), and S93C/C154A (▪). (b) Results of the experiments with S93C/C154S labeled with CPM (•), MDCC (▪), and mBBr (♦). The curves characterizing P450eryF wild-type (○) and its unlabeled S93C/C154S mutant (□) are also shown in this panel for comparison. In these experiments, the ionic strength was adjusted by addition of 3 M KCl in Hepes buffer. The concentration of 1-PB was 40 μM. Other conditions were as in Fig. 1.
In Table 1, we compare the values of Kion, the constant of spin equilibrium of the enzyme-substrate complex at I = 0 (Kspin(0)), and the parameters of Hill equation (S50 and n) for a series of P450eryF variants introduced in this study together with the data on C154I and C154T mutants, which were characterized earlier (29). Kspin(0) given in this table is defined as the ratio of concentrations of high- and low-spin P450 states extrapolated to zero ionic strength:
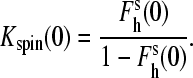 |
(2) |
TABLE 1.
Parameters of the interactions of P450eryF and its mutants with 1-PB
| P450eryF variant | S50, μM | Hill coefficient (n) | ΔFh (%) | Kion (mM) | Kspin(0) |
|---|---|---|---|---|---|
| Wild-type (S93/C154) | 7.9 ± 1.1 | 2.1 ± 0.2 | 29 ± 5 | 850 | 0.25 |
| C154I | 13.2 ± 1.8 | 1.4 ± 0.2 | 21 ± 4 | 1590 | 0.12 |
| C154A | 9.7 ± 0.3 | 2.3 ± 0.1 | 18 ± 2 | 1140 | 0.19 |
| C154T | 9.2 ± 0.1 | 1.8 ± 0.1 | 21 ± 3 | 1100 | 0.52 |
| C154S | 7.6 ± 0.7 | 1.5 ± 0.2 | 57 ± 3 | 340 | 0.75 |
| S93C | 10.0 ± 1.3 | 2.2 ± 0.2 | 16 ± 2 | 1190 | 0.14 |
| S93C/C154S | 8.4 ± 0.7 | 2.2 ± 0.2 | 41 ± 1 | 450 | 0.81 |
| S93C/C154A | 12.1 ± 0.2 | 2.4 ± 0.4 | 9 ± 1 | 2470 | 0.26 |
| S93C-CPM /C154S | 8.4 ± 0.5 | 2.4 ± 0.1 | 30 ± 6 | 600 | 0.61 |
| S93C-MDCC /C154S | 10.2 ± 1.0 | 1.7 ± 0.3 | 31 ± 3 | 1060 | 0.48 |
| S93C-bimane/C154S | 9.3 ± 2.0 | 2.3 ± 0.2 | 36 ± 2 | 350 | 0.54 |
The parameters of 1-PB binding represent the averages of four individual measurements plus/minus confidence intervals calculated for P = 0.95. Parameters of ionic strength dependence represent the results of a single measurement.
For all mutants tested, the substrate dependencies of the high-spin fraction of the enzyme obey the Hill equation with a Hill coefficient (n) indicative of positive cooperativity (Fig. 1, Table 1), which is consistent with that previously reported for the wild-type enzyme (12,29). The replacement of Ser-93 with cysteine did not alter n and only slightly increased the S50 for 1-PB. However, this mutation resulted in almost a twofold decrease in the amplitude of the 1-PB-induced spin shift. Similarly, the substitution of Cys-154 with alanine caused a decrease in the amplitude of the 1-PB-induced spin shift, whereas both S50 and n remained unaffected. Replacement of Cys-154 with serine decreased the Hill coefficient, did not alter S50, and caused a twofold increase in the amplitude of the spin shift.
FIGURE 1.

1-PB binding to P450eryF mutants and CPM-labeled enzyme: wild-type (○), S93C (•), C154S (▿), C154A (▾), S93C/C154S (□), S93C/C154A (▪), and CPM-labeled S93C/C154S mutant (P450eryF-CPM, ⋄). Conditions: 0.1 M Na-Hepes buffer, pH 7.4, 1 mM DTT, 1 mM EDTA, and 2 μM enzyme, 25°C.
The dependencies of the high-spin fraction in the substrate-saturated enzyme on ionic strength are shown in Fig. 2. In both S93C and C154A, the effect of ionic strength on the spin equilibrium is largely suppressed, whereas C154S P450eryF exhibits a considerably enhanced increase in the amplitude of the substrate-induced spin shift at increased ionic strength. Importantly, fitting of the ionic strength dependencies of the spin state for the wild-type P450eryF and each mutant gives a value of the high-spin fraction at infinite ionic strength (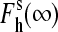 ) close to 100%. Averaging of
) close to 100%. Averaging of 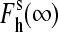 obtained for all P450eryF variants gives a value of 101 ± 11%. Therefore, at infinite ionic strength, where the ion tethers in the vicinity of Cys-154 suggested in our previous study (12) are completely released, the spin equilibrium in the substrate-bound enzyme is completely shifted toward the high-spin state, regardless of the nature of the amino acid residue at position 154.
obtained for all P450eryF variants gives a value of 101 ± 11%. Therefore, at infinite ionic strength, where the ion tethers in the vicinity of Cys-154 suggested in our previous study (12) are completely released, the spin equilibrium in the substrate-bound enzyme is completely shifted toward the high-spin state, regardless of the nature of the amino acid residue at position 154.
Analysis of the data presented in Table 1 reveals an important relationship among the values of S50, n, Kion, and Kspin(0). Comparison of the parameters obtained with various mutants at position 154 exhibits a clear linear correlation between the values of Kion and S50 (Fig. 3 a), so that the increase in Kion is always accompanied by an increase in S50. In terms of our hypothesis, this observation signifies that an increase in the strength of the strategic ion tethers around Cys-154 impedes the interactions of P450eryF with 1-PB. Furthermore, the changes in the value of Kspin(0) are strictly correlated with those of the Hill coefficient (Fig. 3 b). With the sole exception of C154I any replacement of residue 154 that promotes the formation of the high-spin state of the substrate-bound P450eryF at zero ionic strength results in decreased cooperativity of the enzyme with 1-PB. Thus, although all substitutions of residues 93 and 154 probed in our studies resulted in moderate alterations of the parameters of the interactions of the enzyme with 1-PB, the main features observed with the wild-type enzyme (12,29), namely positive cooperativity of 1-PB binding and a well-pronounced effect of ionic strength on the 1-PB-induced spin shift and cooperativity were retained.
FIGURE 3.

Cross correlation between the parameters of 1-PB binding and the effect of ionic strength on the spin equilibrium for a series of P450eryF variants with substitutions at position 154. (a) S50 versus Kion. (b) Hill coefficient, n, versus Kspin(0).
The double mutants S93C/C154A and S93C/C154S reveal additive effects of the single substitutions. S93C/C154A exhibited a higher S50 and lower ΔFh value than the wild-type and each of the respective single mutants (Fig. 1, Table 1). Similarly, the opposite changes in S50 and ΔFh values observed in the respective single mutants were compensatory in the S93C/C154S double mutant, which exhibited 1-PB-binding parameters close to those of the wild-type. Importantly, both S93C/C154S and S93C/C154A double mutants exhibited Hill coefficients close to that of the wild-type enzyme. The effect of the single mutations was also additive with regard to the effect of ionic strength on the spin state of the substrate-saturated enzyme. Due to such compensation of the effects of single substitutions, S93C/C154S exhibited an ionic strength dependency close to that of the wild-type (Fig. 2). Hence, we selected this mutant for further studies.
Labeling of S93C/C154S with SH-reactive probes and characterization of the labeled enzyme
With MDCC and mBBr, the labeling procedure described in Materials and Methods results in preparations with a label/P450 molar ratio of 0.8–1.0 and protein recovery of ∼80–90%. However, in the case of CPM we observed partial bleaching of the protein during its modification, which decreased the recovery of the labeled protein to <50%. This bleaching (heme loss) appears to be activated by light, as the recovery of the labeled P450eryF-CPM was increased considerably if the reaction was carried out in the dark without fluorimetric detection. Formation of a considerable amount of labeled apo-protein required subsequent protein purification in this case (see Materials and Methods).
Detailed characterization of CPM-labeled S93C/C154S P450eryF (P450eryF-CPM) in terms of 1-PB binding (Fig. 1, Table 1) and the effect of ionic strength on the spin equilibrium in the substrate-bound protein (Fig. 2) revealed that covalent attachment of CPM to the mutant had little impact on its characteristics. Parameters of the interactions of the enzyme with 1-PB for P450eryF-MDCC and P450-bimane were very close to those of P450eryF-CPM (Table 1). Thus, our data suggest that the labeling of S93C/C154S with CPM, MDCC, or mBBr does not disturb the structure or conformational dynamics of the enzyme, making the preparations of P450eryF labeled with these probes suitable for further mechanistic studies on enzyme-substrate interactions and cooperativity.
Fluorescence resonance energy transfer in the complexes of 1-PB with S93C/C154S P450eryF labeled with fluorescent probes
The CPM-labeled S93C/C154S P450eryF exhibited a bright fluorescence, having broad excitation and emission bands at 384 and 468 nm, respectively. In the case of P450eryF-MDCC, a broad and asymmetric excitation band was centered around 419 nm with the maximum of emission positioned at 492 nm. Although the intensity of fluorescence of P450eryF-MDCC was comparable to that of P450eryF-CPM, the fluorescence of P450eryF-bimane was considerably less intense. In this case the maximum of excitation was found at 388 nm, whereas the emission maximum was at 466 nm.
FRET in the complex of 1-PB with all three labeled P450eryF preparations was probed by two alternative approaches. In the first setup we titrated 0.3–0.6 μM 1-PB with labeled protein at concentrations increasing from 0 to 20–25 μM. In this setup the maximal concentration of the complex of the substrate with P450eryF was observed at high excess of the enzyme over the substrate and, consistently, this method was selective for the binding of 1-PB at the first (higher-affinity) binding site. In the second setup, which we designate as a dilution approach, a series of emission spectra of the mixture of labeled protein with 1-PB were recorded at a constant molar ratio and simultaneously changing concentrations. Obviously, when the enzyme and substrate were at a 1:1 molar ratio, this approach was also selective for the formation of a binary complex with substrate bound at the higher-affinity site.
In both the titration and dilution setups the changes in the intensity of fluorescence of the FRET donor (1-PB) and acceptor (either CPM, MDCC, or bimane) were followed by recording the spectra of emission from 360 to 600 nm. The excitation wavelength was set at 331 nm. This wavelength, which is close to the midpoint between two maxima of excitation of 1-PB (327 and 343 nm), ensures minimal direct excitation of all three fluorescent probes.
In all experiments the concentration of the enzyme changes considerably during the experiments. Although the absorbance of P450 at the excitation wavelength is low (ɛ331 ≅ 25,000 M−1 cm−1 for the low-spin P450eryF), these experiments required careful correction of the intensity of the excitation light during the titration (see Materials and Methods). In the case of titration of substrate by labeled protein it was also necessary to correct the spectra for direct excitation of FRET acceptors (CPM, MDCC, or bimane) by subtraction of the fluorescence of a corresponding concentration of labeled P450eryF taken alone. This correction is less critical in the case of MDCC, as the intensity of fluorescence of this label upon excitation at 331 nm is considerably lower than that observed with CPM and bimane. Thus, MDCC ensures the highest accuracy of FRET measurements in the titration setup.
Although the comparison of the parameters of 1-PB interactions with three labeled proteins (Table 1) suggests that P450eryF-mBBr represents the best approximation of the properties of the native enzyme, the low efficiency of FRET in P450eryF-bimane complicates studies by static fluorescence techniques. The properties of the other two labeled proteins, P450eryF-CPM and P450eryF-MDCC, are quite similar to each other. However, the above-mentioned advantages of the MDCC probe, together with our preliminary results indicating that, of the three probes tested, P450eryF-MDCC exhibits the lowest sensitivity of fluorescence of the label to hydrostatic pressure, prompted us to choose P450eryF-MDCC for the main part of these studies.
Determination of KD for 1-PB molecule bound at high-affinity binding site
Results of the studies of the interactions of P450eryF-MDCC with 1-PB monitored by FRET using dilution of a 1:1 enzyme-substrate mixture are exemplified in Fig. 4. The spectra shown are normalized to the enzyme concentration, so that in the absence of FRET the spectra are not expected to be affected by the concentration of the mixture. As shown in Fig. 4 a, increasing concentrations of the enzyme-substrate mixture result in changes that are indicative of FRET from 1-PB to MDCC, namely a decrease in the intensity of the 1-PB emission bands located at 378, 396, and 412 nm, concomitant with an increase in the intensity of the emission band of P450eryF-CPM (λmax = 468 nm). Correspondingly, application of principal component analysis to this series of spectra yields the first principal component, which reflects these alterations (Fig. 4 b, long-dashed line) and covers over 99.5% of the total changes in the spectra of fluorescence observed here. The dependence of the amplitude of FRET given by the loading factor for the first principal component on the concentration of P450eryF-CPM (Fig. 4 a, inset) obeys a canonical equation for the equilibrium of formation of enzyme-substrate complex at comparable concentrations of interacting species (42; see page 73, Eq. II-53):
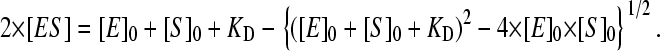 |
(3) |
FIGURE 4.

Determination of KD of the complex of labeled P450eryF S93C/C154S with 1-PB bound at the high-affinity binding site by FRET in a dilution setup. (a) Normalized emission spectra of an equimolar mixture of P450eryF with 1-PB at 0.23, 0.9, 1.4, 2.6, 3.4, 4.9, 7.6, 11, 13, and 16 μM P450eryF-MDCC. Intensity of fluorescence is expressed in arbitrary units (a.u.). The inset shows the results of the same experiment as the plot of the amplitude of FRET given by the loading factor of the first principal component versus the concentration of P450eryF-MDCC. The solid line shows the approximation of this data set by Eq. 3 with KD = 1.2 μM. Conditions were as in Fig. 3. (b) The spectra of the first principal component derived from the experiments with equimolar mixtures of 1-PB with P450eryF-CPM (solid line), P450eryF-MDCC (long-dashed line), and P450eryF-bimane (short-dashed line).
Similar results were obtained with the other two labels, although P450eryF-CPM exhibits a KD value of 0.70 ± 0.3 μM, which is slightly lower than the values obtained with P450eryF-MDCC and P450eryF-bimane (1.24 ± 0.1 and 1.21 ± 0.3 μM, respectively). Fig. 4 b compares the spectra of FRET given by the first principal component derived from the dilution experiments with all three different labels used in this study. The highest increase of fluorescence of the acceptor fluorophore is observed with CPM (Fig. 4 b, solid line), whereas in the case of MDCC and bimane (long- and short-dashed lines, respectively) the changes in the fluorescence of acceptor are smaller. This is consistent with less overlapping of the band of absorbance of the acceptor with the band of emission of the donor (1-PB) for the latter two labels. Although the absorbance band of CPM (λmax = 385 nm) completely overlaps with the main bands of 1-PB emission at 379 and 398 nm, the absorbance band of MDCC is shifted considerably toward longer wavelength (419 nm), which decreases the efficiency of FRET to this donor. Although the band of absorbance of bimane (λmax = 388 nm) has a position close to that of CPM, it is characterized by a very low extinction coefficient (ɛ388 = 5000 M−1 cm−1, as compared to ɛ385 = 33,000 M−1 cm−1 and ɛ319 = 50,000 M−1 cm−1 characteristic of CPM and MDCC, respectively (39), which results in lower efficiency of FRET in the 1-PB/bimane pair than 1-PB/CPM and 1-PB/MDCC.
Similar results were also obtained upon studies of P450eryF-CPM interactions with 1-PB in the titration setup. Titrations of 1-PB with P450eryF-MDCC, P450eryF-CPM, and P450-bimane exhibit changes in fluorescence indicative of FRET from 1-PB to the acceptor fluorophore (data not shown). Consistent with the results found by the dilution approach, the concentration dependency of the intensity of FRET given by the loading factor of the first principal component obeys the equation for the equilibrium of binary complex formation (Eq. 3). These experiments yield the values of 1.0 ± 0.1, 1.4 ± 0.4, and 0.83 ± 0.3 for P450eryF-CPM, P450eryF-MDCC and P450eryF-bimane, respectively. The fact that all three labeled preparations of the enzyme exhibit similar KD values in both dilution and titration setups suggests that the nature of the label has a negligible impact on the interactions of the enzyme with 1-PB.
It should be noted that the dilution setup is preferable for determination of KD by FRET, since no correction for direct excitation of the acceptor is required. However, this approach does not provide an accurate estimate of the amplitude of FRET, as the series of spectra obtained in the dilution setup lacks the point corresponding to the absence of acceptor. The efficiency of FRET estimated from the maximal decrease in 1-PB emission in the titration experiments with P450eryF-CPM and P450-MDCC was estimated to be of 98 ± 5 and 71 ± 11%, respectively.
The mean of the results of all experiments in either dilution or titration setups with all three labeled P450eryF preparations gives a KD of 1.2 ± 0.2 μM, which is fairly consistent with the value of 2.1 ± 0.8 μM reported in our earlier study for the interaction of 1−PB with the wild-type enzyme detected by FRET to the heme (12,29). The difference in these estimates may reflect the effect of C154S and S93C replacements, the impact of chemical modification of the enzyme, or the presence of hydroxypropylcyclodextrin in the reaction media in our earlier experiments. This compound may increase the value of KD due to association with 1-PB, and was omitted in this study.
Determination of KD for 1-PB molecule bound at the low-affinity binding site
Although the studies of the interactions of P450eryF with 1-PB by dilution of a 1:1 enzyme-substrate mixture are specific for determination of KD at the first (higher-affinity) binding site (KD1), dilution experiments at excess substrate can be used to study substrate binding at a lower-affinity site provided that the higher-affinity site is already saturated. According to Eq. 3, at a threefold molar excess of the substrate over the enzyme, 80% saturation of the higher-affinity site (KD = 1.2 μM) is observed at an enzyme concentration of 2 μM. Therefore, at the enzyme/substrate ratio of 1:3 or higher and enzyme concentration above 2 μM, the high-affinity binding site may be considered as occupied by 1-PB, so that FRET will reflect the binding at the second (lower-affinity) site only.
A series of spectra obtained in a FRET dilution experiment with a mixture of P450eryF-MDCC with 1-PB taken at a 3.3-fold molar excess is shown in Fig. 5 a. Here, similar to that observed at a 1:1 molar ratio, increase in the concentration of the enzyme-substrate mixture results in decrease in the amplitude of the 1-PB emission band concomitant with an increase in the fluorescence of MDCC. The dependence of the amplitude of FRET on the protein concentration obeys Eq. 3, exhibiting a KD value of 8.6 ± 0.3 μM. Importantly, increase in the substrate/enzyme molar ratio has no distinguishable effect on the value of KD determined in this way. Thus, the FRET dilution experiments carried out with P450eryF-MDCC and 1-PB at a seven- or eightfold excess of substrate gave a KD of 10.0 ± 0.3 and 9.6 ± 0.4 μM. Averaging these results yields a value of 9.4 ± 0.8 μM for the dissociation constant for 1-PB binding at the low-affinity binding site, provided that the high-affinity site is already occupied by this substrate.
FIGURE 5.

Determination of KD of the complex of labeled P450eryF S93C/C154S with 1-PB bound at the low-affinity binding site by FRET in a dilution setup. (a) Normalized emission spectra of a mixture of P450eryF with 1-PB taken at a 1:3.3 molar ratio at 3.6, 4.7, 5.8, 6.8, 10, 12., 15, 17, 21, 23, and 25 μM P450eryF-MDCC. Intensity of fluorescence is expressed in arbitrary units (a.u.). The inset shows the results of the same experiment as the plot of the amplitude of FRET given by the loading factor of the first principal component versus the concentration of P450eryF-MDCC. The solid line shows the approximation of this data set by the equation for the equilibrium of bimolecular association with KD = 8.6 μM. Conditions were as in Fig. 3. (b) The spectra of the first principal component derived from the experiments with the mixtures of 1-PB with P450eryF-MDCC taken at a molar ratio of 1:3.3 (dashed line) and 1:1 (solid line).
Fig. 5 b shows the first principal component spectrum of FRET in a 1:3.3 P450eryF-MDCC mixture with 1-PB (dashed line) as compared to the spectrum obtained at a 1:1 enzyme-substrate ratio (solid line). With excess substrate the amplitude of the MDCC emission band is considerably lower than in the case of an equimolar enzyme-substrate mixture. This lower efficiency of energy transfer to MDCC fluorophore suggests that the low-affinity binding site is located at a longer distance from MDCC (i.e., from Cys-93 residue of the mutant) than the high-affinity site.
Studies of the interactions of P450eryF-MDCC with 1-PB by absorbance spectroscopy
The ability to resolve 1-PB interactions at each of two binding sites and to determine the values of two dissociation constants provided an important opportunity to reconsider the relationship between the interactions of P450eryF with substrates and the substrate-induced spin shift. A simple calculation using Eq. 3 and KD1 of 1.2 μM shows that the addition of an equimolar concentration of 1-PB to a 10 μM solution of P450eryF-MDCC should result in 70% saturation of the higher-affinity site, whereas the second binding site will remain vacant. Thus, titration of high (≥10 μM) concentration of the enzyme with 1-PB up to a 1:1 enzyme/substrate ratio may be used to probe the changes in the absorbance spectrum of the enzyme caused by the binding of the first substrate molecule. Although the changes in the absorbance spectrum of the enzyme observed in these experiments were extremely small (Fig. 6 a), they revealed a decrease in the amplitude of the Soret band of the low-spin P450eryF concomitant with some broadening of this band upon binding of the first molecule of 1-PB. Attempts to interpret these results in terms of the changes in concentrations of the low-spin and high-spin P450eryF species show no change in the concentration of high-spin P450, but an apparent decrease in concentration of the low-spin P450 (Fig. 6 a, inset). This apparent decrease reflects the changes in the extinction coefficient of the low-spin enzyme caused by the binding of the first molecule of 1-PB. Interestingly, attempts to fit the dependence of these changes on the concentration of substrate to Eq. 3 (Fig. 6 a, inset) yields a dissociation constant of 0.9 ± 0.6 μM, which is consistent with the value of KD1 determined in our FRET experiments (1.2 ± 0.2 μM). However, due to the very small amplitude of the spectral signal and the lack of data for the close-to-saturation region of 1-PB concentrations, the estimate from absorbance spectra is rough and is useful mainly to assess the qualitative consistency of these data with the results of FRET experiments. In contrast, as stated above, the spectral changes observed in the titration of low concentration (0.5–3 μM) of P450eryF-MDCC with concentrations of 1-PB up to 40 μM are indicative of a distinct substrate-induced spin shift (Fig. 6 b).
FIGURE 6.

Changes in the spectra of absorbance of P450eryF-MDCC upon its interactions of 1-PB. (a) A series of the difference spectra derived from the titration of 12.7 μM enzyme with 1-PB. The main panel shows the spectra obtained by subtraction of the spectrum of the substrate-free enzyme from the spectra measured at 2.9 (long-dashed line), 5.8 (medium-dashed line), 8.6 (short-dashed line), and 11 μM (solid line) 1-PB. This experiment was done in a cell with an optical path length of 5 mm. The inset shows the changes in the apparent concentration of the low-spin P450eryF derived from this experiment. The solid line represents the results of the fitting of this data set by Eq. 3 with KD = 0.9 μM. (b) A series of the difference spectra derived from the titration of 2.4 μM enzyme with 1-PB. The main panel shows the spectra obtained by subtraction of the spectrum of the substrate-free enzyme from the spectra measured at 2.6 (solid line), 5.1 (long-dashed line), 7.2 (medium-dashed line), 10 (short-dashed line), 12 (dotted line),14 (dash-dotted line), 18 (dash-double-dotted line), and 23 μM (solid line) 1-PB. The optical path length of the cell used in this experiment was equal to 10 mm. The inset shows the changes in the high-spin fraction of the enzyme derived from this experiment. The solid line represents the results of the fitting of this data set by Eq. 5 (see Discussion) with KD1 = 1.2 and KD2 = 8.5 μM.
Thus, these results confirm our initial conclusion (29) that the binding of the first 1-PB molecule to P450eryF fails to induce any changes in the spin equilibrium of the enzyme. However, recently we demonstrated a prominent effect of ionic strength on the substrate-induced spin shift and cooperativity in P450eryF. To understand the relationship between the effects of ionic strength and the binding of the first substrate molecule to the enzyme it was important to perform experiments at higher salt concentrations. Therefore we compared the effect of ionic strength on the spin equilibrium in substrate-free P450eryF-MDCC withh the enzyme completely saturated with 1-PB, and in P450-MDCC with 1-PB bound only to the high-affinity binding site, which is referred to below as semisaturated enzyme. The effect of salt additions to a concentrated (11–13 μM) equimolar mixture of P450eryF-MDCC with 1-PB is presented in Fig. 7. Although the effect of ionic strength on the spin state of substrate-free enzyme is rather small, addition of KCl to both completely saturated and semisaturated enzyme results in a prominent increase in the high-spin content. At 0.06 M ionic strength (i.e., in 0.1 M Na-Hepes with no KCl added) the content of the high-spin state (Fh) in the semisaturated P450eryF-MDCC is similar to that in the substrate-free enzyme (∼13%). However, increase in ionic strength to 0.8 M increases the value of Fh to 35% in semisaturated enzyme, but only to ∼20% in the substrate-free P450. This observation suggests that the conclusion that the binding of one 1-PB molecule per molecule of P450eryF is insufficient to shift the spin equilibrium of the enzyme is strictly valid only at low ionic strength. A conformational change in P450eryF induced by increased ionic strength makes the first substrate-binding event capable of partial displacement of the spin equilibrium. It is important to note that the shapes of the ionic-strength dependencies of completely saturated (Fig. 7, circles) and semi-saturated enzyme (Fig. 7, squares) are remarkably similar, suggesting that the displacement of the spin equilibrium in both states of the enzyme reflects the same ionic-strength-dependent conformational transition.
FIGURE 7.

Ionic-strength-induced changes in the content of the high-spin state of P450eryF-MDCC in the enzyme saturated with 1-PB (○), and in semisaturated (□) and substrate-free (⋄) enzyme . The resulting curves were obtained by addition of KCl to a 2.6 μM solution of P450eryF-MDCC with no substrate added (⋄) and to the mixtures of P450eryF-MDCC with 1-PB taken at concentrations of 2.9 and 40 μM (○) and 11 and 11 μM, respectively. Solid lines represent the results of the fitting of these data sets by Eq. 1.
DISCUSSION
In this study, we describe the P450eryF S93C/C154S mutant, which reproduces the main properties of the wild-type enzyme and can be modified by CPM, MDCC, and mBBr with virtually no effect on the interactions with 1-PB. Furthermore, the proteins exhibited an extensive FRET to the label upon the binding of 1-PB. The main advantage of FRET from 1-PB to a fluorescent probe as compared to the heme is that the formation of the enzyme-substrate complex is accompanied by a simultaneous decrease in 1-PB fluorescence and increase in the fluorescence of the acceptor fluorophore. This highly distinctive signal considerably broadens the applicability of the method, especially in cases where the intensity of fluorescence of the pyrene fluorophore might be affected by excimerization of 1-PB or changes in the turbidity or refractivity of the medium.
The labeled S93C/C154S preparations, and P450eryF-MDCC in particular, provide an invaluable tool for resolution of two substrate-binding events by FRET and determination of the values of KD1 and KD2 for substrate binding to P450eryF. To our knowledge, this has not been accomplished previously with any allosteric P450. This important result allows us to delineate a mechanistic model of the cooperativity in substrate binding to P450eryF. The interactions of substrate with two binding sites in cytochrome P450 and subsequent spin transitions can be represented schematically as follows:
 . .
|
(4) |
Here E stands for the substrate-free enzyme (P450eryF); the complexes of the substrate bound at each of the two sites are designated by ES and SE; SES stands for the ternary complex with both binding sites occupied. KD1, KD2, KD3, and KD4 are the dissociation constants. The diagonal arrows show spin transitions, and ls and hs correspond to the low- and high-spin P450, respectively. 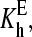
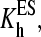
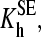 and
and 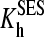 are the constants of spin equilibrium. To simplify the analysis, we introduced an earlier model (29) that suggests independent binding of two substrate molecules at two binding sites. For simplicity we initially hypothesized that substrate binding at each site has no effect on the interactions at the second one, so that KD1 = KD3, KD2 = KD4. According to this simplification, the cooperativity revealed in the substrate-induced spin shift is caused solely by the fact that the displacement of the spin equilibrium is observed in the ternary complex only. Our further studies showed that, although this scheme adequately describes the behavior of the enzyme at low ionic strength, it fails to fit the data obtained at increased salt concentration (12). Furthermore, even in the cases where the experimental data satisfactorily obey the equation derived from this model, fitting of the titration curves often gives a value of KD2 considerably lower than the value of KD1 determined by FRET, which is internally inconsistent (12). This observation suggests that the binding of the first molecule of 1-PB to the enzyme may have an important effect on substrate interactions at the second site, in line with much of the current thinking about P450 cooperativity.
are the constants of spin equilibrium. To simplify the analysis, we introduced an earlier model (29) that suggests independent binding of two substrate molecules at two binding sites. For simplicity we initially hypothesized that substrate binding at each site has no effect on the interactions at the second one, so that KD1 = KD3, KD2 = KD4. According to this simplification, the cooperativity revealed in the substrate-induced spin shift is caused solely by the fact that the displacement of the spin equilibrium is observed in the ternary complex only. Our further studies showed that, although this scheme adequately describes the behavior of the enzyme at low ionic strength, it fails to fit the data obtained at increased salt concentration (12). Furthermore, even in the cases where the experimental data satisfactorily obey the equation derived from this model, fitting of the titration curves often gives a value of KD2 considerably lower than the value of KD1 determined by FRET, which is internally inconsistent (12). This observation suggests that the binding of the first molecule of 1-PB to the enzyme may have an important effect on substrate interactions at the second site, in line with much of the current thinking about P450 cooperativity.
Another extreme case of a two-binding-site model is sequential binding, where the binding at the second binding site is impossible without saturation of the higher-affinity effector site. This model suggests that KD4 ≫ KD1 and KD3 ≫ KD2, so that the pathways leading to the formation of SE may be neglected. To simplify the analysis further, we proceeded with the assumption that the modulation of the spin equilibrium is observed in the ternary complex only and 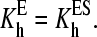 In the most general representation applicable at comparable concentrations of the enzyme and the substrate, the analysis of Scheme 4 with the above simplifications gives the following relationship between the steady-state concentration of the ternary complex ([SES]), that of the complex ES ([ES]), and the total concentration of the enzyme and substrate ([E]0 and [S0], respectively):
In the most general representation applicable at comparable concentrations of the enzyme and the substrate, the analysis of Scheme 4 with the above simplifications gives the following relationship between the steady-state concentration of the ternary complex ([SES]), that of the complex ES ([ES]), and the total concentration of the enzyme and substrate ([E]0 and [S0], respectively):
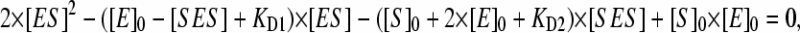 |
where
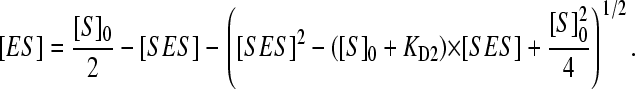 |
(5) |
Although analytical solution of this equation for [SES] is intricate, it can be easily done by a numerical means. To fit the experimental data to the above relationship, we used the nonlinear least-square-fitting module of our SpectraLab program (38).
Using this equation to fit the experimental data on the 1-PB-induced spin shift and fixing the value of KD1 to that found by FRET, we can solve for KD2 to probe the consistency of the above model. Using this approach, we found that Eq. 5 adequately fits all curves of substrate-induced spin shift obtained in this study. An example of such fitting is shown in the inset in Fig. 6 b. Crucial evidence of the applicability of this model is given by the fact that the fitting of the curves obtained with different concentrations of P450eryF-MDCC varying in the range of 0.6–13 μM give similar values of KD2. In these experiments, the mean value of KD2 was estimated to be 10.8 ± 1.7 μM, which is consistent with the value of 9.4 ± 0.8 μM found in our FRET dilution experiments. It is important, that the above equation also provides good fitting for all titration experiments at increased ionic strength obtained in our previous study (12) (data not shown). In this case the value of KD2 is always higher than the value of KD1, which eliminates the inconsistency in our initial simplification. Therefore, we may conclude that the binding of 1-PB at the higher-affinity (effector) binding site radically increases the affinity of the second binding site for this substrate. Although the sequential binding model adequately describes all data obtained in our experiments, the real situation may be intermediate between the two suggested models, being closer, however, to the sequential binding scheme.
Studying the relationship between substrate binding at each of the sites and the substrate-induced spin shift we found that the binding of the first 1-PB molecule has no effect on the spin equilibrium of the enzyme at low ionic strength. Instead, these interactions induce a slight but clearly detectable change in the absorbance spectrum of P450eryF, which is reflected in a decrease in the amplitude and broadening of the Soret band of the low-spin enzyme. These changes apparently reflect changes in the conformation and/or degree of hydration of the heme pocket caused by the binding of the first 1-PB molecule. However, this conclusion appears to be strictly correct only at low ionic strength of the media. As shown in Fig. 7, an increase in salt concentration results in considerable displacement of the spin equilibrium of semisaturated enzyme toward the high-spin state. Thus, an ionic-strength-induced conformational transition in the enzyme makes the first substrate-binding event capable of modulation of the spin equilibrium. This may happen either by promoting the formation of the ES complex (thus compromising the sequential binding scheme), or by making the formation of the SE complex sufficient to modulate the spin equilibrium. Therefore, the decrease in cooperativity observed at increased ionic strength (12) is partially due to the fact that the binding curves for the substrate-induced spin shift at high ionic strength represent the sum of the concentration of the SES, SE, and/or ES complexes.
The above analysis strongly supports the point of view that the phenomenon of cooperativity in P450eryF is based on an effector-induced conformational transition and therefore represents a case of true allostery. In our recent study (12), we presented evidence that this mechanism is based on the dynamics of ion tethers in the vicinity of Cys-154, which determine the conformation of a functionally important cleft formed by α-helices D, E, F, and G and the respective interhelical loops (Fig. 8). Our results obtained with P450eryF mutants in this study allow us to further refine this concept. The clear positive linear correlation between the values of Kion and S50 (Fig. 3 a) suggests that an increase in the strength of the strategic ion tethers around Cys-154 (Fig. 8) impedes the interactions of P450eryF with 1-PB. Furthermore, the changes in the value of Kspin(0) are strictly correlated with those of the Hill coefficient (Fig. 3 b). With the sole exception of C154I, any replacement of residue 154 that promotes the formation of the high-spin state of the substrate-saturated P450eryF at zero ionic strength results in decreased cooperativity of the enzyme with 1-PB.
FIGURE 8.

Models of part of the crystal structures of the complexes of P450eryF with 6-DEB (left) and ketoconazole (right) (28) showing putative charge-pairing contacts in the vicinity of Cys-154, the side chain of which is shown in yellow as a space-filling model. Space filling models also designate Ser-93 (magenta), heme (gray), and substrate (either 6-DEB or ketoconazole) (pale green). Red and green stick models show acidic and basic amino acid residues participating in salt links.
To delineate the relationship between the substrate-induced spin shift and cooperativity and the parameters of residue 154 we analyzed the correlation of S50, n, Kion, and Kspin(0) with such physicochemical properties of this residue as bulkiness, polarity, and hydrophobicity given by several different scales. The correlation was characterized by values of Pearson product-moment correlation coefficient (r). Among the probed scales the best correlation was found between polarity characterized by the free energy of transfer of the side chain from cyclohexane to water (ΔG°w) (43) and Kspin(0) (r = -0.98). Similarly, considerable correlation was found between ΔG°w and the Hill coefficient for the set of all considered P450eryF mutants except C154I (r = 0.96). ΔG°w also reveals a noticeable correlation with the values of S50 and Kion (r = 0.76 in both cases). As the value of ΔG° w characterizes the ability of the side chain to form hydrogen bonds and to interact with water, we may infer that the conformational transition in P450eryF caused by the binding of 1-PB to the effector-binding site promotes a substrate-induced spin shift by a mechanism that implicates the formation of a hydrogen bond involving Cys-154 and/or relocation of some protein-bound water molecule(s) in the proximity of this residue. This change is facilitated when a more polar residue replaces Cys-154, as in C154S. Hence, the requirement for the binding of the substrate to both effector and substrate-binding sites for a substrate-induced spin shift in P450eryF is compromised and cooperativity is decreased. Only C154I deviates from this rule. The cooperativity displayed by this mutant is unusually low, despite the fact that ΔG°w of isoleucine is very high, and, consequently, the mutant exhibits a very low value of Kspin(0). This result might be explained by a specific effect of Cys-154 replacement by the bulky and hydrophobic isoleucine residue on the structure of the enzyme (12).
In addition, the values of S50, and Kion reveal a correlation with the bulkiness of the side chain (44) of residue 154 (r = 0.81 and r = 0.87, respectively). Thus, the highest affinity of P450eryF to 1-PB is observed with the mutants bearing small and polar residues at this position. Hence it appears that the binding of 1-PB to the substrate-binding site of P450eryF is facilitated by increased conformational mobility in the vicinity of Cys-154, which is required for a substrate-induced conformational transition of the enzyme.
Assessment of the exact structural relevance of the above inferences is complicated by the lack of a substrate-free P450eryF structure. However, some idea on conformational dynamics of the enzyme in the proximity of Cys-154 may be derived from the comparative analysis of the available x-ray structures of P450eryF complexes with different substrates—deoxyerythronolide B (6-DEB, Protein Data Bank entries 1oxa and 1jio) (28,45), ketoconazole (entry 1jip) (28), 9-aminophenanthrene (1egy), and androstenedione (1eup) (46). In Fig. 8, we compare the structure of this region in P450eryF complexes with its natural substrate 6-deoxyerythronolide B, 6-DEB (1jio) and with ketoconazole (1jip).
In all known structures of P450eryF, the distance between the sulfur atom of Cys-154 and the epsilon nitrogen atom of Arg-164 is within 3.5–4.0 Å. This observation suggests formation of a S··NH hydrogen bond, the typical length of which is known to be 3.1–3.9 Å (47–49). The latter arginine residue participates in a salt link only in 1jio, forming a salt bridge with Asp-160. In all other structures, Asp-160 interacts with Arg-202, whereas in 1jio this important tether between the E/F loop and the G-helix is formed by the Glu-161/Arg-202 pair (Fig. 8). As a result of this rearrangement, the conformation of the E/F loop and the position of α-helices F and G relative to helices D and E in 1jio differ from those observed in other structures (Fig. 8). It is important to note that the structures of P450eryF complexes with 6-DEB (1jio and 1oxa) are the only ones displaying a water molecule at the distal side of the heme moiety. This water, which is 2.7 Å from the heme plane and 3.6 Å from the iron atom in the 1jio structure, may potentially serve as a heme ligand in the low-spin state of the enzyme-substrate complex. Thus, taking into account that the polarity of residue 154 correlates with both cooperativity and position of spin equilibrium in P450eryF, we have good grounds to believe that the Cys-154-Arg-164 H-bond has strategic importance, and the dynamics of D160-R164, E161-R202, and D160-R202 salt links is fundamental for the mechanisms of spin transition and cooperativity in P450eryF.
Taken together, our results strongly support the point of view that the cooperativity in cytochrome P450eryF represents a classical example of allostery, which involves a conformational transition of the enzyme induced by the binding of the effector. Furthermore, the above analysis emphasizes the importance of further exploration of the dynamics of salt bridges and protein-bound water in P450eryF interactions with substrates and allosteric effectors. We believe that further study of the molecular mechanism of the ligand-induced conformational transition in P450eryF by our new FRET-based approach in combination with site-directed mutagenesis of salt-link-forming residues in proximity of Cys-154 (D160, E161, R164, and R202) and the pressure-perturbation technique will provide us with further details crucial to unraveling the molecular mechanisms and understanding the functional significance of the cooperativity in cytochromes P450.
Acknowledgments
The authors are grateful to Ms. You-Qun He for her help in construction of P450eryF mutants.
This research was supported in part by research grants H-1458 from the Robert A. Welch Foundation, and GM54995 and Center grant ES06676 from the National Institutes of Health.
Alexandra E. Botchkareva's present address is Division of Chemical Enzymology, Dept. of Chemistry, M. V. Lomonosov State University, Moscow, 119899 Russia.
References
- 1.Ekins, S., D. M. Stresser, and J. A. Williams. 2003. In vitro and pharmacophore insights into CYP3A enzymes. Trends Pharmacol. Sci. 24:161–166. [DOI] [PubMed] [Google Scholar]
- 2.Guengerich, F. P. 1999. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol. 39:1–17. [DOI] [PubMed] [Google Scholar]
- 3.Lin, Y., P. Lu, C. Tang, Q. Mei, G. Sandig, A. D. Rodrigues, T. H. Rushmore, and M. Shou. 2001. Substrate inhibition kinetics for cytochrome P450-catalyzed reactions. Drug Metab. Dispos 29:368–374. [PubMed] [Google Scholar]
- 4.Tang, W., and R. A. Stearns. 2001. Heterotropic cooperativity of cytochrome P450 3A4 and potential drug-drug interactions. Curr. Drug Metab. 2:185–198. [DOI] [PubMed] [Google Scholar]
- 5.Shou, M., J. Grogan, J. A. Mancewicz, K. W. Krausz, F. J. Gonzalez, H. V. Gelboin, and K. R. Korzekwa. 1994. Activation of CYP3A4 - evidence for the simultaneous binding of 2 substrates. Biochemistry. 33:6450–6455. [DOI] [PubMed] [Google Scholar]
- 6.Korzekwa, K. R., N. Krishnamachary, M. Shou, A. Ogai, R. A. Parise, A. E. Rettie, F. J. Gonzalez, and T. S. Tracy. 1998. Evaluation of atypical cytochrome P450 kinetics with two-substrate models: evidence that multiple substrates can simultaneously bind to cytochrome P450 active sites. Biochemistry. 37:4137–4147. [DOI] [PubMed] [Google Scholar]
- 7.Ueng, Y. F., T. Kuwabara, Y. J. Chun, and F. P. Guengerich. 1997. Cooperativity in oxidations catalyzed by cytochrome P450 3A4. Biochemistry. 36:370–381. [DOI] [PubMed] [Google Scholar]
- 8.Harlow, G. R., and J. R. Halpert. 1998. Analysis of human cytochrome P450 3A4 cooperativity: construction and characterization of a site-directed mutant that displays hyperbolic steroid hydroxylation kinetics. Proc. Natl. Acad. Sci. USA. 95:6636–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schrag, M. L., and L. C. Wienkers. 2000. Topological alteration of the CYP3A4 active site by the divalent cation Mg2+. Drug Metab. Dispos. 28:1198–1201. [PubMed] [Google Scholar]
- 10.Atkins, W. M., R. W. Wang, and A. Y. H. Lu. 2001. Allosteric behavior in cytochrome P450-dependent in vitro drug-drug interactions: A prospective based on conformational dynamics. Chem. Res. Toxicol. 14:338–347. [DOI] [PubMed] [Google Scholar]
- 11.Davydov, D. R., J. R. Halpert, J. P. Renaud, and G. Hui Bon Hoa. 2003. Conformational heterogeneity of cytochrome P450 3A4 revealed by high pressure spectroscopy. Biochem. Biophys. Res. Commun. 312:121–130. [DOI] [PubMed] [Google Scholar]
- 12.Davydov, D. R., A. E. Botchkareva, S. Kumar, Y. Q. He, and J. R. Halpert. 2004. An electrostatically driven conformational transition is involved in the mechanisms of substrate binding and cooperativity in cytochrome P450eryF. Biochemistry. 43:6475–6485. [DOI] [PubMed] [Google Scholar]
- 13.Schoch, G. A., J. K. Yano, M. R. Wester, K. J. Griffin, C. D. Stout, and E. F. Johnson. 2004. Structure of human microsomal cytochrome P4502C8 - Evidence for a peripheral fatty acid binding site. J. Biol. Chem. 279:9497–9503. [DOI] [PubMed] [Google Scholar]
- 14.Williams, P. A., J. Cosme, D. M. Vinkovic, A. Ward, H. C. Angove, P. J. Day, C. Vonrhein, I. J. Tickle, and H. Jhoti. 2004. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 305:683–686. [DOI] [PubMed] [Google Scholar]
- 15.Hosea, N. A., G. P. Miller, and F. P. Guengerich. 2000. Elucidation of distinct ligand binding sites for cytochrome P450 3A4. Biochemistry. 39:5929–5939. [DOI] [PubMed] [Google Scholar]
- 16.Boek-Dohalska, L., P. Hodek, M. Sulc, and M. Stiborova. 2001. alpha-naphthoflavone acts as activator and reversible or irreversible inhibitor of rabbit microsomal CYP3A6. Chem. Biol. Interact. 138:85–106. [DOI] [PubMed] [Google Scholar]
- 17.Galetin, A., S. E. Clarke, and J. B. Houston. 2003. Multisite kinetic analysis of interactions between prototypical CYP3A4 subgroup substrates: Midazolam, testosterone, and nifedipine. Drug Metab. Dispos. 31:1108–1116. [DOI] [PubMed] [Google Scholar]
- 18.He, Y. A., F. Roussel, and J. R. Halpert. 2003. Analysis of homotropic and heterotropic cooperativity of diazepam oxidation. Arch. Biochem. Biophys. 409:92–101. [DOI] [PubMed] [Google Scholar]
- 19.Koley, A. P., J. T. M. Buters, R. C. Robinson, A. Markowitz, and F. K. Friedman. 1997. Differential mechanisms of cytochrome P450 inhibition and activation by alpha-naphthoflavone. J. Biol. Chem. 272:3149–3152. [DOI] [PubMed] [Google Scholar]
- 20.Koley, A. P., R. C. Robinson, A. Markowitz, and F. K. Friedman. 1997. Drug-drug interactions: effect of quinidine on nifedipine binding to human cytochrome P450 3A4. Biochem. Pharmacol. 53:455–460. [DOI] [PubMed] [Google Scholar]
- 21.Koley, A. P., J. T. M. Buters, R. C. Robinson, A. Markowitz, and F. K. Friedman. 1996. Cytochrome P450 conformation and substrate interactions as probed by CO binding kinetics. Biochimie. 78:706–713. [DOI] [PubMed] [Google Scholar]
- 22.Reed, J. R., and P. F. Hollenberg. 2003. Comparison of substrate metabolism by cytochromes P4502B1, 2B4, and 2B6: relationship of heme spin state, catalysis, and the effects of cytochrome b5. J. Inorg. Biochem. 93:152–160. [DOI] [PubMed] [Google Scholar]
- 23.Hlavica, P., and D. F. V. Lewis. 2001. Allosteric phenomena in cytochrome P450-catalyzed monooxygenations. Eur. J. Biochem. 268:4817–4832. [DOI] [PubMed] [Google Scholar]
- 24.Blanck, J., O. Ristau, A. A. Zhukov, A. I. Archakov, H. Rein, and K. Ruckpaul. 1991. Cytochrome P-450 spin state and leakiness of the monooxygenase pathway. Xenobiotica. 21:121–135. [DOI] [PubMed] [Google Scholar]
- 25.Rein, H., O. Ristau, R. Misselwitz, E. Buder, and K. Ruckpaul. 1979. The importance of the spin equilibrium in cytochrome P-450 for the reduction rate of the heme iron. Acta Biol. Med. Ger. 38:187–200. [PubMed] [Google Scholar]
- 26.Sligar, S. G., D. L. Cinti, G. G. Gibson, and J. B. Schenkman. 1979. Spin state control of the hepatic cytochrome P450 redox potential. Biochem. Biophys. Res. Commun. 90:925–932. [DOI] [PubMed] [Google Scholar]
- 27.Sligar, S. G. 1976. Coupling of spin, substrate, and redox equilibria in cytochrome P450. Biochemistry. 15:5399–5406. [DOI] [PubMed] [Google Scholar]
- 28.Cupp-Vickery, J. R., C. Garcia, A. Hofacre, and K. McGee-Estrada. 2001. Ketoconazole-induced conformational changes in the active site of cytochrome P450eryF. J. Mol. Biol. 311:101–110. [DOI] [PubMed] [Google Scholar]
- 29.Davydov, D. R., S. Kumar, and J. R. Halpert. 2002. Allosteric mechanisms in P450eryF probed with 1-pyrenebutanol, a novel fluorescent substrate. Biochem. Biophys. Res. Commun. 294:806–812. [DOI] [PubMed] [Google Scholar]
- 30.Khan, K. K., H. Liu, and J. R. Halpert. 2003. Homotropic versus heterotropic cooperativity of cytochrome P450eryF: A substrate oxidation and spectral titration study. Drug Metab. Dispos. 31:356–359. [DOI] [PubMed] [Google Scholar]
- 31.Khan, K. K., and J. Halpert. 2002. 7-benzyloxyquinoline oxidation by P450eryF A245T: Finding of a new fluorescent substrate probe. Chem. Res. Toxicol. 15:806–814. [DOI] [PubMed] [Google Scholar]
- 32.Xiang, H., R. A. Tschirret-Guth, and P. R. Ortiz De Montellano. 2000. An A245T mutation conveys on cytochrome P450eryF the ability to oxidize alternative substrates. J. Biol. Chem. 275:35999–36006. [DOI] [PubMed] [Google Scholar]
- 33.Baas, B. J., I. G. Denisov, and S. G. Sligar. 2004. Homotropic cooperativity of monomeric cytochrome P450 3A4 in a nanoscale native bilayer environment. Arch. Biochem. Biophys. 430:218–228. [DOI] [PubMed] [Google Scholar]
- 34.Yoon, M. Y., A. P. Campbell, and W. M. Atkins. 2004. “Allosterism” in the elementary steps of the cytochrome P450 reaction cycle. Drug Metab. Rev. 36:219–230. [DOI] [PubMed] [Google Scholar]
- 35.Davydov, D. R., G. Hui Bon Hoa, and J. A. Peterson. 1999. Dynamics of protein-bound water in the heme domain of P450BM3 studied by high-pressure spectroscopy: Comparison with P450cam and P450 2B4. Biochemistry. 38:751–761. [DOI] [PubMed] [Google Scholar]
- 36.Hui Bon Hoa, G., M. A. Mclean, and S. G. Sligar. 2002. High pressure, a tool for exploring heme protein active sites. Biochim. Biophys. Acta. 1595:297–308. [DOI] [PubMed] [Google Scholar]
- 37.Kornblatt, J. A., and M. J. Kornblatt. 2002. The effects of osmotic and hydrostatic pressures on macromolecular systems. Biochim. Biophys. Acta. 1595:30–47. [DOI] [PubMed] [Google Scholar]
- 38.Davydov, D. R., E. Deprez, G. Hui Bon Hoa, T. V. Knyushko, G. P. Kuznetsova, Y. M. Koen, and A. I. Archakov. 1995. High-pressure-induced transitions in microsomal cytochrome P450 2B4 in solution—evidence for conformational inhomogeneity in the oligomers. Arch. Biochem. Biophys. 320:330–344. [DOI] [PubMed] [Google Scholar]
- 39.Molecular Probes, Inc. 2003. Handbook of Fluorescent Probes and Research Products. Web edition: http://www.probes.com/handbook/. Molecular Probes, Eugene, OR.
- 40.Renaud, J. P., D. R. Davydov, K. P. M. Heirwegh, D. Mansuy, and G. Hui Bon Hoa. 1996. Thermodynamic studies of substrate binding and spin transitions in human cytochrome P-450 3A4 expressed in yeast microsomes. Biochem. J. 319:675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peyser, Y. M., K. Ajtai, T. P. Burghardt, and A. Muhlrad. 2001. Effect of ionic strength on the conformation of myosin subfragment 1-nucleotide complexes. Biophys. J. 81:1101–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Segel, I. H. 1975. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. Wiley-Interscience, New York.
- 43.Radzicka, A., and R. Wolfenden. 1988. Comparing the polarities of the amino acids: Side-chain distribution coefficients between the vapor phase, cyclohexane, 1-octanol, and neutral aqueous solution. Biochemistry. 27:1664–1670. [Google Scholar]
- 44.Zimmerman, J. M., N. Eliezer, and R. Simha. 1968. The characterization of amino acid sequences in proteins by statistical methods. J. Theor. Biol. 21:170–201. [DOI] [PubMed] [Google Scholar]
- 45.Cupp-Vickery, J. R., and T. L. Poulos. 1995. Structure of cytochrome P450eryF involved in erythromycin biosynthesis. Nat. Struct. Biol. 2:144–153. [DOI] [PubMed] [Google Scholar]
- 46.Cupp-Vickery, J., R. Anderson, and Z. Hatziris. 2000. Crystal structures of ligand complexes of P450eryF exhibiting homotropic cooperativity. Proc. Natl. Acad. Sci. USA. 97:3050–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krepps, M. K., S. Parkin, and D. A. Atwood. 2001. Hydrogen bonding with sulfur. Cryst. Growth Design. 1:291–297. [Google Scholar]
- 48.Pal, D., and P. Chakrabarti. 1999. Different types of interactions involving cysteine sulfhydryl group in proteins. J. Biomol. Struct. Dyn. 15:1059–1072. [DOI] [PubMed] [Google Scholar]
- 49.Adman, E., K. D. Watenpaugh, and L. H. Jensen. 1975. NH…S hydrogen-bonds in Peptococcus aerogenes ferredoxin, Clostridium pasteurianum rubredoxin, and Chromatium high potential iron protein. Proc. Natl. Acad. Sci. USA. 72:4854–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]