

Abstract
Human transthyretin (TTR) is an amyloidogenic protein. The pathway of TTR amyloid formation has been proposed based on lines of evidence: TTR tetramer first dissociates into native monomers, which is shown to be a rate-limiting step in the formation of fibrils. Subsequently, the monomeric species partially unfold to form the aggregation intermediates. Once such intermediates are formed, the following self-assembly process is a downhill polymerization. Hence, tertiary structural changes within the monomers after the dissociation are essential for the amyloid formation. These tertiary structural changes can be facilitated by partial denaturation. To probe the conformational changes under the partially denaturing conditions, five independent trajectories were collected for the wild-type (WT) and its pathogenic variants at 300 and 350 K, resulting in simulations that totaled 59 ns. Under these conditions, L55P variant is more labile than the wild-type and V30M variant. We have observed that the D strand of WT-TTR is trapped in two local minima: the native conformation and the amyloidogenic fold that resembles the surface loop of residues 54–55 of L55P variant. In the tetrameric state, the F strand is bent with large separations at the F-F′ interface. This strand becomes flatter in the monomeric state, which may facilitate the formation of new F-F′ interface with possible prolonged hydrogen bonds and/or shift in β-strand register in the fibril state. During the unfolding process, the anticorrelated motion between the strands H and G as well as the strands H and A pulls the H strand out of the inner sheet plane, leading to a more twisted inner sheet. Our simulation has provided important detailed structural information about the partially unfolded state of TTR that may be related to the amyloidogenic intermediates.
INTRODUCTION
Accumulation of amyloid fibrils in tissues or extracellular matrix is the hallmark of amyloid diseases such as Alzheimer's disease and the systemic amyloidoses (for example, immunoglobulin-light-chain diseases and the familial amyloid polyneuropathies that are associated with transthyretin). Up to date, ∼20 human proteins have been found to form amyloid fibrils in vivo (1). These proteins can be divided into two categories based on whether the amyloid fibrils can be converted from a full-length amyloidogenic protein or a fragment of a large precursor protein. For example, Alzheimer's fibrils are formed from amyloid-β peptide, a fragment of amyloid precursor protein; whereas, human transthyretin (TTR) belongs to the category of full-length amyloidogenic protein; however, this does not exclude the fact that TTR fibrils also contain fragments of protein (2). It is likely that these two categories follow different pathways of fibril formation.
In this work, we focus on TTR, which is a plasma protein responsible for the transportation of thyroid hormone. It also binds to retinol-binding protein that in turn associates with vitamin A. The structures of the wild-type and various amyloidogenic single-site mutants have been determined by high-resolution x-ray crystallography (3–5). The native state of TTR is a homotetramer with eight β-strands organized into two sheets, giving rise to a β-sandwich (3) (Fig. 1 a). The eight β-strands are named from A to H and arranged into inner (DAGH) sheet and outer (CBEF) sheet. Two monomeric units form a dimer via extensive hydrogen bonds between the two adjacent H (residues Ser-115–Thr-123) strands and F (residues Ala-91–Ala-97) strands from each monomer (Fig. 1 b). There are five main-chain-main-chain hydrogen bonds between the adjacent H strands, whereas the two F strands are separated more widely, as a result, only two hydrogen bonds (between Glu-89 and Val-94) are formed directly through the main-chain interactions. The remaining hydrogen bonding interactions involve water bridges. Two dimers are related by a crystallographic twofold axis to give a tetramer. A central channel wrapped around by the DAGH (inner) sheets runs through the center of the tetramer and accommodates thyroxin molecules.
FIGURE 1.

(a) Three-dimensional structure of WT-TTR (PDB entry, 1DVQ) in the dimeric form. The eight β-strands are named from A to H. The inner sheet (DAGH) is shown in the front whereas the outer sheet (CBEF) is in the back. (b) Schematic representation of the main-chain hydrogen bonds within WT-TTR monomer as well as at the monomer-monomer interface. The two mutation sites, L55 and V30, are in bold. Arrows point from the hydrogen bond donor to the acceptor.
The pathway of TTR amyloid formation has been proposed based on lines of evidence: TTR tetramer first dissociates into native monomers, which is shown to be a rate-limiting step in the formation of fibrils (6–8). Subsequently, the monomeric species partially unfold to form the aggregation intermediates. Once such intermediates are formed, the following self-assembly process is a downhill polymerization (9). Tetramer dissociation into monomers is necessary but not sufficient to initiate fibril formation because native monomers are nonamyloidogenic unless it is partially denatured (10). Therefore, further tertiary structural changes within the monomers are required. These tertiary structural changes can be facilitated either by partial denaturation (e.g., lowering the pH or the dielectric constant of the medium) (8,11) or by single-point mutation. At pH 7.5 and 37°C, both L55P-TTR (Leu-55→Pro) and V30M-TTR (Val-30→Met) form amyloid protofibrils after two months of incubation, however, V30M-TTR exhibits much smaller amount of protofibrils. By contrast, the wild-type is stable and nonamyloidogenic under the same conditions (12).
The comparison of all TTR crystal structures available in the protein data bank including amyloidogenic variants has led to the conclusion that the structural discrepancies between the wild-type and its variants are nonsignificant (13). However, our molecular dynamics (MD) simulations have revealed substantial differences in the dynamic features of native tetrameric state between the wild-type and its pathogenic variants (14). Our results have shown that the D strand is an intrinsically unstable site, which is consistent with the observation of recent x-ray data that the D strand is trapped in two discrete conformations (15,16). The L55P mutation occurred in the D strand results in not only significant local structural changes but also large global conformational changes in the inner sheets that wrap around the central channel, leading to the opening/closing fluctuation of the channel (14). Our previous simulations on the native monomers under physiological conditions have demonstrated that the secondary structural integrity of the inner sheet is disrupted starting from the “edge strand”, the D strand (17). This may lead to the initiation of the fibril formation. To further explore tertiary structural changes within monomers under partially denaturing condition, we performed multiple MD simulations using generalized Born model (18) at 350 K at neutral pH. These simulations can model localized temperature fluctuations in vivo that may occur under free-radical oxidation or inflammation. The purpose of our simulation is to capture the initial steps of conformational change that may trigger the amyloidogenicity under neutral pH. Furthermore, to completely understand the mutation-dependent differential amyloidogenesis, one needs to investigate the influence of mutation not only on the tetrameric native state but also the monomeric state. In other words, besides the thermodynamic and/or kinetic destabilization of TTR tetramers by single-point mutations, the increased aggregation of mutants may also result from mutations that either slow down folding or accelerate unfolding. Based on the spectroscopic analyses and proteolysis sensitivity studies (19,20), the monomeric intermediates in the amyloid formation are in a partially unfolded state with significant native-like residual structure. Full denaturation (for example, 2 ≤ pH ≤ 3.5) leads to a molten globular state instead of aggregation (19). MD simulations can complement the experimental data to provide detailed structural information for components of the partially unfolded ensemble. Simulations at mildly elevated temperature are appropriate to enhance the sampling in the vicinity of the native-like state. When we performed the unfolding simulations under severer denaturing conditions, 400 K, both wild-type and amyloidogenic variants unfold so quickly that we could not obtain any insights into the mutation-dependent amyloidogenic potential (data are not shown). Therefore, in this work, we focus on the simulations at 300 and 350 K. The simulations at 300 K not only serve as controls, but also provide important information about the flexibility under physiological conditions.
COMPUTATIONAL METHODS
Protocols of MD/GB simulations
The starting point of each simulation is the monomeric form extracted from the x-ray structures (1DVQ, wild-type (21); 1TTC, V30M-TTR (4); and 5TTR, L55P-TTR(5)). The AMBER program (22) and modified parm99 force field (23) were employed to perform the MD simulations. It was found that the refitted main-chain dihedral angles gave better agreement with the ab initio relative energies of alanine tetrapeptide conformation (23). To enhance sampling and speed up the simulations, we used the generalized Born (GB) solvation model implemented in AMBER 7 to describe the aqueous environment (24). The GB model has shown promising performance in various biomolecular systems (24–27). Because GB describes the instantaneous solvent dielectric response, it eliminates the time-consuming step of water reorganization. In addition, due to the lack of viscosity associated with explicit water molecules, the simulation may quickly converge or explore more conformation space. The drawback of this model is that the simulated events take place on a faster timescale than the explicit water MD simulation. Although the water friction is spatially anisotropic, the unfolding trajectories of chymotrypsin inhibitor 2 with implicit solvation model without friction term showed similar behaviors to the simulations with explicit water model (28) because the low (free) energy intermediate states had high populations no matter which solvation model was used. Therefore, implicit solvent is a useful way to study dynamics. The modified parm99 force field combined with the newly optimized GB parameters has been tested on various protein systems (24). Following the designation of Case and co-workers, we used parm99MOD2, GBOBC(I), mBondi2 model (24). After 1000-step minimization, the system was heated slowly from zero to 300 K with the rate of 100 K/5 ps. Additional 5 ps were used to heat the system from 300 to 350K. The time step was 1 fs. The temperature of the system was regulated using the Berendsen coupling algorithm (29) with a coupling constant of 1.0 ps. The SHAKE algorithm was used to constrain the bond length involving H atoms (30). The long-range interaction was truncated at 15 Å. A restraint with force constant equal to 5.0 kcal/(mol Å2) was added to ensure that the protein conformation is close to the x-ray structure during the heating process. During the equilibration, we gradually reduced the restraint force constant to 3.0 kcal/(mol Å2), 1.0 kcal/(mol Å2), and 0.1 kcal/(mol Å2) every 50 ps. After then, a trajectory was collected for 1.8 ns without restraints. The overall center-of-mass translational and rotational motions were removed every 50 steps. Five independent trajectories were collected for each of the wild-type and mutant proteins at 300 and 350 K, resulting in simulations that totaled 54 ns.
Methods of the trajectory analysis
Unfolding yield
We analyzed the secondary structure using the DSSP method (31). The progression of partially unfolding was monitored by the loss of secondary structure content in the inner sheet, DAGH sheet. If a given snapshot loses >40% secondary structure content in the inner sheet, we consider it is in a partially unfolded state. The outer sheet, CBEF sheet, maintains >80% secondary structure content during all of the simulations at 350 K; therefore, we did not include this region for the purpose of monitoring the progression of partially unfolding. We defined unfolding yield as follows 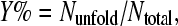 where Ntotal is the total number of snapshots of a given trajectory and Nunfold is the number of snapshots in the partially unfolded states.
where Ntotal is the total number of snapshots of a given trajectory and Nunfold is the number of snapshots in the partially unfolded states.
β-sheet bending and twisting
The local bend angle is defined by the angle between the major axes (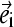 ) of strand residues from i to i + 2 and residues from i + 1 to i + 3 (32). Each residue was represented by three main-chain atoms: N, Cα, and C. Principal component analysis (PCA) (33) was applied to obtain the major axis of three consecutive residues. This major axis represents the best line through the main-chain strand atoms from the N- to C-terminus. To describe the relative orientation of the inner and outer sheet, we employed PCA to obtain the principal plane of each sheet. The plane is defined by the major axis and the minor axis orthogonal to the former. The major axis of the inner sheet was projected to the principal plane of the outer sheet so that the angle formed by the major axis of the outer sheet and the projected axis of the other sheet describes the relative orientation between these sheets. The main-chain C, Cα, and N atoms are included for this calculation. We measured the sheet twisting using the interstrand pair method (34). Because CBEF sheet is antiparallel, for both hydrogen-bonded and nonhydrogen-bonded pairs, the twist angle is defined as the angle formed by the backbone direction vectors of one interstrand pair. For example, residues i and j are located in two adjacent strands and form a pair of main-chain hydrogen bonds. The backbone direction vector of residue i is defined as a vector starting at the middle point of the peptide bond between residues i − 1 and i and pointing to the middle point of the peptide bond between residues i and i + 1. The average value of the twist angle indicates the overall twist of the sheet. An alternative way to define the overall twist is to use the angle calculated from the dot product of the major axes of the edge strands. The PCA method is used to calculate the principal axis for each edge strand.
) of strand residues from i to i + 2 and residues from i + 1 to i + 3 (32). Each residue was represented by three main-chain atoms: N, Cα, and C. Principal component analysis (PCA) (33) was applied to obtain the major axis of three consecutive residues. This major axis represents the best line through the main-chain strand atoms from the N- to C-terminus. To describe the relative orientation of the inner and outer sheet, we employed PCA to obtain the principal plane of each sheet. The plane is defined by the major axis and the minor axis orthogonal to the former. The major axis of the inner sheet was projected to the principal plane of the outer sheet so that the angle formed by the major axis of the outer sheet and the projected axis of the other sheet describes the relative orientation between these sheets. The main-chain C, Cα, and N atoms are included for this calculation. We measured the sheet twisting using the interstrand pair method (34). Because CBEF sheet is antiparallel, for both hydrogen-bonded and nonhydrogen-bonded pairs, the twist angle is defined as the angle formed by the backbone direction vectors of one interstrand pair. For example, residues i and j are located in two adjacent strands and form a pair of main-chain hydrogen bonds. The backbone direction vector of residue i is defined as a vector starting at the middle point of the peptide bond between residues i − 1 and i and pointing to the middle point of the peptide bond between residues i and i + 1. The average value of the twist angle indicates the overall twist of the sheet. An alternative way to define the overall twist is to use the angle calculated from the dot product of the major axes of the edge strands. The PCA method is used to calculate the principal axis for each edge strand.
Isotropically distributed ensemble analysis
We performed isotropically distributed ensemble (IDE) analyses (35) to characterize the concerted molecular motions of the wild-type and its variants. Details of the method have been described previously (35). For completion and clarification, we briefly state the computational protocol here. First, for a protein containing n Cα atoms, we constructed an n × n matrix P with elements 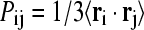 where ri is the position vector of Cα atom i originated from the protein center of mass. The angular bracket denotes averaging over the trajectory. Because the off-diagonal elements correspond to motional covariance, the cross-correlation coefficient (rij) of the motions of atom i and j is defined as
where ri is the position vector of Cα atom i originated from the protein center of mass. The angular bracket denotes averaging over the trajectory. Because the off-diagonal elements correspond to motional covariance, the cross-correlation coefficient (rij) of the motions of atom i and j is defined as 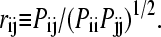 In this calculation, we used only Cα atom to represent a residue. If rij = 1, the Cα atoms of the two residues i and j are motionally highly correlated. In contrast, rij = −1 indicates strong anticorrelated motion. Second, when diagonalizing P we obtain one zero eigenvalue, which reflects the removal of the overall translational motion. For a globular protein, the remaining n − 1 eigenvalues are positive with the largest three corresponding to overall rotational motion whereas the rest correspond to internal motions. An orthogonal set of 3n-dimensional vectors can be constructed from the n-dimensional eigenvectors using
In this calculation, we used only Cα atom to represent a residue. If rij = 1, the Cα atoms of the two residues i and j are motionally highly correlated. In contrast, rij = −1 indicates strong anticorrelated motion. Second, when diagonalizing P we obtain one zero eigenvalue, which reflects the removal of the overall translational motion. For a globular protein, the remaining n − 1 eigenvalues are positive with the largest three corresponding to overall rotational motion whereas the rest correspond to internal motions. An orthogonal set of 3n-dimensional vectors can be constructed from the n-dimensional eigenvectors using 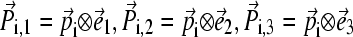 where
where 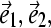 and
and 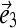 form an orthogonal basis in three-dimensional space. To visualize the motion along a mode
form an orthogonal basis in three-dimensional space. To visualize the motion along a mode 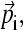 we first project a snapshot R(t) onto the given eigenmode
we first project a snapshot R(t) onto the given eigenmode 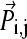 using
using 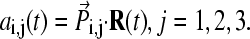 An “end-point” structure is generated by
An “end-point” structure is generated by 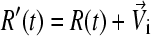 where
where 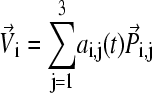 by setting time t = 0.
by setting time t = 0.
RESULTS AND DISCUSSION
Relative stability of the wild-type TTR and its variants
Because the tertiary structural change of TTR monomers is the first step toward fibril formation, it is crucial to characterize these conformational changes at the atomic level. Molecular dynamics simulation complements experimental approaches to probe the conformational changes. Five independent trajectories were collected for each of the wild-type and mutant proteins at 300 and 350 K, resulting in simulations that totaled 54 ns.
The average root mean-square deviations (RMS) from the initial structure over the trajectories as well as the maximum RMS deviation are listed in Table 1. For the simulations at 300 K, it can be seen that the mean RMS deviations of the MD/GB and explicit water simulations are similar, although the latter gave rise to slightly smaller value. Our WT-TTR RMS deviations at 300 K are significantly smaller than those reported by Daggett et al. (Fig. 2 c of Armen et al. (36)) at neutral pH and 310 K. This may be due to the difference in the force fields and the starting structure, given that the temperature effect is small. The large deviations of our simulation were found in the α-helical region and almost all of the loops except CD loop. We have consistently seen large RMS deviations in the α-helical region and the loop connecting to it in our simulations of monomers and tetramers in explicit water (14,17). The AB loop and GH loop are involved in the dimer-dimer interface in the tetrameric form. It is not surprising to see these regions drifting away from the x-ray tetrameric structure after dissociation. The BC loop, helix-F loop, and FG loop exhibit large B-factor in the x-ray structure of the wild-type and V30M-TTR (4), suggesting that these loops are labile. We also see large RMS fluctuations in these regions (data are not shown), which is consistent with the x-ray data.
TABLE 1.
RMS deviation of main-chain atoms and the yield of unfolding
| WT-TTR | V30M-TTR | L55P-TTR | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 300 K
|
300 K
|
300 K
|
|||||||||||
| Trajectory | Mean | SD | Maximum | Y% | Mean | SD | Maximum | Y% | Mean | SD | Maximum | Y% | |
| TIP3P | 1.4 | 0.2 | 2.1 | 1.3 | 0.2 | 1.9 | 1.5 | 0.2 | 2.1 | ||||
| GB | I | 1.8 | 0.4 | 2.9 | 3.2 | 1.8 | 0.2 | 2.5 | 0.4 | 1.8 | 0.2 | 2.3 | 2.4 |
| II | 1.6 | 0.2 | 2.3 | 0.7 | 1.9 | 0.2 | 2.8 | 0.1 | 1.9 | 0.3 | 2.6 | 3.3 | |
| III | 1.5 | 0.2 | 2.3 | 0.2 | 1.9 | 0.2 | 2.5 | 0.2 | 1.8 | 0.2 | 2.5 | 1.7 | |
| IV | 1.8 | 0.2 | 2.5 | 1.7 | 1.8 | 0.2 | 2.4 | 1.0 | 1.7 | 0.2 | 2.6 | 2.1 | |
| V | 2.1 | 0.2 | 2.9 | 2.1 | 1.9 | 0.2 | 2.4 | 0.1 | 2.1 | 0.4 | 3.4 | 9.2 | |
| Average | 1.7 | 0.3 | 2.9 | 1.6 | 1.8 | 0.2 | 2.8 | 0.4 | 1.8 | 0.3 | 3.4 | 3.8 | |
| 350 K
|
350 K
|
350 K
|
|||||||||||
| GB | I | 2.4 | 0.6 | 3.5 | 6.3 | 2.6 | 0.4 | 4.0 | 3.2 | 3.4 | 1.0 | 5.8 | 55.1 |
| II | 3.2 | 1.1 | 5.9 | 37.5 | 3.3 | 0.8 | 5.7 | 11.8 | 2.5 | 0.6 | 4.5 | 19.6 | |
| III | 2.4 | 0.5 | 3.8 | 10.3 | 2.7 | 0.4 | 4.1 | 2.1 | 3.5 | 0.5 | 5.4 | 29.1 | |
| IV | 3.5 | 0.5 | 4.7 | 13.9 | 2.3 | 0.3 | 3.3 | 1.4 | 3.5 | 0.6 | 5.1 | 71.8 | |
| V | 2.9 | 0.4 | 4.7 | 32.3 | 2.6 | 0.4 | 4.0 | 3.4 | 4.2 | 1.9 | 11.1 | 76.6 | |
| Average | 2.9 | 0.8 | 5.9 | 20.1 | 2.7 | 0.6 | 5.7 | 4.4 | 3.4 | 1.2 | 11.1 | 50.4 | |
The mean RMSD, standard deviation (SD) of RMSD, and the maximum RMSD are listed in units of Å. Y% is the yield of unfolding. The five independent MD/GB trajectories are named from I to V. The data of the explicit water MD simulations denoted as TIP3P are from Yang et al. (17).
FIGURE 2.

(a) Unfolding pathway of WT-TTR trajectory II at 350 K. (a) Initial structure. The DAGH sheet is in the front whereas the CBEF sheet is in the back. (b) The 800-ps snapshot. Major conformational changes are in the loop regions with some minor changes in the H strand. The original D strand becomes a surface loop. (c) The 1200-ps snapshot. The D strand that adopts a loop conformation separates from the core of the protein. (d) The 1800-ps snapshot. Most of the A, G, and H strands are disrupted. The inner sheet and outer sheet lose significant amount of native contacts. (b) The final snapshot of the continued 5-ns run of L55P-TTR trajectory II at 350 K. No α-sheet is formed. The outer (CBEF) sheet is intact whereas the inner (DAGH) sheet is severely disrupted. A shift in the β-strand D is illustrated. This three-residue shift along the upstream protein main chain, Leu-58–Thr-60, resembles “the β-slip” observed in the highly amyloidogenic TTR triple mutant G53S/E54D/L55S-TTR (45).
We conducted the unfolding simulations at 350 K. The structures drifted further away from the native state relative to the 300-K simulations, as shown in Table 1. However, not all trajectories lead to partially unfolded states. As defined in the section of computational methods, a given snapshot is considered to be in a partially unfolded state if it loses >40% secondary structure content in the inner sheet. For the wild-type, 37.5% and 32.3% snapshots are partially unfolded in trajectory II and V at 350 K (Table 1) with the maximum RMS deviation reaching 5.9 and 4.7 Å, respectively. Other trajectories have not shown apparent unfolding behaviors. Although the simulations on V30M-TTR at the elevated temperature result in overall similar RMS deviation to the wild-type, none of the trajectories of V30M variant shows obvious unfolding behaviors. Large RMS deviations are mostly seen in the α-helical region and loops rather than the β-sandwich region. The most unstable trajectory, trajectory II, only yields 11.8% unfolded snapshots throughout the simulation. In contrast to V30M-TTR, three of five trajectories of L55P-TTR unfold at 350 K with the maximum RMS deviation up to 11.1 Å in trajectory V (Table 1). The unfolding yields are 55.1, 71.8, and 76.6% for trajectory I, IV, and V, respectively. On average, L55P-TTR simulations resulted in 50.4% unfolding yield, whereas, the simulations on the wild-type and V30M gave rise to 20.1 and 4.4% unfolding yield, respectively. These unfolding simulations demonstrate that L55P-TTR monomer is much more labile than the wild-type and V30M-TTR monomers.
The thermodynamics and kinetics data of TTR tetramers have illustrated that the L55P mutation not only destabilizes the tetrameric native state but also lowers the barrier of tetramer dissociation into monomers (8). In this work, we further reveal that this mutation also destabilizes the monomeric state, explaining why this mutation exhibits severe pathology. However, predictions on the relative stability based on the unfolding yield have to be made with caution because the statistics may not be enough. The previous simulations on both the tetrameric and monomeric forms of TTR in explicit water at 300 K have shown similar flexibility of the wild-type and V30M variant (14,17). However, in this work, the unfolding of V30M-TTR monomer seems more resistant to the elevated temperature than the wild-type. The V30M disease phenotype is milder than that of L55P. The denaturation energetics and kinetics data have shown that the tetrameric native state of V30M-TTR is less stable than the wild-type; in contrast, the dissociation rate of V30M-TTR tetramer into monomers is slower than the wild-type, suggesting that its barrier of dissociation is higher than the wild-type (8). An engineered V30M-TTR monomer exhibits substantially faster aggregation than the wild-type monomer (37). Combining these facts with our simulations, we suspect that the mild phenotype of V30M-TTR may be mainly due to the fast rate of aggregation after the amyloidogenic intermediates are formed rather than the thermodynamics and kinetics of the tetrameric or monomeric states.
The details on the secondary structure analysis along the trajectories are shown in Table 2. The outer sheet, CBEF sheet, maintains >80% of its native secondary structure throughout the simulations. The B and C strands keep even >90% β-sheet contents. However, the inner sheet, DAGH sheet, maintains lower native secondary structure contents than the outer sheet, especially the D and H strands. Specifically, the D strand of the wild-type maintains 65.6 and 43.6% native secondary structure at 300 and 350 K, respectively. The secondary structure content of the H strand of the wild-type is lowered to 70.4% at 350 K. The V30M variant contains slightly higher β-sheet content in the H strand than the wild-type at the elevated temperature. Its secondary structure content within the D strand is lowered to 78.4% at 350 K. The higher temperature simulations of L55P variant yields increasing loss of β-sheet content in the inner sheet. Specifically, the A, G, and H strands contain only 69.9, 74.9, and 61.6% native secondary structure content, respectively. In addition, the α-helical region also exhibits significant loss of secondary structure content, only 66.6 and 65.5% of α-helical content is preserved at 300 and 350 K, respectively. The analysis on the secondary structure stability of the monomeric TTR simulations support our previous simulation results on the native tetrameric forms, where we have identified unstable sites, the D strands and the α-helical region as well as the strands at the monomer-monomer interface (strands H-H′ and F-F′) (14). Overall, L55P variant preserved only 77 and 71% native secondary structure content during the simulations at 300 and 350 K, respectively, whereas, the wild-type and V30M variant maintain at least 10% higher than that of L55P-TTR. The secondary structure analysis demonstrates that the monomeric form of L55P-TTR is less stable than that of the wild-type and V30M variant. Recently, Walsh and co-workers has monitored the temperature-dependent far-ultraviolet and near-ultraviolet circular dichroism (CD) spectroscopy of WT-TTR (38). They have detected small, partially reversible changes in the β-sheet and α-helical region along with major irreversible changes in the loop regions from 75 to 95°C. Minor changes were observed before 75°C. Their observation is in line with our secondary structure analysis during the partial unfolding process at 350 K.
TABLE 2.
Secondary structure analysis
| 300 K
|
350 K
|
|||||
|---|---|---|---|---|---|---|
| Strand | WT | V30M | L55P | WT | V30M | L55P |
| A | 96.9 | 98.3 | 95.5 | 84.3 | 93.8 | 69.9 |
| B | 98.2 | 98.5 | 99.2 | 96.4 | 95.9 | 96.7 |
| C | 95.2 | 94.5 | 98.4 | 93.0 | 94.6 | 92.8 |
| D | 65.6 | 97.3 | 3.4 | 43.6 | 78.4 | 16.3 |
| E | 88.7 | 86.9 | 84.4 | 88.4 | 84.3 | 81.2 |
| Helix | 80.7 | 94.3 | 66.6 | 82.3 | 83.1 | 65.5 |
| F | 92.0 | 89.7 | 83.5 | 90.8 | 86.8 | 80.4 |
| G | 93.2 | 98.6 | 86.4 | 85.5 | 92.8 | 74.9 |
| H | 81.9 | 86.4 | 78.2 | 70.4 | 77.5 | 61.6 |
| Average | 88.0 | 93.8 | 77.3 | 81.6 | 87.5 | 71.0 |
In Fig. 2 a, we present some snapshots along trajectory II of WT-TTR. This trajectory represents a typical unfolding process. During the first 800 ps, the conformation remains nativelike, but after then, the protein undergoes dramatic conformational changes in the following order: 1), the D strand becomes a surface loop and moves apart from the A strand; 2), the H strand becomes shorter, but still keeps good contact with the G strand; 3), the C strand undergoes significant breaking and reforming and, meanwhile, the original D strand that is the current CDE loop, moves closer to the C strand; and 4), the two β-sheets lose most of the native packings. However, the outer sheet, CBEF, maintains native-like conformation although the C strand undergoes breaking/reforming fluctuations. Our results are consistent with earlier spectroscopic data (19) that suggested that the C-strand-loop-D-strand rearrangement led to the formation of a monomeric amyloidogenic intermediate. Recent NMR results also revealed that the C and D strands dislocated from their native edge region in the fibril formation (39). These unfolding simulations support early speculations of the initial conformational changes (toward the amyloid formation) that the separation of the D strand from the core of the protein initiates the conformational changes that could facilitate amyloidogenesis (40,41).
Inspired by the work of Daggett et al. (36,42), which captured the prefibrillar amyloidogenic intermediate, an α-pleated sheet, based on the molecular dynamics simulations of unfolding of TTR, we searched for transient α-sheet intermediates. However, no obvious α-sheet was found in our simulations either at 300 or 350 K. For the wild-type, this is consistent with their simulation results at neutral pH (36). However, for the L55P variant, we expected to observe transient α-sheet intermediates at neutral pH because this variant is amyloidogenic under physiological conditions. In fact, we only found that Ala-120 and Val-121 in the H strand adopted αR and αL conformation simultaneously in 32 of 1800 snapshots of L55P-TTR trajectory II. But none of these 32 snapshots exhibits bifurcated hydrogen bonds. We continued to collect this trajectory for another 5 ns, hoping to observe the formation of α-sheet. However, no α-sheet was observed yet, instead the inner (DAGH) sheet is severely disrupted (Fig. 2 b). It is unlikely that the apart strands can move back to form α-sheet in a longer simulation. We suspect that the difference in the balance of the main-chain dihedral angle and the electrostatic interaction between ENCAD (43) and parm99MOD2/GB (23,24,44) may result in the discrepancy between our simulation and Daggett's work. Interestingly, we observed a shift in the β-strand D (Glu-54–Leu-55 in the native structure) along the upstream protein main chain to Leu-58–Thr-60, resembling “the β-slip” observed in the highly amyloidogenic TTR triple mutant G53S/E54D/L55S-TTR (45).
The wild-type conformation of D strand is trapped in two local minima
The D strand (residues 54–55) was identified as one of the unstable sites based on our structural and dynamic analysis on the native tetramers (14). To characterize the local conformational changes upon unfolding, we calculated the dihedral angle of residue 55 and presented the Ramachandran plot in Fig. 3 a. At 300 K, residue 55 of the wild-type adopts two conformations, antiparallel β-sheet (φ near −139° and ϕ near 135° (42)) and PII(L) (φ near −79° and ϕ near 149° (42)), among which the antiparallel β-sheet conformation dominates. For the simulation conducted at 350 K, the most populated state is shifted from the antiparallel β-sheet to the PII(L) conformation. In contrast, the D strand of V30M variant adopts only one conformation, the antiparallel β-sheet, at 300 K. This conformation still dominates at 350 K even though the PII(L) conformation was sampled to some extent. The D strand of L55P variant adopts the PII(L) conformation at both 300 and 350 K.
FIGURE 3.

(a) Ramachandran plot of residue 55 and the distribution curves of the φ and ϕ angles of this residue in the sampled conformations at 300 and 350 K. The wild-type, V30M-TTR, and L55P-TTR are in black, red, and green, respectively. (b) Distribution of Cα distance between Val-14 and Leu-55 (Pro-55 in L55P-TTR). The solid and dotted lines represent the simulations at 300 and 350 K, respectively.
In addition to the dihedral angle of residue 55, we also calculated the Cα distance between Leu-55 and Val-14 to describe the separation of the D strand from the A strand (Fig. 3 b). A pair of main-chain hydrogen bonds between Leu-55 and Val-14 is present in the wild-type crystal structure. These hydrogen bonds are disrupted by the Leu-55–Pro mutation, leading to a movement of residues 54–55 away from the core of the β-sandwich. We observed that the wild-type exhibited a characteristic two-peak distribution at both 300 and 350 K. At 300 K, the large peak is located near 5.4 Å, which is the normal distance between the D and A strands in the crystal structure and the hydrogen bonds (Leu-55(NH,O)–Val-14(O,NH)) are well maintained. This conformation is corresponding to the well-defined D strand. The second peak is at 7.4 Å, related to a disrupted D strand, where the hydrogen bonds are not presented. The distribution becomes broader at higher temperature. However, the L55P and V30M variants only exhibit one-peak distribution. For V30M-TTR, the peak is situated near the large peak of the wild-type, whereas the peak of L55P is located in the vicinity of the small peak of the wild-type in which case residues 54–55 belong to a surface loop and move away from the A strand. When the temperature is increased, the distributions are broader and the peaks shift to the right. It is apparent that the conformations of the D strand of the wild-type sampled in our simulation are trapped in two conformations, a well-defined D strand and a disrupted D strand. The disrupted D strand of the wild-type was also observed in the crystal structure solved in the P21 space group (15), whereas in the crystal structure of WT-TTRP21212, the D strand is well defined (21). Our simulations show that both conformations are thermally accessible at 300 K, moreover, as temperature increases, the transition toward the disrupted D strand is favored. The D strand of the most amyloidogenic variant, L55P-TTR, is completely disrupted. The leucine to proline mutation may have pushed this mutant one step ahead of the wild-type and other mutants toward the amyloidogenic intermediates, provided that the C-strand-loop-D strand rearrangement is the first conformational change along the pathway of amyloid formation. This result supports earlier speculation that the disruption of the secondary structural integrity through the disorder of the “edge strand” might trigger amyloidogenesis (17,40,41).
Geometry of the β-sheets
When we analyzed the change of intersheet Cα distances with respect to the x-ray structure, we found a common feature for the wild-type and its variants at all temperatures: residues Val-94–Thr-96 located in the F strand moves closer to the inner sheet. As the temperature increases to 350 K, these residues shifted toward the inner sheet up to 2 Å closer than those of the corresponding crystal structure (data are not shown). In the x-ray structures, the F strand bends toward the solvent in the region of Val-94–Thr-96. This observation inspired us to systematically analyze the geometric characters of each protein in terms of the local bend angle of each strand, the sheet twist, and the relative β-sheet orientation. If the extended strand conformation follows a perfect line, the local bend angles will be zero along the strand. However, bending is a pervasive structural distortion. As seen in Fig. 4, the C strand seems to exhibit the largest local bend; however, it is resulted from a β-bulge at Phe-44–Ala-45. The least distorted strand is the B strand with the largest bend angle <10°. The N-terminus of the E strand is bent, whereas the rest of the strand is quite linear. Except the C strand, the B, E, and F strands become flatter with the increase of temperature. Particularly, the three of four local bend angles of the F strand become smaller during the simulation of monomers at 300 and 350 K than those of the native tetrameric state for both the wild-type and its variants. Namely, the F strand becomes flatter in the monomeric state. This strand is at the monomer-monomer interface in the native tetrameric form. The secondary structure of this strand is well maintained during the simulations both at 300 and 350 K as seen in Table 2. Therefore, this change in strand geometry is due to tetramer dissociation into monomer, instead of secondary structural changes. Recent D-H exchange studies of TTR native state and fibrillar state have revealed the difference in the solvent protection factor for the residues in the F strand (39,46). A significant protection was observed for residues 94 and 96 in the fibrillar state (39); in contrast, these residues are relatively unprotected in the native state (46), although a pair of hydrogen bonds between Glu-89 and Val-94 at the F-F′ interface were detected in the x-ray structure (21). Based on the D-H exchange pattern of TTR fibrils, Oloffsson et al. proposed a native-like intermolecular association between strands F-F′ and H-H′ with a possible prolongation of these β-strands and possible shift in β-strand register in the fibrillar state (39). Earlier site-directed spin-labeling results also indicated that in the fibril state, the F and F′ interface was largely unchanged or even somewhat strengthened (47). We suspect that the flatter conformation of the F strand in the monomeric form may facilitate the formation of a novel intermolecular interface with a shift in β-strand register between F and F′ in the fibril state.
FIGURE 4.

Local bend angle of the outer sheet. The average structures of simulation are compared with the native structure (NAT).
The sheet twist was calculated using interstrand pair method (34). If the sheet is a perfect plane, all of the twist angles should be zero. If the sheet is twisted to a certain degree, we expect to obtain a uniform twist angle for all pairs of the interstrand direction vector. The average twist angle observed in the Protein Data Bank is ∼15° (34). However, if sheet twisting is accompanied by bending, a large twist angle may result from both effects. The common feature we have seen in the outer sheet (Fig. 5) is that the strands E and F in the monomeric state twist more uniformly than those in the tetrameric native state. The significant differences between the monomeric state and the tetrameric state are seen in the interstrand pairs of Ile-68/Thr-96 and Lys-70/Val-94. A large bending occurs from Val-94 to Thr-96 in the F strand. As the F strand becomes less bent in the monomeric state as shown in Fig. 4, the twist angles appear uniform across the E and F strands. Overall, the F strand in the monomeric state is flatter, but the E and F strands are slightly more twisted by 2–3° than the tetrameric state. We also list the overall sheet twist calculated using the PCA method in Table 3. Even though the PCA and interstrand pair methods gave different values of sheet twisting, both methods show a similar trend in that the outer sheet is more twisted in the monomeric state than the native state and the twist angle increases with temperature. Because the interstrand pair method only applies to the well-defined β-sheet, we calculated the twist angle of the inner sheet using the PCA method only. As listed in Table 3, the inner sheet exhibits a similar trend as the outer sheet in that it becomes more twisted in the monomeric state and as temperature increases.
FIGURE 5.

Outer sheet twist. The average structures of simulation are compared with the native structure. In the average structure, the overall secondary structure is preserved; as a result, we can analyze the sheet twisting using the average structure. The interstrand pairs are listed as follows: 1), CB strand, Ala-29/Lys-48, Val-30/Gly-47, His-31/Ser-46, Phe-33/Pro-43, Arg-34/Glu-42; 2), BE strand, Val-30/Ile-73, His-31/Glu-72, Val-32/Val-71, Phe-33/Lys-70, Arg-34/Tyr-69, Lys-35/Ile-68; 3), EF strand, Gly-67/Ala-97, Ile-68/Thr-96, Tyr-69/Phe-95, Lys-70/Val-94, Val-71/Val-93, Glu-72/Glu-92, Ile-73/Ala-91.
TABLE 3.
Sheet twist and relative orientation of the inner sheet and outer sheet
| TTR | Trajectory | θR | θI | θo | |
|---|---|---|---|---|---|
| WT | Nat. | 30.73 | 31.52* | 38.99* | 19.07† |
| 300 K | 31.18 | 33.85 | 42.86 | 19.48 | |
| 350 K | 35.59 | 41.65 | 43.56 | 20.71 | |
| V30M | Nat. | 30.15 | 31.57 | 37.64 | 18.86 |
| 300 K | 31.98 | 34.43 | 41.41 | 19.85 | |
| 350 K | 33.89 | 40.37 | 46.40 | 21.98 | |
| L55P | Nat. | 27.75 | 31.48 | 38.76 | 18.66 |
| 300 K | 30.90 | 35.60 | 41.60 | 19.51 | |
| 350 K | 25.43 | 51.02 | 44.84 | 21.52 | |
We compared the tetrameric native structure (Nat.) with the average structures of the simulations at 300 and 350 K. The relative β-sheet orientation is described by θR. The inner and outer sheet twist angle is designated by θI and θo, respectively.
PCA method. We used the strands B and F to calculate the overall twist of the outer sheet whereas the inner sheet twisting is calculated using the A and H strands.
The overall sheet twist calculated with the interstrand pair method. The average value of the twist angles of all interstrand pairs throughout the sheet is presented. All the angles are in units of degree.
We also calculated the relative orientation between the inner sheet and the outer sheet. By superimposing the outer sheet of the average structure of the simulation with the x-ray structure, we found that the inner sheet rotated clockwise in the plane of the sheet. The out-of-plane rotation is not as obvious as the in-plane motion. As seen in Table 3, the monomeric state of the wild-type and V30M variant exhibit ∼4° of clockwise rotation at 350 K with respect to the corresponding tetrameric native state. The L55P-TTR monomer gives rise to ∼3° clockwise rotation during the simulations at 300 K. However, at 350 K, the inner sheet in-plane movement is dominated by translational motion toward the original monomer-monomer interface, accompanied by slightly anticlockwise rotation.
Correlated motions
It is interesting to characterize the correlated motions during the partially unfolding process. For most of the covariance matrix analysis, such as quasiharmonic analysis, a prerequisition is to superimpose the structure with a reference set to eliminate the overall rotation; however, this protocol is not unambiguous when the protein is partially unfolded because the rigid body motion and the internal motion are not separable in this case (35). In the IDE analysis, it is not necessary to separate the overall rotation from the internal motion before diagonalizing matrix P. The separability of these two motions can be expressed as follows:
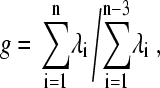 |
where λi is the eigenvalue of the ith mode, whereas n is the total number of mode, and n − 3 is the total number of mode excluding the three rotational modes. The separability varies from 114.1 for L55P-TTR at 300 K to 37.2 at 350 K, indicating that the largest internal mode contains some feature of rigid body motion when the protein partially unfolds.
Here we present IDE analysis for the trajectories collected at 350 K. In terms of the cross-correlation coefficient matrix, the wild-type and its variant show some similar features. However, the anticorrelated motions are more obvious for L55P-TTR monomer. Anticorrelated motions tend to tear the protein apart; therefore, we have observed the evident anticorrelated manner in the L55P variant that exhibits the largest unfolding yield. For the simulations of native states, it has been found that regions of secondary structure move in a correlated manner (48). As seen in Fig. 6 a, the strands C, B, E, and F that form the outer sheet move in a correlated manner; however, the strands A and H as well as the strands G and H in the inner sheet move in an anticorrelated manner. When the largest internal mode is excluded from the cross-correlation coefficient matrix, the anticorrelated motion is not evident any more (data are not shown). This suggests that the anticorrelated motions are dominant by the first largest internal mode. By visualizing the largest internal mode (Fig. 6 b), we found that along this mode, the strands A and G in the inner sheet move toward the outer sheet, whereas the intrasheet strand H move in the opposite direction, toward the solvent. This motion results in a more twisted inner sheet. Our observation of IDE analysis is consistent with the PCA analysis on the sheet twisting. Hence, the dominant internal mode during the unfolding process is to pull the H strand out of the plane of inner sheet.
FIGURE 6.

(a) Cross-correlation coefficient matrix between Cα atoms of L55P-TTR monomer sampled at 350 K. The most correlated are in red, whereas the most anticorrelated are in blue. The secondary structure is labeled at the bottom and the right. The strands are named from A to H and the helix is denoted by h. (b) The L55P-TTR Cα displacements along the largest internal mode at 350 K. The x-ray structure is in yellow. The end structure, R′(t = 0), generated from the IDE analysis is in red. The β-strands are named from A to H. The DAGH sheet is in the front whereas the CBEF sheet is in the back. The arrows schematically show the direction of the movement.
CONCLUSIONS
Under the partially denaturing conditions, L55P variant is more labile than the wild-type and V30M variant. We have observed that the D strand of WT-TTR is trapped in two local minima: the native conformation and the amyloidogenic fold that resembles the surface loop of residues 54–55 of L55P variant. In the tetrameric state, the F strand is bent with large separations at the F-F′ interface. This strand becomes flatter in the monomeric state, which may facilitate the formation of new F-F′ interface with possible prolonged hydrogen bonds and/or shift in β-strand register in the fibril state. During the unfolding process, the anticorrelated motion between the strands H and G as well as the strands H and A pulls the H strand out of the inner sheet plane, leading to a more twisted inner sheet. Our simulation has provided important detailed structural information about the partially unfolded state of TTR that may be related to the amyloidogenic intermediates. In principle, the protocol developed by Forman-Kay and co-workers to combine the experimental spectroscopic data and computational approaches to characterize the ensembles of structures representing the unfolded state of an SH3 domain (49) can be applied to study the partially unfolded state of TTR monomers and compared with our simulations.
Acknowledgments
S. Huo is very grateful to Prof. Ranjan Mukhopadhyay for fruitful discussions. We thank Prof. D. A. Case for providing the newly developed GB parameters before they are published. We thank the National Center of Supercomputing Applications and the Scientific Computing and Visualization group at Boston University for providing part of the computational resources.
This work was supported by Petroleum Research Fund (PRF No. 39205-G4) and partially supported by National Institutes of Health (1R15 AG025023-01) and National Science Foundation Major Research Instrumentation (DBI-0320875). The molecular graphics images were generated using the Chimera (50) package from the Computer Graphics Laboratory, University of California, San Francisco, CA (supported by National Institutes of Health P41 RR-01081).
Rafael Bruschweiler's present address is National High Magnetic Field Laboratory, Rm. B234, 1800 E. Paul Dirac Dr., Tallahassee, FL 32310.
References
- 1.Sacchettini, J. C., and J. W. Kelly. 2002. Therapeutic strategies for human amyloid diseases. Nat. Rev. Drug Discov. 1:267–275. [DOI] [PubMed] [Google Scholar]
- 2.Hamilton, J. A., and M. D. Benson. 2001. Transthyretin: a review from a structural perspective. Cell. Mol. Life Sci. 58:1491–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blake, C. C., M. J. Geisow, S. J. Oatley, B. Rerat, and C. Rerat. 1978. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J. Mol. Biol. 121:339–356. [DOI] [PubMed] [Google Scholar]
- 4.Hamilton, J. A., L. K. Steinrauf, B. C. Braden, J. Liepnieks, M. D. Benson, G. Holmgren, O. Sandgren, and L. Steen. 1993. The x-ray crystal structure refinements of normal human transthyretin and the amyloidogenic Val-30→Met variant to 1.7-A resolution. J. Biol. Chem. 268:2416–2424. [PubMed] [Google Scholar]
- 5.Sebastiao, M. P., M. J. Saraiva, and A. M. Damas. 1998. The crystal structure of amyloidogenic Leu55 → Pro transthyretin variant reveals a possible pathway for transthyretin polymerization into amyloid fibrils. J. Biol. Chem. 273:24715–24722. [DOI] [PubMed] [Google Scholar]
- 6.Hammarstrom, P., F. Schneider, and J. W. Kelly. 2001. Trans-suppression of misfolding in an amyloid disease. Science. 293:2459–2462. [DOI] [PubMed] [Google Scholar]
- 7.Schneider, F., P. Hammarstrom, and J. W. Kelly. 2001. Transthyretin slowly exchanges subunits under physiological conditions: a convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein Sci. 10:1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hammarstrom, P., X. Jiang, A. R. Hurshman, E. T. Powers, and J. W. Kelly. 2002. Sequence-dependent denaturation energetics: a major determinant in amyloid disease diversity. Proc. Natl. Acad. Sci. USA. 99(Suppl. 4):16427–16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hurshman, A. R., J. T. White, E. T. Powers, and J. W. Kelly. 2004. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry. 43:7365–7381. [DOI] [PubMed] [Google Scholar]
- 10.Jiang, X., C. S. Smith, H. M. Petrassi, P. Hammarstrom, J. T. White, J. C. Sacchettini, and J. W. Kelly. 2001. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 40:11442–11452. [DOI] [PubMed] [Google Scholar]
- 11.Colon, W., and J. W. Kelly. 1992. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry. 31:8654–8660. [DOI] [PubMed] [Google Scholar]
- 12.Lashuel, H. A., Z. Lai, and J. W. Kelly. 1998. Characterization of the transthyretin acid denaturation pathways by analytical ultracentrifugation: implications for wild-type, V30M, and L55P amyloid fibril formation. Biochemistry. 37:17851–17864. [DOI] [PubMed] [Google Scholar]
- 13.Hornberg, A., T. Eneqvist, A. Olofsson, E. Lundgren, and A. E. Sauer-Eriksson. 2000. A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J. Mol. Biol. 302:649–669. [DOI] [PubMed] [Google Scholar]
- 14.Lei, M., M. Yang, and S. Huo. 2004. Intrinsic versus mutation dependent instability/flexibility: a comparative analysis of the structure and dynamics of wild-type transthyretin and its pathogenic variants. J. Struct. Biol. 148:153–168. [DOI] [PubMed] [Google Scholar]
- 15.Wojtczak, A., P. Neumann, and V. Cody. 2001. Structure of a new polymorphic monoclinic form of human transthyretin at 3 A resolution reveals a mixed complex between unliganded and T4-bound tetramers of TTR. Acta Crystallogr. D Biol. Crystallogr. 57:957–967. [DOI] [PubMed] [Google Scholar]
- 16.Hornberg, A., A. Olofsson, T. Eneqvist, E. Lundgren, and A. E. Sauer-Eriksson. 2004. The beta-strand D of transthyretin trapped in two discrete conformations. Biochim. Biophys. Acta. 1700:93–104. [DOI] [PubMed] [Google Scholar]
- 17.Yang, M., M. Lei, and S. Huo. 2003. Why is Leu55→Pro55 transthyretin variant the most amyloidogenic: insights from molecular dynamics simulations of transthyretin monomers. Protein Sci. 12:1222–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Still, W. C., A. Tempczyk, R. C. Hawley, and T. Hendrickson. 1990. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 112:6127–6129. [Google Scholar]
- 19.Lai, Z., W. Colon, and J. W. Kelley. 1996. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry. 35:6470–6482. [DOI] [PubMed] [Google Scholar]
- 20.Liu, K., H. S. Cho, H. A. Lashuel, J. W. Kelly, and D. E. Wemmer. 2000. A glimpse of a possible amyloidogenic intermediate of transthyretin. Nat. Struct. Biol. 7:754–757. [DOI] [PubMed] [Google Scholar]
- 21.Klabunde, T., H. M. Petrassi, V. B. Oza, P. Raman, J. W. Kelly, and J. C. Sacchettini. 2000. Rational design of protein human transthyretin amyloid disease inhibitors. Nat. Struct. Biol. 7:312–321. [DOI] [PubMed] [Google Scholar]
- 22.Pearlman, D., D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham, S. DeBolt, D. Ferguson, G. Seibel, and P. A. Kollman. 1995. AMBER, a package of computer programs for applying molecular mechanics, normal-mode analysis, molecular-dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91:1–41. [Google Scholar]
- 23.Simmerling, C., B. Strockbine, and A. E. Roitberg. 2002. All-atom structure prediction and folding simulations of a stable protein. J. Am. Chem. Soc. 124:11258–11259. [DOI] [PubMed] [Google Scholar]
- 24.Onufriev, A., D. Bashford, and D. A. Case. 2004. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. 55:383–394. [DOI] [PubMed] [Google Scholar]
- 25.Tsui, V., and D. A. Case. 2000. Molecular dynamics simulations of nucleic acids with a generalized Born solvation model. J. Am. Chem. Soc. 122:2489–2498. [Google Scholar]
- 26.Tsui, V., and D. A. Case. 2000. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers. 56:275–291. [DOI] [PubMed] [Google Scholar]
- 27.Cornell, W., R. Abseher, M. Nilges, and D. A. Case. 2001. Continuum solvent molecular dynamics study of flexibility in interleukin-8. J. Mol. Graph. Model. 19:136–145. [DOI] [PubMed] [Google Scholar]
- 28.Lazaridis, T., and M. Karplus. 1997. “New view” of protein folding reconciled with the old through multiple unfolding simulations. Science. 278:1928–1931. [DOI] [PubMed] [Google Scholar]
- 29.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 30.Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion for a system with constraints: molecular dynamics of n-alkanes. J. Comp. Phys. 23:327–341. [Google Scholar]
- 31.Kabsch, W., and C. Sander. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 32.Daffner, C., G. Chelvanayagam, and P. Argos. 1994. Structural characteristics and stabilizing principles of bent beta-strands in protein tertiary architectures. Protein Sci. 3:876–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chatfield, C., and A. J. Collins. 1980. Introduction to multivariate analysis. Chapman and Hall, London, UK and New York, NY.
- 34.Ho, B. K., and P. M. Curmi. 2002. Twist and shear in beta-sheets and beta-ribbons. J. Mol. Biol. 317:291–308. [DOI] [PubMed] [Google Scholar]
- 35.Prompers, J. J., and R. Bruschweiler. 2002. Dynamic and structural analysis of isotropically distributed molecular ensembles. Proteins. 46:177–189. [DOI] [PubMed] [Google Scholar]
- 36.Armen, R. S., D. O. Alonso, and V. Daggett. 2004. Anatomy of an amyloidogenic intermediate: conversion of beta-sheet to alpha-sheet structure in transthyretin at acidic pH. Structure (Camb). 12:1847–1863. [DOI] [PubMed] [Google Scholar]
- 37.Reixach, N., S. Deechongkit, X. Jiang, J. W. Kelly, and J. N. Buxbaum. 2004. Tissue damage in the amyloidoses: transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. USA. 101:2817–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chung, C. M., L. H. Connors, M. D. Benson, and M. T. Walsh. 2001. Biophysical analysis of normal transthyretin: implications for fibril formation in senile systemic amyloidosis. Amyloid. 8:75–83. [DOI] [PubMed] [Google Scholar]
- 39.Olofsson, A., J. H. Ippel, S. S. Wijmenga, E. Lundgren, and A. Ohman. 2004. Probing solvent accessibility of transthyretin amyloid by solution NMR spectroscopy. J. Biol. Chem. 279:5699–5707. [DOI] [PubMed] [Google Scholar]
- 40.Kelly, J. W., and P. T. Lansbury. 1994. A chemical approach to elucidate the mechanism of transthyretin and B protein amyloid fibril formation. Amyloid: Int. J. Exp. Clin. Invest. 1:186–205. [Google Scholar]
- 41.Serpell, L., G. Goldstein, I. Dacklin, E. Lundgren, and C. C. F. Blake. 1996. The “edge strand” hypothesis: prediction and test of a mutational “hot spot” on the transthyretin molecule associated with FAP amyloidogenesis. Amyloid: Int. J. Exp. Clin. Invest. 3:75–85. [Google Scholar]
- 42.Armen, R. S., M. L. DeMarco, D. O. Alonso, and V. Daggett. 2004. Pauling and Corey's alpha-pleated sheet structure may define the prefibrillar amyloidogenic intermediate in amyloid disease. Proc. Natl. Acad. Sci. USA. 101:11622–11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levitt, M., M. Hirshberg, R. Sharon, and V. Daggett. 1995. Potential energy function and parameters for simulations of the molecular dynamics of proteins and nucleic acids in solution. Comput. Phys. Commun. 91:215–231. [Google Scholar]
- 44.Wang, J. M., P. Cieplak, and P. A. Kollman. 2000. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21:1049–1074. [Google Scholar]
- 45.Eneqvist, T., K. Andersson, A. Olofsson, E. Lundgren, and A. E. Sauer-Eriksson. 2000. The beta-slip: a novel concept in transthyretin amyloidosis. Mol. Cell. 6:1207–1218. [DOI] [PubMed] [Google Scholar]
- 46.Liu, K., H. S. Cho, D. W. Hoyt, T. N. Nguyen, P. Olds, J. W. Kelly, and D. E. Wemmer. 2000. Deuterium-proton exchange on the native wild-type transthyretin tetramer identifies the stable core of the individual subunits and indicates mobility at the subunit interface. J. Mol. Biol. 303:555–565. [DOI] [PubMed] [Google Scholar]
- 47.Serag, A. A., C. Altenbach, M. Gingery, W. L. Hubbell, and T. O. Yeates. 2001. Identification of a subunit interface in transthyretin amyloid fibrils: evidence for self-assembly from oligomeric building blocks. Biochemistry. 40:9089–9096. [DOI] [PubMed] [Google Scholar]
- 48.Ichiye, T., and M. Karplus. 1991. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 11:205–217. [DOI] [PubMed] [Google Scholar]
- 49.Choy, W. Y., and J. D. Forman-Kay. 2001. Calculation of ensembles of structures representing the unfolded state of an SH3 domain. J. Mol. Biol. 308:1011–1032. [DOI] [PubMed] [Google Scholar]
- 50.Pettersen, E. F., T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng, and T. E. Ferrin. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612. [DOI] [PubMed] [Google Scholar]