

Abstract
Voltage gated potassium channels open and inactivate in response to changes of the voltage across the membrane. After removal of the fast N-type inactivation, voltage gated Shaker K-channels (Shaker-IR) are still able to inactivate through a poorly understood closure of the ion conduction pore. This, usually slower, inactivation shares with binding of pore occluding peptide toxin two important features: i), both are sensitive to the occupancy of the pore by permeant ions or tetraethylammonium, and ii), both are critically affected by point mutations in the external vestibule. Thus, mutual interference between these two processes is expected. To explore the extent of the conformational change involved in Shaker slow inactivation, we estimated the energetic impact of such interference. We used κ−conotoxin-PVIIA (κ−PVIIA) and charybdotoxin (CTX) peptides that occlude the pore of Shaker K-channels with a simple 1:1 stoichiometry and with kinetics 100-fold faster than that of slow inactivation. Because inactivation appears functionally different between outside-out patches and whole oocytes, we also compared the toxin effect on inactivation with these two techniques. Surprisingly, the rate of macroscopic inactivation and the rate of recovery, regardless of the technique used, were toxin insensitive. We also found that the fraction of inactivated channels at equilibrium remained unchanged at saturating κ−PVIIA. This lack of interference with toxin suggests that during slow inactivation the toxin receptor site remains unaffected, placing a strong geometry-conservative constraint on the possible structural configurations of a slow inactivated K-channel. Such a constraint could be fulfilled by a concerted rotation of the external vestibule.
INTRODUCTION
Inactivation is a widely spread functional process in voltage gated potassium channels, a family of membrane proteins broadly distributed among living organisms. By determining the functional availability of potassium channels, this process has a direct impact on a number of physiological responses, ranging from K+ uptake and homeostasis in bacteria to setting the firing rate in neurons and endocrine cells. Voltage gated K-channels activate in response to sustained depolarization and then undergo one or more inactivation processes that interrupt ion conduction. The physiological behavior of different cell types depends largely on the harmonious balance between activation and inactivation in their own inventory of K-channels. Shaker K-channels, the prototypical voltage gated K-channels, are endowed with at least two types of inactivation: a fast N-type and a slow C-type inactivation, as they were first described (1). The N-type inactivation is produced after channel activation by the intracellular amino end of the protein that moves to occlude the permeation pathway (2–4). For the slow inactivation we have a less precise picture. It is believed that it involves a concerted conformational change at the external entrance that reduces potassium ions permeability at the pore (5–9). Such conformational change is prevented by external application of tetraethylammonium (TEA)—a well-known K-channel's pore blocker—and appears to be accelerated by decreased pore occupancy by permeant ions ((10–12); but see Klemic et al. (13)). Slow inactivation in Shaker K-channels may involve more than a single conformation, and thus two different, but not necessarily conflicting, views about the extension of the structural change during inactivation have been proposed. One maintains that it is a very localized constriction, or collapse, of the external opening of the ion conduction pore (5,6,14), whereas the other states that it is a more widespread movement that extends up to the voltage sensor (15,16) and even displaces a large volume of solvent at the intracellular mouth of the pore (17).
Small peptide toxins from a wide variety of origins bind with high specificity to the external mouth of K-channels, the same functional locus where the slow inactivation is presumably taking place. Marine snail toxins (κ−PVIIA), α−KTx from scorpion (Agitoxin II and charybdotoxin (CTX)), sea anemone (ShK, from Stichodactyla helianthus and BgK from Bunodosoma granulifera), and snake (Dendrotoxin) have functionally converged to a mechanism of inhibition that is remarkably similar: A lysine together with an aromatic residue ∼7 Å apart form a functional dyad that is critical for binding ((18,19); but see Mouhat et al. (20)). At least for scorpion and snail toxins it has been shown that they bind to K-channels with a 1:1 stoichiometry, compete with external TEA, and are sensitive to the degree of occupancy of the pore—indicating that they occlude the ion conduction pathway (18,21–25).
Not surprisingly, mutations of a large number of residues in the external vestibule are critical for both slow inactivation and scorpion and snail toxin binding. Blue arrows in Fig. 1 A point to the residues in the H5-S6 region that appear to be important for C-type inactivation in Shaker K-channels. These residues expand a region extending from the C-terminus of S5 to the N-terminus of S6. Taking the crystal structure of the K-channel from Streptomyces lividans (KcsA) as a structural template (26), we modeled the architecture of the Shaker K-channel pore domain to evaluate the spatial arrangement of these residues. Fig. 1 B shows in blue that most of these residues are confined to a compact area around the external pore entrance. This area overlaps with the receptor for peptide toxins from scorpion or marine snail (shown in red, residues shown to be important for scorpion toxin, in yellow are those for conotoxin, and in orange are those residues important for both toxins; (27–30)). Because the high degree of overlap between the functional areas for slow inactivation and toxin binding, mutual interaction between both phenomena is expected.
FIGURE 1.

Spatial overlapping between slow inactivation and peptide toxin binding. (A) Amino acid sequence of the H5 and S6 segments of Shaker K-channels. Red, yellow, and orange arrows point to residues important for slow toxin binding whereas blue arrows indicate residues important for slow inactivation. (B) Possible position of residues involved in slow inactivation (blue) and in peptide toxin binding (red, yellow, and orange) according to a homology modeling of the Shaker sequence on the KcsA structure template (www.expasy.org/spdv). Red residues have been shown to be important for the binding of α−KTx scorpion toxins (AgTx II and CTX). Yellow residues are important for κ−PVIIA binding, whereas orange residues are important for both types of toxins. Important peptide toxin binding residues are defined as those in which nonconservative mutations promote >8-fold changes in affinity. These data were taken from Goldstein et al. (27), Ranganathan et al. (28), and Gross and Mackinnon (47) for α-KTx and Scanlon et al. (29) and Jacobsen et al. (30) for κ−PVIIA. Residues important for slow inactivation were those in which conservative mutations promote >3-fold changes in the inactivation kinetics. Data for the slow inactivation were from (1) Ortega-Saenz et al. (49), (2) Perez-Cornejo (50), (3) Starkus et al. (7), (4) Heginbotham et al. (51), (5) Liu et al. (6), (6) Larsson and Elinder (48), and (7) Hoshi et al. (1) and Ogielska and Aldrich (52). Molecular rendition was by POVRAY 3.5 (www.povray.org).
Consistent with expectations, Koch and colleagues (31) found that in a few inactivation mutants of Shaker-IR (M448K and T449S) recorded in whole Xenopus oocytes, the rate of putative C-type inactivation is slower when κ−PVIIA is present in the external side of the channel, suggesting that toxin binding and inactivation are mutually exclusive. However, in outside-out patches, κ−PVIIA showed no apparent effect on the inactivation kinetics of wild-type Shaker-IR (32). Moreover, Agitoxin II application to outside-out patches appears to block inactivated channels nearly as well as it blocks closed channels (6). These discrepancies can be reconciled if i), the pore mutants M448K and T449S promote new inactivated conformations, and ii), inactivation behaves dissimilarly in the different recording configurations. For example, in outside-out patches, external TEA slows inactivation of Shaker-IR channels in a behavior consistent with a mutually exclusive mechanism (10), whereas in whole oocytes, it has a very modest effect on the inactivation kinetics inconsistent with such mechanisms (13,33; D. Naranjo, unpublished).
κ−PVIIA and CTX have an important advantage as a probe for interactions between peptide toxin binding and slow inactivation in Shaker K-channels: its binding-unbinding kinetics is 100-fold faster than that of slow inactivation. Thus, inactivation can occur while the toxin binding is at near equilibrium. We studied the effect of κ−PVIIA binding on slow inactivation of Shaker-IR K-channels at toxin concentrations in which most of the channels were blocked. Both the onset of slow inactivation and the recovery were studied in macroscopic currents in whole oocytes and in outside-out patches, and both were almost unaffected by the toxin. Also, inactivation at equilibrium, measured as the voltage dependence of channel availability, was not significantly modified. As with κ−PVIIA, the onset and the recovery from inactivation remained unchanged in the presence of CTX. These results suggest that the geometry of the peptide toxin receptor is not significantly altered during slow inactivation and, somewhat surprisingly, suggest that occupancy by permeant ions pore region proximal to the bound toxin is unaffected, or is not very relevant for slow inactivation. To account for this lack of interference with toxin binding, we propose that inactivation involves a concerted rotational movement of the vestibule.
MATERIALS AND METHODS
Salts were purchased from Merck Chile S.A. (Santiago, Chile). Gentamicin, EGTA, HEPES, and bovine serum albumin were from Sigma-Aldrich (St. Louis, MO). Type II collagenase was from Worthington Biochemical Corporation (Freehold, NJ). κ−conotoxin-PVIIA was a gift from Dr. Martin Scanlon (3D Center, University of Queensland, St. Lucia, Australia), and CTX was purchased from Alomone Labs (Jerusalem, Israel). Xenopus leavis females were purchased from local providers and kept in accordance with the Guide for the Care and Use of Laboratory Animals (1996, National Academy of Sciences, Washington, D.C.). Shaker B Δ(6–46)—here called Shaker-IR—was heterologously expressed in Xenopus oocytes (2). Details of the methods of in vitro RNA synthesis and injection are described elsewhere (34). External solution for two-electrode voltage clamp was composed of (in mM) 96 NaCl, 2 KCl, 0.3 CaCl2, 1 MgCl2, 10 HEPES-NaOH, pH 7.4. The voltage- and current-electrode were filled with a solution of 3 M KCl, 1 mM EGTA, and 5 mM HEPES-KOH, pH 7.0. For patch clamp, the pipette solution was (in mM) 80 KF, 20 KCl, 1 MgCl2, 10 EGTA, and 10 HEPES-KOH, pH 7.4, and the external recording solution was 115 NaCl, 1 KCl, 0.2 CaCl2, 1 MgCl2, and 10 HEPES-NaOH, pH 7.4.
Details of the performed methods for electrophysiological measurements are described elsewhere (24). For the two electrode voltage clamp experiments (TEVC), to minimize electrode polarization upon sustained voltage pulses, the amount of K-channel cRNA injected was graded to obtain expression levels yielding ionic currents <2 μA. Voltage pulse protocols were applied by means of a Digidata 1200 B interface under the control of pClamp 5.5 or pClamp 7 software (Axon Instruments, Foster City, CA). Electrode potentials were measured at the end of each experiment, and membrane voltage was corrected according to the shift in the voltage-sensing electrode. The corrected voltages were typically within ±5 mV from the initial value. Additionally, to track the drift in the voltage electrode during the long measurements of voltage dependent availability at equilibrium (see Fig. 5), a brief protocol to measure the conductance versus voltage relationships (activation curves) in control conditions was applied. These measurements were repeated at the beginning of the measurements, between long voltage pulse protocols, and at the end of the experiment. The electrode shift was estimated from the displacement of the activation curve in the voltage axis and found to agree within ±2 mV with the displacement of the voltage-sensing electrode. The outside-out patch-clamp experiments (OOPC) were done according to Garcia et al. (24), with the difference that toxins were applied directly to the bath from stocks of 10 μM. Because inactivation kinetics gets faster upon excision due to a methionine oxidation (35), we usually waited 3–5 min until the inactivation kinetics stabilization before measurements.
FIGURE 5.

Voltage dependent availability of Shaker-IR K-channels in the presence and absence of 1.5 μM κ−PVIIA. (A) With TEVC, 1-min conditioning pulses ranging from −60 to 0 mV separated by increments of 5 mV were applied before a 1 s test pulse to +50 mV. (B) Plots of the current in response to the test pulse voltage. Current amplitudes were normalized to the maximum current elicited at each experimental condition. Voltage dependent availability is plotted in filled circles for control (top) and in the presence of 1.5 μM κ−PVIIA (bottom). Open circles plot the relative oocyte conductance before the control voltage excursion (top) and after the voltage excursion with toxin was taken (see Materials and Methods). To calculate the conductance, we assumed a reversal potential for K+ as −100 mV and a maximal conductance of 0.015 mS. Continuous lines on top of the experimental estimates are fits to a Boltzman function of the form Y = 1/(1 + e(V−Vo)/kv). For the voltage dependent availability, the fits parameters of this experiments were Vo = −30 mV, kv = 3.1 mV−1 for the control measurements and Vo = −30.5 mV, kv = 3.3 mV−1 for the measurements in the toxin presence. For the normalized conductances, the fit parameters were Vo = −12 mV and kv = −8.3 mV−1 before the control experimental run and Vo = −10 mV and kv = −9.0 mV−1 after the toxin run. These estimations gave values of ΔV = −18 mV and ΔV = −20.1 for the control and toxin run, respectively.
TEVC traces were usually filtered to 1 kHz with the output filter of the amplifier, digitized at 0.25–10 kHz. Data analysis and curve fitting were done with Clampfit 8.0 (Axon Instruments) and Microcal Origin 3.5 and 4.1 (Microcal, Northampton, MA). With either electrophysiological technique, a two exponential function was usually required to obtain acceptable fits to the inactivation time course, with time constants ∼1 s and 5–6 s for the fast and slow component, respectively. For the whole oocytes experiments, the slow component was always dominant, whereas the fast component prevailed in outside-out patches. To simplify the analysis, all relaxations were fit with a single exponential function, which in all cases closely resembled the dominant component seen in each experimental condition. Mostly with TEVC technique, we found variability in the inactivation time constant in oocytes from different frogs. A source for such variability in the kinetics could be originated by a variable amount of slow activating outward chloride currents. In general, for a larger contribution of the outward current, the kinetics of inactivation was faster.
Kinetic analysis
Both open and inactivated Shaker K-channels are competent to bind peptide toxins with a bimolecular stoichiometry according to the following simplified scheme,
where [T] is the toxin concentration and O is the unblocked-open and is the only conducting state, whereas I, O·T, and I·T are the nonconducting unblocked-inactivated, blocked-open, and blocked-inactivated, respectively. The constants kono and koni are the toxin association rates to open and inactivated channels, respectively, whereas koffo and koffi are the corresponding dissociation rates. Rates ki and kr are the inactivation and recovery rates in the unblocked channel, whereas kib and krb correspond to the rates for the toxin-blocked channel. Based on Scheme 1 , we solved the following equation system to describe the time course of the decay of ionic currents:
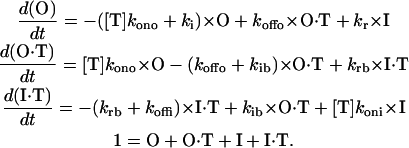 |
SCHEME1.

Setting O = 1/(1+[T]/KDc), O T = 1 − O, and I·T = 0 as initial conditions, where KDc = 60 nM is the toxin dissociation constant for the closed channels, the time course of the decay of ionic currents was calculated with the Runge-Kutta algorithm (www.myphysicslab.com). To obtain the different kinetics parameters, we complied with the following strategy: i), we imposed values for kono = 50 μM−1s−1 and koffo = 25 s−1 (32); ii), ki and kr were determined from the single exponential fits to the decays in the absence of toxin by using the expressions ki = (1 − ss)/τ and kr = ss/τ, where τ and ss were the relaxation time constant and the steady-state fractional residual current, respectively; iii), in agreement with the results shown in Fig. 4 A, the rate constant of the recovery in the presence of the toxin, krb, was assumed to be identical to kr; iv), kib and koni were obtained by fitting the decay traces in the presence of the toxin; and v), koffi was calculated assuming microscopic reversibility according to koffi = (ki·koni·krb·koffo)/(kono·kib·kr). This is an extremely oversimplified scheme. On one hand, the assumption of microscopic reversibility is untested and, on the other hand, slow inactivation appears to involve more than a single conformational state or mechanism ((13,15,16); see Figs. 3 and 4). Because inactivation kinetics spans from milliseconds to tens of seconds, for simplicity we deal mostly with the conformation(s) with kinetics that dominates the decay and recovery in the timescale of several seconds after the depolarizing pulse.
FIGURE 4.

Recovery from inactivation in the absence and in the presence of 0.5 μM κ−PVIIA. (A) Currents elicited with a two-pulse protocol from −90 mV holding voltage to 0 mV with variable interpulse interval at -90 mV for whole oocytes (left) and excised patches (right). In both techniques, the prepulse was ∼15 s long, whereas the test pulse was 400 ms long and ∼15 s long for TEVC and OOPC, respectively. (B) Same protocols as in A in the presence of 0.5 μM κ−PVIIA. (C) Recovery from the slow inactivation measured 200 ms into the test pulse was indistinguishable from controls in TEVC experiments. Values were normalized by the peak amplitude of the prepulse, and the solid line was traced with a two-exponential function with time constants of 0.1 and 2.0 s. Each plotted value corresponds to the average taken from four oocytes to which a full protocol was applied with and without toxin. In OOPC, recovery was fitted to a single exponential that resembled the slow component of the recovery in the TEVC experiments. Each plotted value corresponds to seven control patches and three patches with toxin.
FIGURE 3.
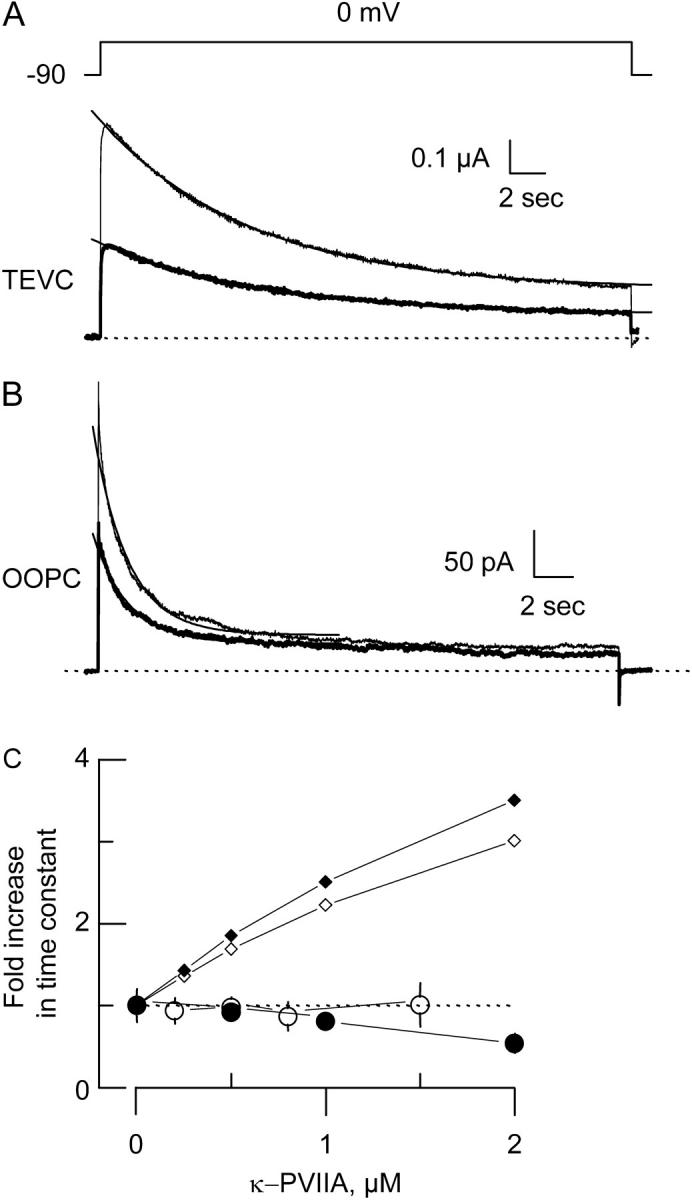
κ−PVIIA does not alter slow inactivation kinetics. (A and B) TEVC (A) and OOPC (B) current traces in the absence (thin lines) and in the presence (thick lines) of 0.5 μM κ−PVIIA elicited by ∼15 s pulses to 0 mV from a holding voltage of −90 mV. Each trace corresponds to an average of 6–10 individual traces. Single exponential fits drawn on top of the experimental traces were extrapolated to the beginning of the pulse. For TEVC traces, the time constants were 4.3 s and 4.7 s for control and toxin traces, respectively, whereas for OOPC they were 0.88 s and 0.9 s for control and toxin traces, respectively. For curve fitting details, see legend to Table 1. Traces were leak subtracted, and dotted lines correspond to the zero current level. (C) Normalized changes in the time constant of slow inactivation. Open and solid circles correspond to the averages of experimental values from 4 to 10 different patches or oocytes. Open and solid small diamonds are the expected time constant changes from Scheme 1, in which toxin is unable to bind to the inactivated state for TEVC and OOPC, respectively. For TEVC, this expectancy was obtained by assuming that in Scheme 1 the complex I·T has a very low probability of existence by multiplying koni and kib by 10−4 and koffi and krb by 104, respectively. For OOPC, kr = 0.076 s−1 and ki = 0.62 s−1, whereas krb=104·kr and kib=10−4·ki, and koni= 10−4·kono.
RESULTS
Slow inactivation in the presence of κ−PVIIA
We have previously shown that with TEVC or OOPC, the voltage pulse used to activate Shaker-IR K-channels also destabilizes κ−PVIIA binding (24). After channel activation, the toxin-receptor equilibrium relaxes from a voltage independent binding regime to closed channels to a lower affinity and voltage dependent, binding equilibrium to open channels. The dissociation constant is KDc = 60 nM for the closed channels and KDo(V = 0) of ∼0.5 μM with an effective valence, zδ = 0.6 for the open channels (24,29,32,36). Fig. 2 A shows TEVC traces in response to an activating voltage pulse to 0 mV in the absence and in the presence of 0.5 μM κ−PVIIA, a concentration near the KDo for that voltage. Application of the toxin decrease the amplitude of K-currents by ∼50% and produces an apparent delay of the activation. Only a small part of this delay can be attributed to a slower activation of the channels; most of it reveals the relaxation to the lower toxin affinity binding equilibrium after channel opening (24,32,36). The time course of this relaxation was obtained by performing a point-by-point ratio between the traces in the presence of the toxin and the corresponding control ((29); Fig. 2 B). On top of the experimental data, a single exponential function with a time constant of 23 ms and a steady-state fractional current of 0.55 was drawn. Assuming little inactivation during the pulse, from this relaxation we obtain kono = 40 s−1μM−1 and koffo = 23 s−1 (upper branch of the kinetic Scheme 1). These results are similar to those measured previously from macroscopic currents in whole oocytes and outside-out patches and with single channel analysis (24,32). Thus, within the 200 ms pulse to 0 mV, the toxin binding to open channels has reached steady state.
FIGURE 2.
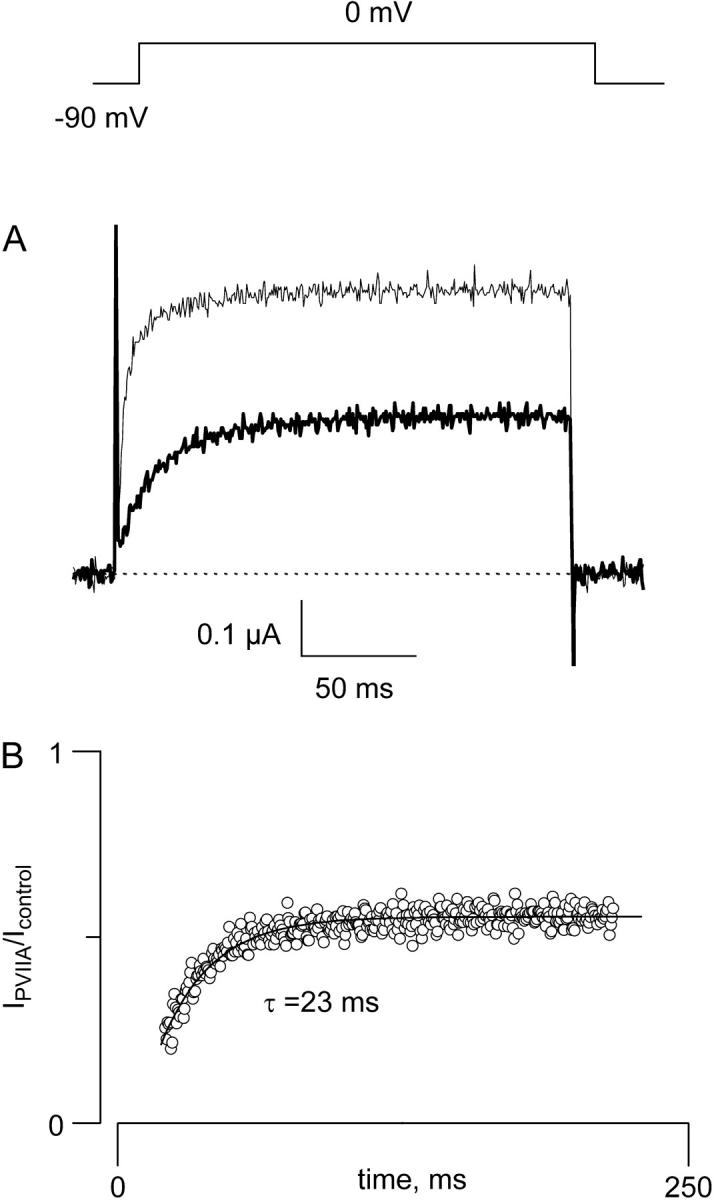
Toxin binding equilibrium is much faster than inactivation kinetics. (A) Two-electrode voltage clamp current traces were elicited by a 200 ms pulse to 0 mV from a holding voltage of −90 mV in the absence (thin trace) and in the presence (thick trace) of 0.5 μM κ−PVIIA. Traces were leak subtracted. (B) Point-by-point ratio between the trace in the presence of the toxin and the control trace shown in A to show that κ−PVIIA is destabilized by the voltage pulse used to open the Shaker voltage gated K-channel (see Garcia et al. (24)). The continuous trace is the fit to a single exponential function with time constant of 23 ms and a steady-state component of 0.55.
Does κ−PVIIA modify inactivation kinetics of Shaker-IR K-channels? We tried to answer this question using TEVC and OOPC techniques because slow inactivation behavior appears functionally different in these two experimental procedures. In classical experiments, assayed with OOPC, external TEA appears to interfere in a fashion strictly competitive with the kinetics of Shaker slow inactivation (see, for example, Choi et al. (10)), whereas with TEVC slow inactivation proceeds at ∼4-fold slower speed, and external TEA, even al saturating concentrations, has only modest effects on the slow inactivation kinetics ((13,37); D. Naranjo, unpublished). At present, we do not know the origin for such discrepancy, but as we show below regarding κ−PVIIA, such discrepancy does not appear to be significant.
With 15 s long voltage pulses, slow inactivation of Shaker-IR K-channels becomes apparent. Fig. 3 A shows responses to a voltage pulse to 0 mV from a holding potential of −90 mV in the absence and in the presence of 0.5 μM of κ−PVIIA (thick traces). Although inactivation kinetics can be very complex (see, for example, Panyi et al. (8), Ogielska et al. (9), Olcese et al. (15), and Loots and Isacoff (16)) here to compare the slow inactivation decay, single exponential functions with a steady-state constant were fit to individual traces (represented by a solid line on top of the traces; see Materials and Methods). For this batch of oocytes, the average time constant for inactivation of control traces in TEVC was 4.5 ± 0.9 s (n = 12 oocytes), whereas in the presence of 0.5 μM κ−PVIIA was 4.6 ± 0.5 s (n = 8 oocytes) (Table 1). This value remained relatively constant at toxin concentrations up to 1.5 μM. Although having faster inactivation kinetics in OOPC, determinations are qualitatively similar to TEVC. By fitting a single exponential to the first 7 s into the voltage pulse, we obtained time constants of 1.44 ± 0.11 s (n = 10 patches) for control and 1.32 ± 0.13 (n = 7 patches) in the presence of 0.5 μM κ−PVIIA. Fig. 3 C plots the relative effects of the toxin on the time constants (open circles for TEVC and solid circles for OOPC). On OOPC the inactivation time constant became a significant 40% smaller with 2 μM toxin. We do not understand this speeding effect in OOPC and why it was not observed in TEVC. It is possibly due to methionine oxidation after patch excision (35) or to a predicted synergistic interaction between pore blocking peptides and slow inactivation (see Discussion). Small diamonds (open for TEVC and solid for OOPC) are the expected increase in time constants if κ−PVIIA cannot bind to the inactivated state and the blocked channel cannot inactivate—a condition equivalent to a mutually exclusive competitive model ((10); see figure legend for details). This large departure from a mutually exclusive interaction indicates that the effect of toxin binding on slow inactivation rate(s), if any, is small.
TABLE 1.
Kinetic parameters of inactivation at 0 mV
| Whole oocytes
|
Outside-out patch
|
|||
|---|---|---|---|---|
| Control | 0.5 μM κ−PVIIA | Control | 0.5 μM κ−PVIIA | |
| Time constant(s) | 4.5 ± 0.9 | 4.6 ± 0.5 | 1.44 ± 0.11 | 1.32 ± 0.13 |
| Fractional steady state | 0.21 ± 0.04 | 0.25 ± 0.04 | 0.11 ± 0.011 | 0.15 ± 0.012 |
| n | 12 | 8 | 10 | 7 |
Summary of the single exponential fits to the current decay in the absence and in the presence of 0.5 μM κ−PVIIA applied to the bath. The first 200 ms into the 0 mV pulse were excluded because at this time, the toxin has not yet reached equilibrium in either TEVC or in OOPC (see Fig. 2). In whole oocytes experiments, fits included ∼14 s into the trace, whereas in OOPC, the fits only includes 7 s subsequent to the first 200 ms. The fractional steady-state currents were calculated from the ratio of the current measured at 15 s divided by the current measured at 200 ms. n represents the number of oocytes or excised patches for TEVC and OOPC, respectively.
What is the effect of slow inactivation on κ−PVIIA binding? Because Scheme 1 is energy conservative, a similarly small reciprocal effect should be expected on κ−PVIIA binding due to inactivation. To estimate the energetic impact of the slow inactivation on toxin binding, the kinetics of slow inactivation in TEVC was modeled and fitted to Scheme 1 (see Materials and Methods for details on the fitting strategy and Table 2 for results). As expected, the values for the resulting rate constants changed by at most 50%, indicating that inactivation has minor effects on toxin binding (ΔΔG ∼ 0.2 kcal/mol). Bearing in mind the simplistic nature of Scheme 1 and the complexity of the inactivation process, this conclusion is premature. However, two predictions can be made to test the validity of Scheme 1: recovery and steady-state inactivation should be toxin insensitive. The following sections deal with both predictions.
TABLE 2.
Inactivation rate constants in whole oocytes
| Control
|
In 0.5 μM κ−PVIIA
|
||
|---|---|---|---|
| Average ± SE | Average ± SE | ||
| kono (μM−1s−1) | 50.0 | koni (μM−1s−1) | 28.3 ± 7.5 |
| koffo (s−1) | 25.0 | koffi (s−1) | 14.0 ± 5.4 |
| ki (s−1) | 0.187 ± 0.013 | kib (s−1) | 0.234 ± 0.074 |
| kr (s−1) | 0.049 ± 0.0042 | krb (s−1) | 0.054 ± 0.016 |
| Ki | 0.26 ± 0.0082 | Kib | 0.37 ± 0.190 |
| κ−PVIIA binding constants | |||
| Open channels
|
Inactivated channels
|
||
| KDo (μM) | 0.5 | KDi (μM) | 0.68 ± 0.35 |
Kinetic and rate constants that describe Scheme 1 applied to the TEVC relaxation curves as those shown in Fig. 3. Values are the average of four oocytes to which the relaxations in the presence and in the absence of toxin were compared. The strategy to do the fitting is described in Materials and Methods. Ki and Kib, the apparent inactivation equilibrium constants at 0 mV, were calculated from kr/ki and krb/kib, respectively, whereas KDo and KDi were calculated from koffo/kono and koffi/koni, respectively. A value of ΔΔG = +0.2 kcal/mol results from the change in binding energy of the toxin to inactivated channels relative to the binding to open channels.
Recovery from the slow inactivation in the presence of κ−PVIIA
The recovery from inactivation was measured with a two-pulse protocol. With TEVC a 15 s conditioning pulse to 0 mV was followed by a short test pulse to 0 mV after a variable interpulse interval at −90 mV (Fig. 4, left panels). For OOPC, the protocol consisted of two identical 14 s pulses separated by the variable interval (Fig. 4, right panels). Fig. 4, A and B, shows several superimposed traces in response to the conditioning and test pulses in the presence (Fig. 4 A) and in the absence (Fig. 4 B) of 0.5 μM κ−PVIIA. For TEVC, the recovery appears to be composed of at least two phases in both experimental conditions, suggesting more than a single inactivated state; ∼50% of the recovery has already occurred after the first 500 ms interval, indicating a fast component, whereas a slower phase dominates the kinetics of the recovery afterwards. On the other hand, with OOPC, the recovery was monotonic. Fig. 4 C plots normalized recoveries in the absence and in the presence of 0.5 μM toxin for several oocytes (left) and membrane patches (right). For the TEVC data, the normalized recoveries were almost identical, with the same two-exponential function describing reasonably well both data sets: a time constant of ∼0.1 s, accounting for ∼65% of the recovery, and a 2 s time constant for the remainder. For OOPC recording, single exponential functions with time constant resembling the slow component seen in TEVC were fitted to the average recovery measurements. The average of the time constants for the individual recoveries measured 1.72 ± 0.33 s for the seven control patches and 1.55 ± 0.14 s for three toxin patches (not significantly different at the 0.05 level). In summary, results from whole oocytes and isolated membrane patches indicate that in transit to the slow inactivated conformation, channels visit at least two inactivated states, none of them having rates of recovery affected by the toxin.
κ−PVIIA and the near steady-state slow inactivation
Results of Fig. 4 show that the slow inactivation involves a complex set of kinetic states. Then, to assess the effect of the toxin on the steady-state inactivation it is necessary to get as close as is experimentally possible to the equilibrium. We composed a two-part protocol to test for these effects. The first part consisted of one min long pulses between −60 and 0 mV with 5 mV increments to produce inactivation. After this long conditioning pulse, a test pulse to +50 mV was applied to estimate the fraction of channels that remained available (see schematic protocol in Fig. 5 A, top). We were unable to get the outside-out patches to last enough to survive the whole protocol, thus data in this section are confined to TEVC recordings. Traces in Fig. 5 A show the current relaxation produced by the conditioning pulses followed by the test pulses in the absence (top traces) and in the presence of 1.5 μM κ−PVIIA, an excess of toxin that blocks ∼75% of the open channels at 0 mV (bottom traces). It appears that due to the complex relaxation of the inactivation kinetics, at the end of these long conditioning pulses channels did not reach yet to a steady-state inactivation regime. With this proviso, the fractional current at the test pulse is plotted in Fig. 5 B as filled circles for control (top) and in the presence of 1.5 μM toxin (bottom). The activation curves obtained from records acquired in the absence of toxin at the beginning (top) and at the end of the experiments after removal of the toxin (bottom) are plotted in open circles (see Materials and Methods). These activation curves were computed to estimate the drift of the voltage electrode produced by the recurring sustained voltage pulses in this long experimental protocol. Single Boltzman distributions functions were fitted to both inactivation and activation data. To compensate for effects of electrode polarization, the shift of the voltage dependent availability was computed relative to the midpoint of the activation curve (ΔV in Fig. 5 B). For 12 control oocytes, ΔV was −19.0 ± 3.3 mV, whereas it was −17.9 ± 1.8 mV for four oocytes to which full voltage excursion with and without toxin could be performed. On the other hand, the effective valences for inactivation were 6.7 ± 0.9 for control and 8.0 ± 0.3 for data in the presence of toxin. No significant differences were found between both experimental conditions for both parameters compared.
Within the uncertainties produced by drift of the voltage electrodes during the long pulse experiments, the effect of saturating concentrations of κ−PVIIA on slow inactivation could be at most a shift of ±5 mV in the near steady-state voltage dependent availability. If slow inactivation is fully coupled to opening and 14 is the total charge movement of the voltage sensor (38–40), then a shift of ±5 mV in the voltage dependent availability that could be well resolved in our experimental conditions would be equivalent to ∼1.6 Kcal/mol of channels. Because the inactivation appears not to be fully coupled to opening, as single channel experiments suggest (32), this number could be an upper limit to the energetic impact on the slow inactivation by κ−PVIIA binding.
CTX does not interfere with the kinetics of slow inactivation in TEVC
To test if the lack of an effect of κ−PVIIA on Shaker-IR slow inactivation was particular to this toxin or a more general phenomenon, we assayed slow inactivation in the presence of CTX, a well-characterized pore blocker scorpion toxin of the α−KTx family (41). The association and dissociation rate constants for the binding of CTX to open Shaker-IR are 50 μM−1s−1 and 7.3 s−1, respectively, similar to those of κ−PVIIA in Table 2 (42). Fig. 6 A shows 400 ms traces obtained at 0 mV in the absence (thin trace) and in the presence (thick trace) of 150 nM CTX, a concentration near the KDo for CTX at this voltage. After the activation of the ionic currents, the trace with CTX presents a small decrement in amplitude (shaded area under the trace). This decrement is produced because, contrary to κ−PVIIA, CTX binds with twofold lower affinity to the closed channels than it binds to the open conformation at zero voltage (D. Naranjo, unpublished). This type of reverse state dependent is also seen with κ−PVIIA at high external potassium concentration (see Boccaccio et al. (25)). In contrast to the relaxation in Fig. 2 B for κ−PVIIA, the point-by-point ratio between the CTX trace and the control trace has a downward direction. This relaxation was fitted to a single exponential function with time constant of 60 ms and a steady-state inhibition of 0.55 (Fig. 6 B). Assuming little inactivation, from this relaxation we obtain association and dissociation rate constants of 50 μM−1s−1 and 9.3 s−1, respectively, in agreement with Goldstein and Miller (42). Thus, we consider that binding of CTX has equilibrated within 400 ms. Because of the kinetics similarities with κ−PVIIA, we used the same pulse protocols to test slow inactivation with CTX. Fig. 6 C shows the effect of 150 nM CTX on potassium currents elicited by a 15 s depolarization to 0 mV. At this timescale, the state dependent affinity of CTX is shown as a brief hump at the beginning of the trace (thick line). After that, inactivation kinetics proceeds similar to the control trace (thin line). Although CTX increased the time constant by 17% ± 8% (n = 5 oocytes), this is a modest increment compared to the 70% expected for a mutually exclusive interaction with slow inactivation. Fig. 6 D summarizes measurements of the recovery from inactivation measured in the presence of 150 nM CTX with the two-pulse protocol as in Fig. 4 for κ−PVIIA (inset). The solid line was drawn with the same time constants used to fit TEVC data in Fig. 4 C to show that CTX did not change significantly the rate of recovery. Thus as κ−PVIIA, the effect of CTX on slow inactivation is very small.
FIGURE 6.

Slow inactivation in the presence of 150 nM CTX. (A) Two-electrode voltage clamp current traces elicited by a 400 ms pulse to 0 mV from a holding voltage of −90 mV in the absence (thin trace) and in the presence (thick trace) of 150 nM CTX. In addition to the inhibitory effect of CTX on the potassium channels, it introduced a downward component to the ionic current due to the lower affinity that the toxin has for the closed channels (shaded area). This effect is reminiscent of the κ−PVIIA blockade of Shaker K-channels in symmetrical potassium concentrations (25). Traces were leak-subtracted. (B) Point-by-point ratio of the toxin and control traces. The solid line is a fit of a single exponential function with time constant of 60 ms plus a steady-state component of 0.55, an inhibition level equivalent to a KDo = 183 nM. The extrapolation of the trace to the beginning of the voltage pulse yields to 0.64 as the extent of the unblocked closed channels, equivalent to a KDc = 267 nM. (C) Responses to a 15 s pulse to 0 mV from a holding voltage of −90 mV in the absence (thin trace) and in the presence (thick trace) of 150 nM CTX. Note the hump in the toxin trace corresponding to the relaxation to the higher affinity toxin-channel equilibrium in the open state. Single exponential fits to the decays of the traces shown gave time constants of 6.5 s and 7.1 s for the control and the toxin traces, respectively. Traces were leak subtracted. (D) Recovery from inactivation. (Inset) A two-pulse protocol was applied to the oocyte in the presence of 150 nM CTX as in Fig. 4. The recovered currents were measured 400 ms into the test pulse and normalized by the peak amplitude of the prepulse. The solid line is a two exponential function with time constants of 0.1 and 2.0 s as in Fig. 4 superimposed to the experimental points with an offset of 0.09. Error bars are standard error of five oocytes at CTX concentrations ranging from 150 to 700 nM.
DISCUSSION
Blocking 30–75% of the channels with fast equilibrating κ−PVIIA has negligible effects on slow inactivation onset and recovery when recorded in whole oocytes. Although inactivation kinetics in outside-out patches deviates from that observed in TEVC, it is minimally altered by the toxin (Figs. 3 and 4). The effect of this toxin on steady-state inactivation also seems to be within the detection limits of the TEVC technique (Fig. 5). In addition, compared with the expected results for a mutually exclusive interaction, slow inactivation and recovery seems to be marginally sensitive to CTX in TEVC also (Fig. 6). According to Scheme 1 (see Materials and Methods), these peptide toxins should bind to the inactivated channels as well as they do to the open channels (see Table 2). Liu and colleagues (6) used AgTx II, whose unbinding kinetics from Shaker-IR is much slower than that of inactivation, to measure toxin binding to either closed or previously inactivated Shaker K-channels. Fast applications of AgTx II in both experimental conditions inhibited nearly the same fraction of channels (6). Thus fast unbinding toxins, such as CTX and κ−PVIIA, do not modify inactivation (koffo/kono ∼ koffi/koni) and inactivation does not alter the binding of slow toxins as AgTx II (kr/ki ∼ krb/kib). Although it is not rigorous to compare toxins assayed in different branches of the same scheme to provide experimental support for the microscopic reversibility assumed in Scheme 1, its validity rests on the fact that CTX, κ−PVIIA, and AgTx II share the same blocking mechanism on the pore of Shaker-IR.
Results shown here are somewhat surprising given the important superposition of the residues that are important for the peptide toxin binding and those important for slow inactivation (see Fig. 1). Nevertheless, in the conceptual framework provided by Scheme 1, this type of result was previously anticipated by work with both AgTx II scorpion toxin, as mentioned above, and κ−PVIIA, as discussed next. Single Shaker K-channel recording in the presence of high concentration of κ−PVIIA showed that the fraction of null traces (without opening) was not modified in the presence of the toxin (32). If the fraction of null traces indeed corresponds to channels residing in the inactivated state, it was suggested that the toxin does not modify the steady-state inactivation. The results shown in Fig. 5 validate such an interpretation for outside-out patches. However, we have to keep in mind that these types of results are inconsistent with those of maurotoxin, another pore blocker peptide toxin that is unable to block Na+ currents passing through the inactivated channels as it does in open channels (10). This discrepancy could be attributed to the existence of more than a single conformation responsible for slow inactivation (13,15–17).
Pore occupancy
In addition to negative interference as would be a mutually exclusive interaction between pore blockade and inactivation, we also expect some degree of positive interference since both pore occlusion by toxins and slow inactivation antagonize with the occupancy of the pore by permeant ions (11,12,22–24,43,44). If toxin-blocked channels inactivate with the same rate as toxin free channels, as we observed in whole oocytes, then it is possible that the occupancy of the pore is not largely decreased in the toxin-occluded channel. This is a somewhat surprising conclusion because if the bound toxin does not sterically prevent the conformational change leading to inactivation, it should increase the inactivation rate by decreasing pore occupancy. According to Scheme 1, such a synergistic effect should be reciprocal on toxin binding by inactivation. A hint that this synergistic interaction could be occurring is the significant decrease by ∼40% in the inactivation time constant observed in excised patches at 2 μM toxin (Fig. 3 C). However, peptide toxins are not very sensitive to the occupancy of the pore; a twofold and a fourfold increase in the stability of the channel-toxin complex was obtained upon total removal of internal potassium for the κ−PVIIA/Shaker and the α−KTx/MaxiK systems, respectively (22,24). These modest stabilization effects are equivalent to 0.4 and 0.8 kcal/mol, respectively, energy values that are well below our limiting resolution of 1.6 kcal/mol imposed by the experiments in Fig. 5. We cannot rule out that the lack of interference observed in whole oocytes actually results from a mutual cancellation between two antagonizing processes: competitive exclusion and synergistic reduction in pore occupancy. However, the expected synergic stabilization of the inactivated state would be undetectable in steady-state inactivation experiments.
How large is the conformational change in the external vestibule?
We estimate the energetic impact of toxin binding on the slow inactivation to be at most 1.6 kcal/mol (Fig. 5). Moran (45) correlated experimental coupling energies, generated by mutation cycle analysis, with the crystallographic distances between residues for a number of interacting protein surfaces. Coupling energy is maximal (1.2–6.5 kcal/mol) for distances shorter than 4 Å, decreasing to be negligible for distances longer than 12 Å. Molecular dynamics simulations on the interaction of Shaker with AgTx II, a peptide similar to CTX, resulted in 15–17 side chain contacts that are 4 Å or less apart from each other (46). Although it is conceivable that for such a large number of contacts different regions of the toxin receptor move in a compensatory way such that the energetic balance of the interaction with Agtx II is minimally altered, it is difficult to imagine that the same energetic balance occurs for two types of peptide as different as α−KTxs and conotoxins are. Alternatively we can suggest that i), all movable residues during inactivation are located >12 Å away from the toxin receptor surface, or ii), they all move in a geometry conservative manner with respect to the toxin binding site. Although (i) is in agreement with recent estimations of large changes in the volume of the internal vestibule but negligible effect in the external vestibule during slow inactivation (17), it is not consistent with the large changes in solvent exposure proposed for residues 448 and 449 in the hot spot of the interaction surface of pore occluding toxins (28,30,34). For example, Cd2+ binds with a rate 45,000 faster to slow inactivated Shaker–IR having a cysteine in position 449 (5). In addition, with mutations M448C, T449C, and P450C, Liu and colleagues (6) observed in the slow inactivated channel increments of ∼100-, ∼1,000-, and ∼10,000-fold in the rate of cysteine modification by hydrophilic methanethiosulfonate derivatives, respectively. These increments in the modification rate were increasingly larger as the α-carbon of the residues was located farther away form the pore. Together with the lack of the ability of AgTx II to distinguish from inactivated channels, these results prompted Yellen and co-workers to propose that slow (or C) inactivation was a localized constriction of the pore internal to the toxin binding receptor. Today, in light of the structure of KcsA emerged from the MacKinnon group (26), it is apparent that those residues extend into a significant 20%–30% of the toxin receptor area and may involve 3–6 contact pairs with AgTx II and possibly with CTX (see Eriksson and Roux (46)). Because α−KTx scorpion toxins probably contact all four subunits and at least two and possibly four residues 449 simultaneously (34,46,47), it imposes a strong geometrically conservative constraint on the conformational change involved in slow inactivation. As proposed earlier, such a constraint could be solved assuming a simple rotational movement of the vestibule that would twist the pore linings, as shown in Fig. 7, changing ion conduction properties without varying the toxin binding site (32,48).
FIGURE 7.

A geometry conservative conformational change may underlie slow inactivation. Voltage gated K-channel is seen from the external side perpendicular to the plane of the membrane, with the axis of symmetry in the pore. The oval represents a peptide toxin, which can bind to both conformations. The rotational movement of the vestibule would twist pore lining, changing the ion conduction properties resulting from slow inactivation.
Acknowledgments
We thank Victoria Prado for technical help, Martin Scanlon for the generous gift of κ−PVIIA, Alan Neely for insightful discussions and careful reading of the manuscript, and Paul Brehm, Chris Miller, and Irwin Levitan for providing equipment essential for the experiments in this article. C.O. and V.G. are FONDECYT doctoral fellows.
This work was partially funded by FONDECYT 1030285 and 1020899 and DIPUV 30-2004.
References
- 1.Hoshi, T., W. N. Zagotta, and R. W. Aldrich. 1991. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 7:547–556. [DOI] [PubMed] [Google Scholar]
- 2.Hoshi, T., W. N. Zagotta, and R. W. Aldrich. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538. [DOI] [PubMed] [Google Scholar]
- 3.Demo, S. D., and G. Yellen. 1991. The inactivation gate of the Shaker K+ channel behaves like an open-channel blocker. Neuron. 7:743–753. [DOI] [PubMed] [Google Scholar]
- 4.Zhou, M., J. H. Morais-Cabral, S. Mann, and R. Mackinnon. 2001. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. [DOI] [PubMed] [Google Scholar]
- 5.Yellen, G., D. Sodickson, T. Y. Chen, and M. E. Jurman. 1994. An engineered cysteine in the external mouth of a K+ channel allows inactivation to be modulated by metal-binding. Biophys. J. 66:1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu, Y., M. E. Jurman, and G. Yellen. 1996. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 16:859–867. [DOI] [PubMed] [Google Scholar]
- 7.Starkus, J. G., L. Kuschel, M. D. Rayner, and S. H. Heinemann. 1998. Macroscopic Na+ currents in the “nonconducting” Shaker potassium channel mutant W434F. J. Gen. Physiol. 112:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Panyi, G., Z. F. Sheng, L. W. Tu, and C. Deutsch. 1995. C-type inactivation of a voltage-gated K+ channel occurs by a cooperative mechanism. Biophys. J. 69:896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogielska, E. M., W. N. Zagotta, T. Hoshi, S. H. Heinemann, J. Haab, and R. W. Aldrich. 1995. Cooperative subunit interactions in C-type inactivation of K channels. Biophys. J. 69:2449–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi, K. L., R. W. Aldrich, and G. Yellen. 1991. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc. Natl. Acad. Sci. USA. 88:5092–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopez-barneo, J., T. Hoshi, S. H. Heinemann, and R. W. Aldrich. 1993. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1:61–71. [PubMed] [Google Scholar]
- 12.Baukrowitz, T., and G. Yellen. 1996. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+ channel. Science. 271:653–656. [DOI] [PubMed] [Google Scholar]
- 13.Klemic, K. G., G. E. Kirsch, and S. W. Jones. 2001. U-type inactivation of Kv3.1 and Shaker potassium channels. Biophys. J. 81:814–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molina, A., P. Ortega-Saenz, and J. Lopez-Barneo. 1998. Pore mutations alter closing and opening kinetics in Shaker K+ channels. J. Physiol (Lond.). 509:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olcese, R., R. Latorre, L. Toro, F. Bezanilla, and E. Stefani. 1997. Correlation between charge movement and ionic current during slow inactivation in Shaker K+ channels. J. Gen. Physiol. 110:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loots, E., and E. Y. Isacoff. 1998. Protein rearrangements underlying slow inactivation of the Shaker K+ channel. J. Gen. Physiol. 112:377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang, X. J., G. C. L. Bett, X. Y. Li, V. E. Bondarenko, and R. L. Rasmusson. 2003. C-type inactivation involves a significant decrease in the intracellular aqueous pore volume of Kv1.4 K+ channels expressed in Xenopus oocytes. J. Physiol (Lond.). 549:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dauplais, M., A. Lecoq, J. X. Song, J. Cotton, N. Jamin, B. Gilquin, C. Roumestand, C. Vita, C. L. C. deMedeiros, E. G. Rowan, A. L. Harvey, and A. Menez. 1997. On the convergent evolution of animal toxins—conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J. Biol. Chem. 272:4302–4309. [DOI] [PubMed] [Google Scholar]
- 19.Menez, A. 1998. Functional architectures of animal toxins: a clue to drug design? Toxicon. 36:1557–1572. [DOI] [PubMed] [Google Scholar]
- 20.Mouhat, S., M. De Waard, and J. M. Sabatier. 2005. Contribution of the functional dyad of animal toxins acting on voltage-gated Kv1-type channels. J. Pept. Sci. 11:65–68. [DOI] [PubMed] [Google Scholar]
- 21.Miller, C. 1988. Competition for block of a Ca-2+-activated K+ channel by charybdotoxin and tetraethylammonium. Neuron. 1:1003–1006. [DOI] [PubMed] [Google Scholar]
- 22.Mackinnon, R., and C. Miller. 1988. Mechanism of charybdotoxin block of the high-conductance, Ca-2+-activated K+ channel. J. Gen. Physiol. 91:335–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park, C. S., and C. Miller. 1992. Interaction of charybdotoxin with permeant ions inside the pore of a K+ channel. Neuron. 9:307–313. [DOI] [PubMed] [Google Scholar]
- 24.Garcia, E., M. Scanlon, and D. Naranjo. 1999. A marine snail neurotoxin shares with scorpion toxins a convergent mechanism of blockade on the pore of voltage-gated K channels. J. Gen. Physiol. 114:141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boccaccio, A., F. Conti, B. M. Olivera, and H. Terlau. 2004. Binding of κ−conotoxin PVIIA to Shaker K+ channels reveals different K+ and Rb+ occupancies within the ion channel pore.. J. Gen. Physiol. 124:71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doyle, D. A., J. M. Cabral, R. A. Pfuetzner, A. L. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. Mackinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- 27.Goldstein, S. A. N., D. J. Pheasant, and C. Miller. 1994. The charybdotoxin receptor of a shaker K+ channel—peptide and channel residues mediating molecular recognition. Neuron. 12:1377–1388. [DOI] [PubMed] [Google Scholar]
- 28.Ranganathan, R., J. H. Lewis, and R. Mackinnon. 1996. Spatial localization of the K+ channel selectivity filter by mutant cycle-based structure analysis. Neuron. 16:131–139. [DOI] [PubMed] [Google Scholar]
- 29.Scanlon, M. J., D. Naranjo, L. Thomas, P. F. Alewood, R. J. Lewis, and D. J. Craik. 1997. Solution structure and proposed binding mechanism of a novel potassium channel toxin κ-conotoxin PVIIA. Structure. 5:1585–1597. [DOI] [PubMed] [Google Scholar]
- 30.Jacobsen, R. B., E. D. Koch, B. Lange-Malecki, M. Stocker, J. Verhey, R. M. Van Wagoner, A. Vyazovkina, B. M. Olivera, and H. Terlau. 2000. Single amino acid substitutions in κ−conotoxin PVIIA disrupt interaction with the Shaker K+ channel.. J. Biol. Chem. 275:24639–24644. [DOI] [PubMed] [Google Scholar]
- 31.Koch, E. D., B. M. Olivera, H. Terlau, and F. Conti. 2004. The binding of κ-conotoxin PVIIA and fast C-type inactivation of Shaker K+ channels are mutually exclusive. Biophys. J. 86:191–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naranjo, D. 2002. Inhibition of single Shaker K channels by κ−conotoxin-PVIIA. Biophys. J. 82:3003–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holmgren, M., P. L. Smith, and G. Yellen. 1997. Trapping of organic blockers by closing of voltage-dependent K+ channels—evidence for a trap door mechanism of activation gating. J. Gen. Physiol. 109:527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naranjo, D., and C. Miller. 1996. A strongly interacting pair of residues on the contact surface of charybdotoxin and a Shaker K+ channel. Neuron. 16:123–130. [DOI] [PubMed] [Google Scholar]
- 35.Chen, J. G., V. Avdonin, M. A. Ciorba, S. H. Heinemann, and T. Hoshi. 2000. Acceleration of P/C-type inactivation in voltage-gated K+ channels by methionine oxidation. Biophys. J. 78:174–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terlau, H., A. Boccaccio, B. M. Olivera, and F. Conti. 1999. The block of Shaker K+ channels by κ−conotoxin PVIIA is state dependent. J. Gen. Physiol. 114:125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang, Y. S., Y. Y. Yan, and F. J. Sigworth. 1997. How does the W434F mutation block current in Shaker potassium channels? J. Gen. Physiol. 109:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Islas, L. D., and F. J. Sigworth. 1999. Voltage sensitivity and gating charge in Shaker and Shab family potassium channels. J. Gen. Physiol. 114:723–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aggarwal, S. K., and R. Mackinnon. 1996. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 16:1169–1177. [DOI] [PubMed] [Google Scholar]
- 40.Seoh, S. A., D. Sigg, D. M. Papazian, and F. Bezanilla. 1996. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 16:1159–1167. [DOI] [PubMed] [Google Scholar]
- 41.Miller, C. 1995. The charybdotoxin family of K+ channel-blocking peptides. Neuron. 15:5–10. [DOI] [PubMed] [Google Scholar]
- 42.Goldstein, S. A. N., and C. Miller. 1992. A point mutation in a Shaker K+ channel changes its charybdotoxin binding-site from low to high affinity. Biophys. J. 62:5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dauplais, M., B. Gilquin, L. D. Possani, G. Gurrola-Briones, C. Roumestand, and A. Menez. 1995. Determination of the three-dimensional solution structure of noxiustoxin: analysis of structural differences with related short-chain scorpion toxins. Biochemistry. 34:16563–16573. [DOI] [PubMed] [Google Scholar]
- 44.Goldstein, S. A. N., and C. Miller. 1993. Mechanism of charybdotoxin block of a voltage-gated K+ channel. Biophys. J. 65:1613–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moran, O. 2002. Modelling of the interaction of κ−conotoxin PVIIA to the Shaker potassium channel. Biophys. J. 82:628A–629A. [DOI] [PubMed] [Google Scholar]
- 46.Eriksson, M. A. L., and B. Roux. 2002. Modeling the structure of Agitoxin in complex with the Shaker K+ channel: a computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys. J. 83:2595–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gross, A., and R. Mackinnon. 1996. Agitoxin footprinting the Shaker potassium channel pore. Neuron. 16:399–406. [DOI] [PubMed] [Google Scholar]
- 48.Larsson, H. P., and F. Elinder. 2000. A conserved glutamate is important for slow inactivation in K+ channels. Neuron. 27:573–583. [DOI] [PubMed] [Google Scholar]
- 49.Ortega-Saenz, P., R. Pardal, A. Castellano, and J. Lopez-Barneo. 2000. Collapse of conductance is prevented by a glutamate residue conserved in voltage-dependent K+ channels. J. Gen. Physiol. 116:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez-Cornejo, P. 1999. H+ ion modulation of C-type inactivation of Shaker K+ channels. Pflueg. Arch. Eur. J. Physiol. 437:865–870. [DOI] [PubMed] [Google Scholar]
- 51.Heginbotham, L., Z. Lu, T. Abramson, and R. Mackinnon. 1994. Mutations in the K+ channel signature sequence. Biophys. J. 66:1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ogielska, E. M., and R. W. Aldrich. 1998. A mutation in S6 of shaker potassium channels decreases the K+ affinity of an ion binding site revealing ion-ion interactions in the pore. J. Gen. Physiol. 112:243–257. [DOI] [PMC free article] [PubMed] [Google Scholar]