

Abstract
By means of high-intensity 532 nm laser pulses, a photochemical conversion of the initial B570 state of bacteriorhodopsin (BR) to a stable photoproduct absorbing maximally at ≈620 nm in BR suspensions and at ≈610 nm in BR films is induced. This state, which we named F620, is photochemically further converted to a group of three products with maximal absorptions in the wavelength range from 340 nm to 380 nm, which show identical spectral properties to the so-called P360 state reported in the literature. The photoconversion from B570 to F620 is most likely a resonant two-photon absorption induced step. The formation of F620 and P360 leads to a distinguished photo-induced permanent optical anisotropy in BR films. The spectral dependence of the photo-induced anisotropy and the anisotropy orientations at the educt (B570) and product (F620) wavelengths are strong indicators that F620 is formed in a direct photochemical step from B570. The chemical nature of the P360 products probably is that of a retro-retinal containing BR, but the structural characteristics of the F620 state are still unclear. The photo-induced permanent anisotropy induced by short laser pulses in BR films helps to better understand the photochemical pathways related to this transition, and it is interesting in view of potential applications as this feature is the molecular basis for permanent optical data storage using BR films.
INTRODUCTION
Bacteriorhodopsin (BR) is a retinal protein which was discovered in the archaebacterium Halobacterium salinarum (1). BR is isolated from H. salinarum in the form of so-called purple membrane (PM) patches, which consist of BR and lipids only. Inside the PM the BR molecules are arranged in a two-dimensional hexagonal crystalline lattice. BR is the key protein to the halobacterial photosynthetic capabilities. Its biological function is that of a light-driven proton pump. The photochemical changes of the molecule are summarized in the so-called photocycle scheme which comprises the spectroscopically distinguishable intermediates of BR (Fig. 1). They are commonly represented by a single letter code where the index represents the absorption maximum. In the initial photoreaction, B570 is converted to the short-living J625 state and proceeds to the K state. During these initial steps, the isomerization from all-trans to 13-cis retinal occurs. A cyclic sequence of thermal conversions leading back to the B570 state starts. The retinal conformation of the B570 and O640 state is all-trans, and that of all other states in the core part of the photocycle is 13-cis. The retinal isomerization in combination with other well-defined conformational changes in BR during the photocycle results in the transport of a proton across the BR. During the proton movement through the proton channel, a reversible protonation/deprotonation of the Schiff base group occurs which forms the linkage between the retinylidene residue and lysine-216. This reversible deprotonation and reprotonation causes the distinguished visible and fully reversible photochromic shift from purple to yellow.
FIGURE 1.

Scheme of the photocycle of BR. Absorption of a photon (hν) converts B to J from where the molecule cycles thermally through a sequence of spectroscopically distinguishable intermediates, which are given by their common single letter abbreviations. The isomer state of the retinylidene residue is represented by roman letters for all-trans (O, B) and italic letters for 13-cis (K, L, M, N, D). Underlined letters indicate the Schiff base linkage between the retinylidene residue and the protein moiety deprotonated (MI, MII). The P contains 9-cis retinal and is photochemically reached from O. Q is then formed in a thermal reaction from P. The absorption maxima are given as subscripts.
Upon keeping BR in the dark for a while, it relaxes to the dark-adapted state (2), which is a mixture of the B and D states. Photochemical excitation of the O state enables the formation of the 9-cis retinal containing P state, which thermally relaxes to Q390 (3).
Irradiation of BR suspension with high-intensity 532 nm laser pulses causes a nonreversible color shift to blue, and upon extended exposure a yellowish color remains. The obtained material was named laser-induced blue membrane (LIBM) (4–7). Obviously LIBM has nothing in common with so-called blue membrane (8–10), which is obtained from PM, e.g., by deionization.
The latest model for the LIBM formation was published by Masthay et al. (5). Upon photoexcitation of BR in the initial B state by high light intensities in a two-photon reaction or two sequential single-photon reactions, a state named P360 is formed which absorbs maximally in the ultraviolet (UV). This state would appear colorless or yellowish to the naked eye. The formation of P360 triggers conformational changes inside the BR trimer, and due to an altered interaction of the BRs inside a trimer the two BRs which were not photochemically converted change their absorption and turn to a blue state named P605. This model is summarized by the following scheme (taken in modified form from Masthay et al. (5)).
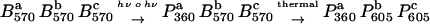 |
The superscripts indicate the three BRs inside a trimer. The process can be repeated and P605 can be photochemically converted to P360. Finally a trimer comprising three P360 states may remain.
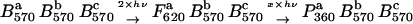 |
We analyzed the effect of high-intensity laser pulses on BR in polymeric films. We found that a distinguished permanent optical anisotropy is induced. But the model shown above could not explain the experimental results, in particular not the spectral dependence of the photo-induced permanent optical anisotropy. We included BR suspensions into our research and derived a new model which is consistent with the experimental results obtained from suspensions as well as BR films. We believe that the photoproduct P360 is not formed directly from B570. The primary photoproduct is a red-shifted state, which we named F620. In a secondary photochemical reaction of the F620 state, three products with absorption maxima around 360 nm are formed, which show identical spectral properties to the P360 state. Interaction between the BRs in the trimer is not excluded but probably is of minor importance for this process.
METHODS AND MATERIALS
BR films and BR suspensions
Wild-type BR in PM form (BR-WT) was received from MIB (Munich Innovative Biomaterials, Munich, Germany) and used without further purification. BR suspensions were diluted with water to the desired concentrations. BR films on glass substrates were prepared on a Film Applicator 2100 (Byk Gardner, Geretsried, Germany). BR films and BR suspensions were light adapted before all experiments.
Photochemical conversions with nanosecond laser pulses
Nanosecond high-intensity laser pulses from a frequency-doubled Nd:YAG laser (Infinity 40-100, Coherent, Dieburg, Germany) were used to induce nonreversible photo-induced changes in BR. Laser pulses of 3 ns duration and 532 nm wavelength at a repetition rate of 10 Hz were used throughout the experiments.
UV-Vis spectra
Where UV-Vis absorption spectra were to be taken inside an optical setup, a fiber-coupled charge-coupled device (CCD) array spectrophotometer (Instaspec IV, Oriel, Darmstadt, Germany) was used. In addition a conventional spectrophotometer (Uvikon 922, Kontron, Eching, Germany) was employed.
Measurement of permanent photo-induced anisotropy
Two different macroscopic setups were used: one using polychromatic light and a diode array detector and a second one using 532 nm laser light. In the first setup, a Xe-lamp (60,000 series, Oriel) was collimated and the output intensity was reduced by neutral density filters not to induce photochromic changes in the BR films during measurement. A rotatable linear polarizer and a fiber coupled CCD array spectrophotometer (Instaspec IV, Oriel) with adjustable gating time were used to record polarization-dependent absorption spectra. In the second setup, a Nd:YAG CW laser Verdi (532 nm, Coherent) with polarized output, a rotatable λ-half plate (λ/2) and a calibrated photodiode were employed. Data obtained at 532 nm were mathematically converted to the corresponding values at 570 nm by multiplying by the factor 1.29, which arises from the ratio between the molar extinction coefficients ɛ568nm = 63,000 M−1 cm−1 and ɛ532nm = 49,000 M−1 cm−1 for PM (7).
Microsopic sample points were analyzed in a microscope H 600 AM 50 (Hund, Wetzlar, Germany) equipped with a band pass filter (568 nm), a rotatable linear polarizer, and a monochrome CCD camera Orca C4742-95 (Hamamatsu, Herrsching, Germany) with nearly linear intensity response.
Analysis of linear dichroism
Photo-induced anisotropy was calculated from the measurement of the absorption change OD parallel (‖) and perpendicular (⊥) to the axis of the linear polarized actinic light in dependence on the energy exposure E, i.e., ▵OD‖ (E) = OD‖ (initial) − OD‖ (E) and ▵OD⊥ (E) = OD⊥ (initial) − OD⊥ (E). As the BR films are fully isotropic in the beginning, OD‖ (initial) = OD⊥ (initial) =: OD0 holds. The dichroism of a particular film is proportional to ▵OD = ▵OD‖ − ▵OD⊥. As the ▵OD value achievable depends on the amount of BR molecules per area, the dichroic factor DCF = ▵OD/OD0 is used for characterization.
In the case of the microscopic samples, the intensity of the background and the maximum intensity of the sample points were measured by means of the calibrated CCD camera with linear response. Line profiles through the sample spots were drawn and the values measured in the nonirradiated area used as the background intensity Iback and the maximal transmitted intensity in the sample spot used as Imax. The relative ▵ODrel was then derived from ▵ODrel = log (Iback/Imax).
RESULTS
Absorption changes in BR suspensions upon pulsed laser irradiation
Irradiation of BR suspensions with intense 532 nm laser pulses cause a color shift of the suspension to blue, and upon extended exposure a yellowish color remains. The initial absorption band decreases, and the absorption in the red wing increases. For lower energies, a first isosbestic point around 606 nm and a second less expressed at ≈418 nm is found. The formation of some products in the UV with maxima around 360 nm is observed too.
The exposures were done using quartz cuvettes of 1 cm depth and 1 cm width equipped with a miniaturized magnetic stirrer. The cuvettes contained 3 ml BR suspension, which means that 3 cm2 of the input window were filled with BR. The laser beam did not cover the whole surface of the cuvette. The exposed area was 0.238 cm2. The energies given in Fig. 2 are corrected by this factor, i.e., the measured energies were divided by the area ratio factor of 12.63.
FIGURE 2.

Spectral changes induced by irradiation of BR-WT suspension (OD570 = 1.3) with 3 ns high-intensity laser pulses of 532 nm wavelength at a repetition rate of 10 Hz. The total energy densities exposed to the cuvette are given on the right. The uppermost curve corresponds to the initial absorption profile. With increasing exposure, the absorption curves with less intense absorption at 570 nm are obtained.
The spectral changes observed comprise a decrease of absorption at 570 nm and a proportional increase in the red wing of the absorption band as well as in the UV (Fig. 2). For a more quantitative evaluation, the spectra in Fig. 2 were corrected for light scattering caused by the size of the PM patches, which are in the same order of magnitude as visible wavelengths. In the next step, a numerical fit of the absorption band before laser exposure was made. A reasonable fit is obtained using an asymmetric double Gaussian band. The found wavelength of maximal absorption and the returned form parameters were kept constant for the further fits. For the red-shifted absorption band, the same form parameters were applied. The maximal absorption wavelength as well as the relative contribution were obtained from the fitting procedure (Table 1). From this fit a wavelength of maximal absorption of ≈620 nm is obtained. Numerical fits using 605 nm as the maximal wavelength for the red-shifted absorption band returned an insufficient approximation for our experimental results.
TABLE 1.
Numerical analysis of the absorption bands contributing to the spectra given in Fig. 3
| B state
|
F state
|
|||
|---|---|---|---|---|
| Energy (J/cm2) | Amplitude (au) | λ (nm) | Amplitude (au) | λ (nm) |
| 0 | 1.14 | 566 | 0 | 0 |
| 2.4 | 0.99 | 566 | 0.08 | 622 |
| 4.9 | 0.87 | 566 | 0.16 | 618 |
| 9.8 | 0.65 | 566 | 0.28 | 617 |
| 14.6 | 0.49 | 566 | 0.32 | 617 |
| 19.5 | 0.38 | 566 | 0.34 | 619 |
| 24.4 | 0.32 | 566 | 0.34 | 618 |
| 29.3 | 0.25 | 566 | 0.31 | 620 |
| 39 | 0.2 | 566 | 0.24 | 621 |
| 48.8 | 0.15 | 566 | 0.18 | 623 |
| 60.9 | 0.13 | 566 | 0.12 | 625 |
| 73.1 | 0.09 | 566 | 0.08 | 626 |
Dependence of the absorption changes in BR suspensions induced by pulsed laser irradiation on the total energy flux
The light-dependent absorption changes at the wavelength derived from the fitting of the absorption bands (Table 1) are plotted in Fig. 3. Whereas the absorption at 566 nm decreases constantly with increasing energy absorbed by the sample, the absorption at 620 nm first increases and then decreases after passing through a maximum. The absorption at 340 nm increases slowly without any observable maximum. In the LIBM model proposed by Masthay et al. (5), the formation of P360 is accompanied by the formation of P605. Both products should appear with identical kinetics and reciprocal proportional to the B570 decrease. From the kinetic behavior observed in Fig. 3, a sequential reaction seems to be more likely to happen because only in this case an intermediate is observed which first increases and then decreases. This intermediate state in our experiment must be the product which absorbs maximally at 620 nm, which we name F620. The question arises of whether this state either thermally or photochemically is converted to the P360 products absorbing in the UV.
FIGURE 3.

Energy dependence of the absorption changes at selected wavelengths in BR suspension (data taken from Fig. 2). The wavelengths plotted correspond to the maximal absorptions of the B and F state. The absorption increase in the UV is represented at 340 nm. The data points taken from Fig. 2 are represented by squares. The lines arise from a numerical fit of the data assuming two sequential photonic reactions.
Absorption changes in BR films upon pulsed laser irradiation
Similar experiments as described for BR suspensions were repeated with BR films. Different from the BR suspensions, no diffusion of the BR molecules occurs during light exposure. The micrometer-sized PM particles are immobilized in the polymeric matrix of the BR films. No translation can occur, and even rotation of the BR molecules themselves is suppressed because of the two-dimensional crystalline nature of the PMs.
To distinguish between a thermal and a photochemical reaction from F to P360, we measured the changes in the absorption spectra of BR films treated with high-energy laser pulses. For ≈12 h after exposure to high-intensity laser pulses at room temperature (Fig. 4), no measurable changes except the dark adaptation of the B state are observed. From this experiment, we conclude that there is no thermal pathway from F620 to P360 but a photochemical one.
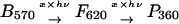 |
FIGURE 4.

Thermal absorption changes observed upon exposure of BR film with high-power 3 ns 532 nm laser pulses. Dark adaptation of unconverted B state but no changes in the red wing of the absorption curves are observed. This indicates that the F state, which absorbs maximally around 620 nm, is thermally stable and not thermally converted to the P360 state.
Analyzing the absorption properties of the BR films after exposure to the laser pulses in more detail showed that the selective bleaching of chromophore species parallel to the polarization of the actinic light is preferred, and permanent photo-induced optical anisotropy is induced (Fig. 5). There are several differences observed compared with the experiment in suspension. First, well-defined isosbestic points are observed in the BR films at ≈608 nm as well as at ∼458 nm (Fig. 5, A and B). Second, the photoreactions parallel (▵OD‖; Fig. 5 A) and perpendicular (▵OD⊥; Fig. 5 B) to the polarization of the actinic light are different in efficiency, and both reach a saturation value, and the energy dependence of both shows the same characteristics (Fig. 5 C, left axis). On the right axis in Fig. 5 C, the photo-induced anistropy is plotted. The induced permanent optical anisotropy observed in the BR films of course cannot be observed in BR suspensions due to the lack of immobilization of the BR molecules. Third, the energies to reach similar optical changes are significantly less in BR films than in BR suspensions.
FIGURE 5.

Spectral changes induced by irradiation of a BR-WT film with 3 ns high-intensity laser pulses of 532 nm wavelength at a repetition rate of 10 Hz. The initial absorption of the BR films was OD570 = 0.9. Changes of the optical density (A) colinear (‖) and (B) perpendicular (⊥) to the polarization of the actinic laser pulses. The energy densities are given in the plots. (C) Absorption changes at 561 nm in dependence on the total energy flux in parallel (‖) and perpendicular (⊥) orientation. The resulting relative optical anisotropy is given on the right axis.
An equivalent analysis of the absorption bands was made as described above for suspensions (Table 2). For the numerical treatment, the data from Fig. 5 A were used. The B state absorption is blue shifted compared to BR in suspension by ≈5 nm. The F state is observed at an absorption of ≈610 nm. This is ≈10 nm blue shifted compared to the suspension. The absolute values for B and F states are both blue shifted in the BR film, but the spectral shift between both is almost identical and ≈50 nm. This indicates that the photochemistry in BR suspensions and in BR films is the same and matrix effects are of minor importance. The F state population does not pass through a maximum as observed with BR suspensions. The absorption spectra taken from the BR films do not show the UV absorbing states because the optical fiber equipped with the spectrometer only poorly transmitted UV light. The formation of the UV absorbing species is seen more clearly in Fig. 2 (suspension) and Fig. 4 (film).
TABLE 2.
Numerical analysis of the absorption bands contributing to the spectra given in Fig. 5 A
| Parallel
|
||||
|---|---|---|---|---|
| B state
|
F state
|
|||
| Energy (J/cm2) | Amplitude (au) | λ (nm) | Amplitude (au) | λ (nm) |
| 0 | 0.77 | 561 | 0 | – |
| 0.3 | 0.73 | 561 | 0.04 | 572 |
| 0.6 | 0.69 | 561 | 0.07 | 576 |
| 0.9 | 0.67 | 561 | 0.07 | 587 |
| 1.3 | 0.65 | 561 | 0.08 | 598 |
| 1.7 | 0.59 | 561 | 0.11 | 603 |
| 2.2 | 0.52 | 561 | 0.14 | 604 |
| 2.7 | 0.45 | 561 | 0.15 | 606 |
| 3 | 0.41 | 561 | 0.18 | 607 |
| 3.5 | 0.35 | 561 | 0.2 | 608 |
| 4.2 | 0.32 | 561 | 0.22 | 609 |
| 4.9 | 0.27 | 561 | 0.23 | 607 |
| 5.1 | 0.26 | 561 | 0.24 | 609 |
| 5.9 | 0.26 | 561 | 0.25 | 611 |
| 6.6 | 0.23 | 561 | 0.25 | 610 |
| 7.1 | 0.23 | 561 | 0.26 | 611 |
Angular and spectral dependence of the photo-induced optical anisotropy
The angular dependence of the photo-induced anisotropy of the BR films has been analyzed in more detail. The spectral dependence of the angular absorption has been measured. It is shown for the wavelengths 560 nm (Fig. 6 A) where the B state absorbs maximally in BR films. The maximal anisotropy is observed in the maximum of the difference spectra derived from the spectra in Fig. 5 A at 639 nm. In Fig. 6 C the spectral dependence of the anisotropy is plotted. The wavelength dependence reflects the degree and orientation of the optical anisotropy changes beyond 600 nm.
FIGURE 6.

Photo-induced anisotropy in a BR film with an initial optical density of OD570 = 0.9 after exposure to 10 J/cm2 of 532 nm pulses. (A) 561 nm. (B) 640 nm. (C) Spectral dependence of the dichroism factor.
The angular dependence of the absorption was fitted using Malus' law. The angle of the chromophore orientation of the BR in a trimer is 120°. The photoconversion probability, PCPeff, of the three chromophores of a single BR trimer together is proportional to
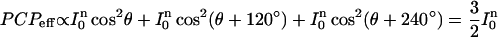 |
(1) |
where n characterizes the number of mechanistically necessary photons. Because the absorption probability is totally independent on the angle even for a single BR trimer, the total chromophore population can be considered isotropically angular oriented. The photoselection process then produces an anisotropic population of the initial B state B(θ) ∝1 − cos2(θ) = cos2(θ + 90°) and generates a population of the F state F(θ) ∝ cos2(θ). The long axes of the angular distributions of B570 and F620 are expected to be 90° rotated in this model.
A least-square fit of the function f1(θ) = C11 × (1 − cos2(θ)) + C12 versus the data from Fig. 5 A and Table 2 the values C11 = 0.18 and C12 = 0.65 for absorption changes parallel to the actinic light were obtained. C11 represents the maximal absorption and θ the angle between the polarization of the actinic light and the direction of the measurement. C12 represents the offset value of isotropic background. The corresponding data in Fig. 5 B were fitted with the function f2(θ) = C21 × cos2(θ) + C22. The values C21 = 0.05 and C22 = 0.10 for the absorption perpendicular to the actinic light (Fig. 5 B) were returned.
The anisotropy induced at 570 nm is as expected. Parallel to the actinic light (0°) the maximal bleaching is induced, and perpendicular to it very little absorption changes occur. At 639 nm the opposite situation is observed. An increase of 639 nm absorption and only a little change compared to the initial absorption is observed. This finding perfectly matches the theoretical model and is a strong indicator that the F620 product is formed by direct photochemical conversion from B570.
In the model proposed by Masthay et al. (5), the actinic light converts a B state molecule to P360. Due to conformational changes, the other two BR molecules inside the BR trimer change their absorption and form P605 molecules. The remaining anisotropic absorption at 570 nm and the increase in absorption at 605 nm should be parallel oriented (Fig. 7).
FIGURE 7.

Scheme explaining the spectral dependence of the anisotropy changes. Initially the 561 nm chromophore population is isotropically distributed, represented by a vertical and horizontal contribution. No population of a state absorbing at 620 or 605 nm exists initially. (A) Model proposed here where F620 is formed photochemically from B570. (B) In the model proposed in the literature (see Czégé and Reinisch (4) and Masthay et al. (5)), P360 is formed from B570. The generated P605 absorption is perpendicular to the B570 loss and P360 formation. The orientation of the photo-induced red-shifted absorption increase allows differing between models A and B.
In view of the spectral dependence of the optical anisotropy observed in BR films, only the model of a direct photochemical formation of B570 to F620 is consistent with the experimental data.
Is the B570 → F620 transition a two-photon reaction?
The photoconversion of the B state of BR has been intensely investigated (11–13). The dominating reaction at lower intensities is a single-photon reaction which triggers the isomerization of the retinylidene group and the start into the photocycle. The photochemical formation of F620 cannot be a simple single-photon reaction but might be either a resonant two-photon reaction or a sequence of two single-photon reactions where a secondary reaction of any of the early photointermediates of BR is involved.
All the experiments up to now have been done with 3 ns pulses. This means that even the formation of the K state is within the length of the pulses and from there a photochemical reaction back to B is well known (14).
The kinetic nature of the process can be characterized by the power dependence of the photoconversion. Using the method of initial rates and assuming that the rate is first order in the optical density OD and nth in the power P, the kinetic equation (15), with t the total irradiation interval in seconds, f the repetition rate of the laser in Hz, Δtpulse the pulse length, and k the rate constant, the following equation results:
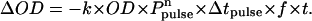 |
(2) |
Dividing this equation by OD and replacing Etotal for Ppulse × Δtpulse × f × t and assuming for the initial 20% of the photoconversion process OD ≈ ODinitial, the equation is obtained:
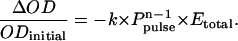 |
(3) |
The linear least-square fit of the data in the double-logarithmic plot in Fig. 8 yields a slope of n = 1.60, indicating that the photoconversion process has a two quantum character.
FIGURE 8.

Power dependence of the photochemical conversion of B570 with high-intensity laser pulses. A BR film (OD570 = 1.0) was irradiated with high-intensity laser pulse trains of 20 pulses (3 ns, 20 Hz, 532 nm) of varying total energies. The OD was measured at 532 nm and converted to 570 nm, and ΔOD was calculated as ODinitial − ODII concerning the polarization plane alignment of write- and readout beam.
This result is consistent with data from the literature on the irradiation of BR suspensions (5,7,13) where a power dependence with the range n = 1.5–1.8 was found. A sequential biphotonic process mediating the photoconversion process in BR suspensions is suggested due to the Goeppert-Mayer values (16) measured, which are in suspensions in the range of δ532 = 1.0 × 106 GM (5). It should be mentioned that there may be and probably are more than one photoconversion of the B state or early photointermediates of BR which show a two-photon absorption character. For the two-photon absorption at lower light intensities, a value of 290 GM was reported (17). This is still a high value as for a polyene, a value of only ≈10 GM is expected (16). The high values observed in suspension probably are due to a resonant two-photon absorption process. More information on the exact nature of the photoprocess requires femtosecond experiments.
Direct photoconversion of the B state versus involvement of early intermediate states
In Masthay et al. (5), a scheme is proposed where in a first reaction BR is turned from the B state to P360 in a two-photon reaction. Due to resulting changes in the interaction of the BRs inside the trimer, the two other BRs change to P605, a state which should still contain all-trans retinal. The key to understanding this photochemical conversion is an understanding of the primary photoreaction. Upon absorption of a single photon, B570 is excited to B*/Bfast and from there the J625 ground state is reached. The mechanism of the F620 formation might be either that in a two-photon absorption B is directly converted to F620 or the primary photoreaction is identical to the conventional photocycle and a second photon absorbed by one of the early intermediates, in particular B*/Bfast, J625, or K590, then leads to the formation of F620.
CONCLUSION AND PERSPECTIVE
We have analyzed the irreversible photochemically induced changes which occur upon exposure of BR to nanosecond light pulses which cause the formation of LIBM. The comparison of BR suspension and BR films revealed the following:
Analysis of the spectral changes shows that a), in PM suspension a red-shifted state is formed which absorbs maximally at ≈620 nm. Further products with maximal absorptions in the UV are also formed, and b), in BR films in principle the same photoreactions are observed; however, due to the matrix entrapment all states appear ≈10 nm blue shifted.
Analysis of the kinetics of the photoproduct formation shows that a), in PM suspensions, a sequential process B570 → F620 → P360 is much more likely than a parallel process P360 ← B570 → P605, and b), in BR films the same process may be assumed.
Analysis of the time-dependent absorption changes in BR films after pulsed laser exposure shows that F620 is thermally stable and no thermal reaction to P360 occurs.
Analysis of the wavelength dependence of the photo-induced optical anistropy in BR films shows that B570 is photochemically converted to F620.
Analysis of the intensity dependence of the B → F transition shows that it is very likely that it is a resonant two-photon process. If photointermediates of BR are involved only the very early ones, i.e., before the K state is reached, need to be considered.
Based on these results, we suggest the modified photocycle model from Fig. 1 to include the two-photon absorption induced formation of F620 from the B state and the photochemical conversion of F620 to P360 (Fig. 9).
FIGURE 9.

Extended version of the photocycle scheme for BR. The state F620 is obtained upon resonant two-photon absorption from the B570 state. From the F620 state, the P360 state is reached in a second photochemical step.
There are still several questions which need further analysis. First, what is the exact nature of the F620 state as far as the configuration of the retinylidene residue and the protonation states of amino acids like Asp-85 etc. are concerned. Second, the photochemical reaction from F620 to P360 needs to be analyzed in more detail. The P360 products need to be characterized in more detail too. Potential contributions from dark-adapted BR in the D548 state need to be analyzed. Experiments with femtosecond pulse excitation may be a suitable tool.
Acknowledgments
This work was supported by the German Ministry for Education and Research (BMBF, FKZ 13N8196).
References
- 1.Oesterhelt, D., and W. Stoeckenius. 1971. Rhodopsin-like protein from the purple membrane of halobacterium halobium. Nat. New Biol. 233:149–152. [DOI] [PubMed] [Google Scholar]
- 2.Hofrichter, J., E. R. Henry, and R. H. Lozier. 1989. Photocycles of bacteriorhodopsin in light- and dark-adapted purple membrane. Studies by time-resolved absorption spectroscopy. Biophys. J. 56:695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popp, A., M. Wolperdinger, N. Hampp, C. Bräuchle, and D. Oesterhelt. 1993. Photochemical conversion of the O-intermediate to 9-cis retinal containing products in bacteriorhodopsin films. Biophys. J. 65:1449–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Czégé, J., and L. Reinisch. 1991. Photodestruction of bacteriorhodopsin. Photochem. Photobiol. 53:659–666. [Google Scholar]
- 5.Masthay, M. B., D. M. Sammeth, M. C. Helvenston, C. B. Buckman, W. Li, M. J. Cde-Baca, and J. T. Kofron. 2002. The laser-induced blue state of bacteriorhodopsin: mechanistic and color regulatory roles of protein-protein interactions, protein-lipid interactions, and metal ions. J. Am. Chem. Soc. 124:3418–3430. [DOI] [PubMed] [Google Scholar]
- 6.Balashov, S. 1995. Photoreactions of the photointermediates of bacteriorhodopsin. Isr. J. Chem. 35:415–428. [Google Scholar]
- 7.Govindjee, R., S. P. Balashov, and T. G. Ebrey. 1990. Quantum efficiency of the photochemical cycle of bacteriorhodopsin. Biophys. J. 58:597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kimura, Y., A. Ikegami, and W. Stoeckenius. 1984. Salt and pH-dependent changes of the purple membrane absorption spectrum. Evidence for changes in conformation of the protein. Photochem. Photobiol. 40:641–646. [DOI] [PubMed] [Google Scholar]
- 9.Chang, C. H., R. Jonas, S. Melchiore, R. Govindjee, and T. G. Ebrey. 1986. Mechanism and role of divalent cation binding of bacteriorhodopsin. Biophys. J. 49:731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang, C. H., R. Jonas, R. Govindjee, and T. G. Ebrey. 1988. Regeneration of blue and purple membranes from deionized bleached membranes. Photochem. Photobiol. 47:261–265. [Google Scholar]
- 11.Nuss, M. C., W. Zinth, W. Kaiser, E. Koelling, and D. Oesterhelt. 1985. Femtosecond spectroscopy of the first events of the photochemical cycle in bacteriorhodopsin. Chem. Phys. Lett. 117:1–7. [Google Scholar]
- 12.Polland, H. J., M. A. Franz, W. Zinth, W. Kaiser, E. Koelling, and D. Oesterhelt. 1986. Early picosecond events in the photocycle of bacteriorhodopsin. Biophys. J. 49:651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moltke, S., V. Alexiev, and M. P. Heyn. 1995. Kinetics of light-induced intramolecular charge transfer and proton release in bacteriorhodopsin. Isr. J. Chem. 35:401–414. [Google Scholar]
- 14.Balashov, S. P., E. S. Imasheva, R. Govindjee, and T. G. Ebrey. 1991. Quantum yield ration of the forward and back light reactions of bacteriorhodopsin at low temperature and photosteady-state concentration of the bathoproduct K. Photochem. Photobiol. 54:955–961. [Google Scholar]
- 15.Chizhov, I. V., M. Engelhard, A. V. Sharkov, and B. Hess. 1992. Two quantum absorption of ultrashort laser pulses by the bacteriorhodopsin chromophore. In Structures and Functions of Retinal Proteins. J. L. Rigaud, editor. John Libbey Eurotext Ltd., London. 171–173.
- 16.Birge, R. R. 1983. One-photon and two-photon excitation spectroscopy. In Ultrasensitive Laser Spectroscopy. D. S. Kliger, editor. Academic Press, New York. 109–174.
- 17.Birge, R. R., and C.-F. Zhang. 1990. Two-photon double resonance spectroscopy of bacteriorhodopsin. Assignment of the electronic and dipolar properties of the low-lying 1Ag*− -like and 1Bu*+ -like π, π* states. J. Chem. Phys. 92:7178–7195. [Google Scholar]