

Abstract
The pH dependence of the structure of the main Na+/H+ antiporter NhaA of Escherichia coli is studied by continuous-wave (CW) and pulse electron paramagnetic resonance (EPR) techniques on singly spin-labeled mutants. Residues 225 and 254 were selected for site-directed spin labeling, as previous work suggested that they are situated in domains undergoing pH-dependent structural changes. A well-defined distance of 4.4 nm between residues H225R1 in neighboring molecules is detected by a modulation in double electron-electron resonance data. This indicates that NhaA exists as a dimer, as previously suggested by a low-resolution electron density map and cross-linking experiments. The modulation depth decreases reversibly when pH is decreased from 8 to 5.8. A quantitative analysis suggests a dimerization equilibrium, which depends moderately on pH. Furthermore, the mobility and polarity of the environment of a spin label attached to residue 225 change only slightly with changing pH, while no other changes are detected by CW EPR. As antiporter activity of NhaA changes drastically in the studied pH range, residues 225 and 254 are probably located not in the sensor or ion translocation sites themselves but in domains that convey the signal from the pH sensor to the translocation site.
INTRODUCTION
A better understanding of the formation of well-defined homo- and heterooligomers as well as larger aggregates of membrane proteins could enhance our insight into regulatory processes (1) and lead to improved strategies for membrane protein crystallization (2). Such understanding is currently hindered by a relative lack of characterization techniques that can quantify partial aggregation and that give detailed structural information on the aggregates. A combination of site-directed spin labeling (for reviews see, e.g., Hubbell and Altenbach (3), Hubbell et al. (4), and Steinhoff (5)) with the measurement of small magnetic dipole-dipole interactions between electron spins at distances of 2–6 nm by pulse electron paramagnetic resonance (EPR) (6,7,8) should be well suited for studies of protein aggregates as it can characterize local structure without depending on long-range order. We have recently demonstrated that the four-pulse double electron-electron resonance (DEER) experiment (9) can provide structural information on integral membrane proteins (10). Combined with triangulation techniques (11), the experiment was used to assess structural differences between monomers and trimers of plant light harvesting complex IIb (12)—a case where both the monomer and trimer are stable in detergent micelles and can be separated from each other. In many cases, however, oligomerization of membrane proteins has to be treated as a thermodynamic equilibrium, and the dependence of that equilibrium on external parameters is of interest. This situation implies that not only the structure of the oligomer but also the average number of protein molecules in the oligomer have to be determined.
Pulse electron double resonance techniques such as four-pulse DEER are suited for this purpose since the modulation depth in the signal depends on the number of remote spins coupled to the observer spin (13). We have recently given the corresponding equations for the case where spin-to-spin distances are distributed and a contribution from spins in neighboring oligomers or clusters has to be considered (14). In this study, we estimate the precision of determining the average number of coupled spins by measurements on a number of authentic biradicals and a triradical (15) and apply the approach to pH-dependent oligomerization of the sodium/proton antiporter NhaA of Escherichia coli.
NhaA is the main one of two antiporters of E. coli that accomplish specific exchange of Na+ or Li+ for H+ (16). This electrogenic antiporter, which exhibits a stoichiometry of 2H+/Na+ (17), is indispensable for ion homeostasis in E. coli and has homologs in many other enterobacteria (18). Among all Na+/H+ exchangers, NhaA of E. coli is the best characterized. Two-dimensional crystals diffracting at 4 Å resolution could be obtained, and analysis of electron density projection maps (19) and of a reconstructed three-dimensional map (20) revealed that NhaA forms dimers in crystals, with each monomer containing 12 tilted transmembrane helices. Coexpression of pairs of conditional lethal mutants demonstrated functional complementation, which, together with cross-linking data, strongly suggests that NhaA also functions as a dimer (21). Like many other Na+/H+ antiporters, NhaA is regulated by pH (18,22). It is essentially inactive below pH 7, and the Na+ efflux rate increases by three orders of magnitude between pH 6.5 and 8.5 (23). Cross-linking between loops VIII-IX, which are located in the interface between the individual NhaA molecules in the dimer, causes a dramatic change in the pH response for a short and rigid linker and no effect for a long and flexible linker (21). This suggests that a functional interaction between NhaA molecules is involved in the pH response.
From a mechanistic point of view, the pH response could be explained by pH-dependent dimerization, by a pH-dependent change of the structure in the interface between the two molecules (monomers), or by a conformational change within one of the two monomers that depends on the presence of certain residues in the nearby second monomer. By measuring the degree of dimerization as a function of pH and distances between residues in different monomers, we attempt to decide between these possible mechanisms. We concentrate on two residues that appear to be involved in the pH response. The first of these residues, His-225, is located at the periplasmic end of transmembrane domain VIII. The mutation H225R was shown to change, but not eliminate, pH dependence of NhaA antiporter activity (24). The second residue, Val-254 in loop VIII-IX, was selected as its mutation to Cys causes an acidic shift of the pH response. Furthermore, nearby Lys-249 in the same loop is a site of pH-dependent proteolytic cleavage by trypsin (25). Since susceptibility to this cleavage increases in the pH range of activation of NhaA, one may assume that loop VIII-IX becomes more accessible to trypsin by a conformational change that is related to the pH response of the antiporter.
The article is organized as follows: First, continuous-wave (CW) EPR is used to study possible pH-dependent changes of these residues in their mobility and accessibility to water- and lipid-soluble paramagnetic quenchers. Second, calibration of measurements of the average number of spins within the same oligomer or aggregate is discussed briefly. Third, four-pulse DEER is applied to test for pH-dependent changes in the distances H225R1/H225R1′ and V254R1/V254R1′ in dimers and in the degree of dimerization. Finally, the results are discussed in terms of the most likely mechanism of pH response.
MATERIALS AND METHODS
Preparation and labeling of NhaA
The generation of nhaA alleles encoding single Cys NhaA molecules used in this study has already been described (21,26). For overexpression the resulting single Cys NhaA encoding plasmids were transformed into E. coli TA16. TA16 is nhaA+nhaB+lacIQ and otherwise isogenic to TA15 (23). Cells were grown, membranes were prepared, and NhaA was solubilized and purified by Ni-nitrilotriacetic acid affinity chromatography as described (27). Before elution of the single Cys NhaA molecules (in 20 mM Tris-HCl, pH 7.9, containing 500 mM NaCl, 300 mM imidazole, 10% glycerol (v/v), 0.02% β-D-dodecylmaltoside (w/v)), the protein was reacted with MTSSL spin label (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl)-methanethiosulfonate (Toronto Research Chemicals, Toronto, Canada). For this purpose, washing buffer (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 5 mM imidazole, 10% glycerol (v/v), 0.1% β-D-dodecylmaltoside (w/v)) containing 1 mM MTSSL was applied to the column followed by overnight incubation at 4°C (hereafter we will refer to the spin-labeled residue as R1). Afterwards, unbound label was removed by washing the column with washing buffer. The bond of MTSSL to the protein was stable over the few hours required for sample preparation, and measurements throughout the pH range from 5.8 to 8. No free label was observed in the CW EPR spectra. After elution the protein was reconstituted into proteoliposomes with a composition of 67% phosphatidylethanolamine, 23.2% phosphatidylglycerol, and 9.8% cardiolipin at a lipid/protein ratio of 20:1 (w/w) as described (28). Finally, the proteoliposomes were washed twice with 50 mM KPi and pH 7.5 and resuspended in 10 mM KPi, pH 5.8 or 8.0, 100 mM KCl, and 5 mM MgCl2 to yield a NhaA concentration of 50–100 μM. Proteoliposomes were frozen and stored in liquid nitrogen until use.
Four-pulse DEER measurements
Four-pulse DEER measurements were performed at a Bruker Elexsys 580 spectrometer (Bruker Biospin GmbH, Karlsruhe, Germany) equipped with a 3 mm split-ring resonator under conditions of strong overcoupling (Q ≈ 100) at a temperature of 50 K. Before insertion into the probehead, the samples were shock-frozen in liquid nitrogen to avoid crystallization of water. The pulse sequence (π/2)ν1 − τ1 − (π)ν1 − t′ − (π)ν2 − τ1 + τ2 − t′ − (π)ν1 − τ2 − echo was used. The π/2- and π-pulses at the observer frequency ν1 had equal pulse lengths of 32 ns to ensure equal excitation bandwidths, whereas the π-pulse at the pump frequency ν2 had a length of 12 ns to maximize modulation depth while still keeping the two excitation bands separate. These conditions maximize sensitivity of the experiment (29). The long interpulse delay was τ2 = 2000 ns with an initial value of t′ = 80 ns and an increment Δt′ = 8 ns. To suppress proton modulation, data were added for eight equidistant values of τ1 between 200 and 256 ns. A phase cycle +x/−x was applied to the first pulse, and the two signals were subtracted. The pump frequency ν2 (typically 9.33 GHz) was set to the center of the resonator mode and to coincide with the global maximum of the nitroxide spectrum. The observer frequency ν1 was set to the local maximum at the low field edge of the spectrum (ν1 − ν2 = 65 MHz). Accumulation times for the data sets varied between 8 and 14 h. Data were analyzed for dipolar evolution times t = t′ − τ1 ≥ 0.
Four-pulse DEER data analysis
Distance distributions and average numbers of spins were obtained from the dipolar time evolution data by the home-written MatLab (The MathWorks, Natick, MA) program package DeerAnalysis 2004 (freely available at http://www.mpip-mainz.mpg.de/∼jeschke/distance.html), which is based on algorithms explained in Jeschke et al. (14) and Weese (30). The background contribution from spin labels in other dimers was fitted either by a three-dimensional distribution (spatial background correction) corresponding to a functional dependence exp(−kt) in time domain or by a two-dimensional distribution (planar background correction) corresponding to exp(−kt2/3). Measurements on single mutants of a monomeric protein had shown that for membrane proteins inserted into liposomes, such a two-dimensional distribution fits data significantly better than a three-dimensional one. Realistic resolution of the distance distributions was estimated from the dependence of the root mean square deviation of simulated from experimental data for Hermite interpolation with different numbers of sampling points. The smallest number of sampling points for which there was no significant deviation of fitted from experimental data was selected. Final distance distributions P(r) were then obtained by Tikhonov regularization in distance domain, incorporating the constraint P(r) > 0. The regularization parameter was adjusted to obtain the realistic resolution determined before. Modulation depths were determined from the level of the fitted dipolar evolution function. As fitting is performed for background-corrected data, these values pertain to the average number of spins in the same NhaA molecule or dimer, excluding spins in remote molecules or dimers.
CW EPR measurements
CW EPR spectra were recorded with homemade X-Band EPR spectrometers equipped with a dielectric cavity (Bruker, Ettlingen, Germany) for room-temperature (T ≈ 293 K) measurements or with an AEG TE-103 cavity for low-temperature (T ≈ 170 K) measurements. The magnetic field was monitored using a Bruker B-NM 12 B-field meter; microwave power was determined using an HP 430C bolometer. The NhaA samples were loaded into quartz capillaries with an inner diameter of 1 mm. At room temperature, 10 scans with a scan width of 12.8 mT (microwave power <0.6 mW, modulation amplitude <0.33 mT) were averaged. At 170 K, 10 or 20 scans (12.8 mT scan width, microwave power <0.4 mW, modulation amplitude 0.23 mT) were accumulated.
For power saturation experiments in the presence of oxygen or chromium oxalate (CROX), the samples were loaded into gas-permeable TPX capillaries. The samples were deoxygenated by passing nitrogen around the sample capillary. For oxygen accessibility experiments, nitrogen was replaced by air. Saturation curves were determined from the peak-to-peak amplitudes of the center line measured at seven different incident microwave power levels in the range from 0.5 to 80 mW. The saturation behavior of the samples was parametrized by the quantity P1/2, which is defined as the power level of the incident radiation at which the amplitude of the saturated line is half of the amplitude in the absence of saturation. Values for this parameter were calculated from fitting the function
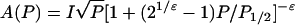 |
(1) |
to the experimental amplitudes A(P) according to the method of Altenbach et al. (31). The scaling factor I and the measure of the saturation homogeneity ɛ were adjustable parameters. The quantity ΔP1/2 was calculated from the difference in P1/2 values in the presence and absence of the relaxing agent. The ΔP1/2 values were divided by the peak-peak line width and normalized by the same quantity of a 2,2-diphenyl-1-picrylhydrazyl (DPPH) standard sample to obtain the dimensionless accessibility parameter Π.
Simulation of CW EPR spectra
CW EPR powder spectra were simulated based on the method described previously (32). The motion of proteins is strongly restricted for temperatures <200 K. Below that temperature, the apparent hyperfine splitting was found to deviate no more than 2% from the rigid limit value determined at 80 K, and the EPR line shape resembles that of a powder spectrum. The values of the g- and hyperfine-tensor components were taken from a sample for which no dipolar broadening is expected: gxx = 2.0084, gyy = 2.0065, gzz = 2.0026, Axx = 0.51 mT, Ayy = 0.40 mT. To account for unresolved hyperfine interactions and relaxational broadening, the calculated powder stick spectrum was convoluted by a field-independent line-shape function (48% Lorentzian and 52% Gaussian with widths of 0.351 and 0.387 mT, respectively). Dipolar interaction is considered by additional convolution with a Pake pattern (32,33). The width of the distance distribution was fixed to 0.2 nm.
RESULTS
Local mobility-CW EPR spectra at room temperature
The room-temperature spectra (T = 293 K) at pH 5.8 of the singly spin-labeled NhaA mutants are depicted in Fig. 1. The spectrum of V254R1 (dotted line) indicates a more restricted motion of the bound spin label compared to H225R1 (solid line). This is revealed by a shift of the low field peak of V254R1 from ∼342.0 mT to 340 mT and a reciprocal line width (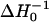 ) of ∼0.16 × 10−4 T−1 compared to 0.22 × 10−4 T−1 at position 225. These values indicate an exposed surface position of the nitroxide at position 225 and a more buried location of the nitroxide at position 254.
) of ∼0.16 × 10−4 T−1 compared to 0.22 × 10−4 T−1 at position 225. These values indicate an exposed surface position of the nitroxide at position 225 and a more buried location of the nitroxide at position 254.
FIGURE 1.

Comparison of normalized room temperature CW EPR spectra (T = 293 K) of mutants H225R1 (solid line) and V254R1 (dotted line) recorded at pH 5.8. Differences in the signal/noise ratio are due to different concentrations of the samples.
We recorded the room temperature EPR spectra of H225R1 and V254R1 at two additional pH values to determine whether changes of pH cause conformational changes. In contrast to the spectra of the V254R1 (Fig. 2 b), the data of H225R1 reveal slight but significant alterations of the mobility of the spin label side chain (Fig. 2 a). The nitroxide becomes successively more immobile as the pH is shifted from 5.8 to 7.0 (dashed line) and 8.0 (dotted line). This is represented by a peak shift toward 342.0 mT and a corresponding slightly more pronounced dent in the high field region between 348.5 mT and 349.0 mT.
FIGURE 2.
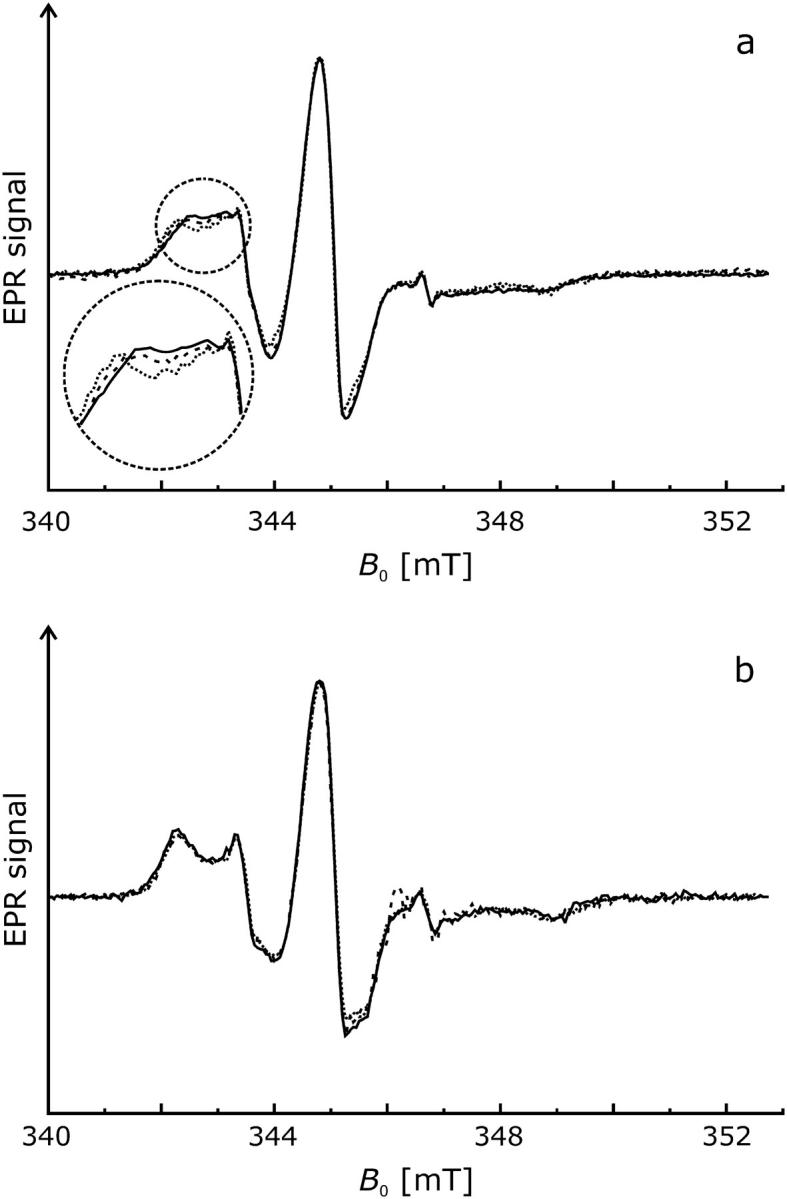
Effect of pH on CW EPR spectra of mutants H225R1 and V254R1 (T = 293 K). Normalized spectra are shown for pH 5.8 (solid lines), pH 7.0 (dashed lines), and pH 8.0 (dotted lines). (a) H225R1. (b) V254R1.
Polarity and proximity-CW EPR spectra at low temperature
The existence of a pH-induced conformational change affecting position 225 is confirmed by low temperature EPR spectroscopy (T = 170 K). Increasing the pH from 5.8 to 8.0 causes a shift of the low field peak minimum to lower field strengths and, consequently, an increase of the Azz value by ∼5 mT. This is evidence for a movement of the R1 side chain into a more polar environment (Fig. 3). In contrast, the change of pH does not affect the Azz value determined for V254R1, which indicates that the environmental polarity of the nitroxide attached to position 254 is not influenced by pH (data not shown).
FIGURE 3.

Effect of pH on the low temperature CW EPR spectrum (T = 170 K) of NhaA mutant H225R1. Normalized spectra are shown for pH 5.8 (solid lines) and pH 8.0 (dotted lines).
Low temperature EPR spectra of H225R1 and V254R1 were also analyzed for possible dipole-dipole interactions, which would be manifested in CW EPR spectra if dimerization leads to interspin distances H225R1/H225R1′ or V254R1/V254R1′ shorter than 2 nm. However, none of the low-temperature spectra of the singly spin-labeled NhaA mutants exhibits significant dipolar line broadening. Best fits of the spectra according to the method described by Steinhoff et al. (32) are obtained with interspin distance values equal to or exceeding (2.0 ± 0.1) nm (data not shown), which corresponds to the upper detection limit of the CW EPR technique. Therefore, we conclude that there is no significant fraction of NhaA with interspin distance values of H225R1 and V254R1 shorter than 1.8 nm. No evidence for dimerization is obtained by CW EPR.
Accessibility measurements by CW EPR
To obtain semiquantitative information on the location of residues H225R1 and V254R1 relative to the membrane, we use power saturation of CW EPR spectra to determine the accessibility parameter Π of the nitroxide side chains for freely diffusing paramagnetic quenchers (31,34). Molecular oxygen and water-soluble CROX are suitable quenchers because of their sizes and solubility properties. Molecular oxygen has a low concentration in the tightly packed protein interior, whereas both the solubility and the diffusion coefficient are high in the lipid bilayer. Therefore, for spin label side chains located in the protein interior, the collision frequency with oxygen and the accessibility parameter ΠOxygen are low, whereas these are high if the spin label faces the bilayer. Likewise, measurements of the collision frequency with the water-soluble CROX allows determination of the location of the R1 side chain with respect to the aqueous phase (31,35). In principle, it has to be taken into account that the collision frequency of the R1 side chain with the negatively charged CROX is a function of both accessibility and local electrostatic potential. However, at the relatively high salt concentrations used here the electrostatic potential is strongly screened, so that accessibility has the dominating influence on the collision frequency.
The results of the accessibility analysis are displayed in Table 1. The low ΠCROX values of 0.01 and 0.02 at pH 5.8 imply low collision frequencies with CROX for nitroxides at both positions 225 and 254. This suggests that both nitroxide groups are either facing the phospholipid bilayer or are buried within the protein monomer or dimer. Also, for both residues, ΠOxygen values of ∼0.4 indicate intermediate oxygen accessibility at pH 5.8, suggesting that the R1 side chain is in contact with the apolar environment of the bilayer. Increasing pH from 5.8 to 8.0 does not significantly affect the accessibility of both nitroxides.
TABLE 1.
Accessibility of nitroxide groups in NhaA mutants H225R1 and V254R1
| pH 5.8
|
pH 8.0
|
|||
|---|---|---|---|---|
| Mutant | Πoxygen | ΠCROX | Πoxygen | ΠCROX |
| NhaA-H225R1 | 0.44 | 0.02 | 0.48 | 0.04 |
| NhaA-V254R1 | 0.40 | 0.01 | 0.27 | 0.02 |
Π, orientation of the spin-labeled side chains with respect to the protein, lipid, or aqueous phase (see text) and is normalized by the quantities of a DPPH standard sample (Π = (P1/2/lw − P1/2(N2)/lw(N2) × lw(DPPH)/P1/2(DPPH)).
P1/2, power level of the incident radiation at which the amplitude of the saturated line is half of the amplitude in absence of saturation.
lw, line width.
Πoxygen, measured in equilibrium with air and scaled to pure oxygen.
ΠCROX, measured in the presence of 50 mM CROX.
Calibration of spin counting by DEER measurements
For sufficiently dilute samples, primary DEER data can be separated into contributions due to spins within the same nanoobject as the observer spin and due to spins in other nanoobjects. This requires that the size of the nanoobject, in our case a membrane protein monomer or oligomer, must be within the distance range accessible to DEER. For membrane proteins, we find that this range extends from 1.75 to 6 nm. In this situation, the modulation depth Δ for the contribution from spins within the same nanoobject depends on the average number 〈n〉 of spins in that nanoobject as
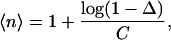 |
(2) |
where the parameter C depends on the excitation profile of the pump pulse and spectral line shape of the nitroxide (13,14). For measurements performed with the same microwave resonator and pulses of the same lengths on nitroxides with similar Azz values, C can be considered a constant. For calibration of that constant across the distance range of interest, we used dilutions in o-terphenyl of the rigid biradicals 3 from Jeschke et al. (36) with an average distance 〈r〉 = 1.96 nm and 1 (〈r〉 = 2.84 nm), 2a (〈r〉 = 3.65 nm), and 2b (〈r〉 = 5.02 nm) from Godt et al. (15), the flexible biradical 4 (〈r〉 = 1.96 nm) from Jeschke et al. (36), and the rigid symmetric triradical 3 (〈r〉 = 3.40 nm) from Godt et al. (15). We find a correlation coefficient of 0.9665. The calibration was tested by independent measurements on rigid biradicals 2a and 2b (15) and the ultralong biradical shown in Fig. 4 A of Jeschke et al. (29) (〈r〉 = 7.6 nm), all diluted in perdeuterated o-terphenyl. In these test measurements, we find 〈n〉 = 2.08, 1.99, and 2.03, respectively, in good agreement with the expectation of 〈n〉 = 2.
FIGURE 4.

Normalized DEER time-domain data for mutants H225R1 (bottom) and V254R1 (top, shifted by 0.2) at pH 8. The dashed lines indicate smooth decaying signals as expected for a homogeneous distribution.
Evidence for specific oligomerization by four-pulse DEER
Pulse EPR distance measurements using the four-pulse DEER sequence were performed for both mutants, H225R1 and V254R1. Primary time-domain DEER data at pH 8.0 are compared in Fig. 4. For a homogeneous distribution of the spin labels in three dimensions or in a lipid membrane, one would expect a smoothly decaying signal as indicated by the dashed lines. Clearly, the experimental data contain additional contributions at short times. These contributions correspond to label-to-label distances within a nano-object composed of several NhaA molecules that must be smaller than the upper limit of DEER distance measurements of 6 nm.
To quantify the label-to-label distance distribution, one has to separate the contribution of labels within the same nanoobject from the contribution of labels in neighboring objects. The latter contribution corresponds to the smooth decay and is of the form exp(−kt) for a homogeneous spatial distribution of spins in three dimensions and of the form exp(−kt2/3) for a homogeneous planar distribution as expected for nanoobjects distributed in a membrane. For singly labeled mutants of Na+/proline transporter PutP in liposomes, we have found that the decay conforms to a planar distribution (D. Hilger, H. Jung, H.-J. Steinhoff, and G. Jeschke, unpublished), whereas singly labeled mutants of plant light-harvesting complex IIb monomers solubilized in detergent micelles conform to a spatial distribution (12). In the case at hand, a planar distribution is more likely but cannot be verified independently by control experiments. We therefore analyze our data for the cases of both planar and spatial distributions (Fig. 5).
FIGURE 5.

DEER distance distributions P(r) obtained from the time-domain data in Fig. 4 by Tikhonov regularization in the range from 1 to 8 nm with a regularization parameter of 100. (a) H225R1 with planar background correction. (b) H225R1 with spatial background correction. (c) V254R1 with planar background correction. The dotted line marks the lower limit of distances that are detectable by DEER with our setup. (d) V254R1 with spatial background correction.
For H225R1 the distance distribution does not strongly depend on the choice of the background distribution. Less noise-related baseline artifacts are observed with the planar distribution (Fig. 5 a) compared to the spatial distribution (Fig. 5 b), which suggests that the planar distribution is the better model. The dominant peak in the distribution is at (4.36 ± 0.04) nm and has a width of ∼0.3 nm, as found by fitting the data with a distance distribution consisting of a single Gaussian peak. The occurrence of one relatively narrow peak strongly suggests formation of an oligomer with well-defined structure. This oligomer cannot be composed of more than four NhaA molecules, as this would imply more than one peak in the distance distribution H225R1/H225R1′. For a tetramer a single peak would only be expected if it had tetrahedral symmetry, which can be excluded for an integral membrane protein. The distance distribution is thus consistent only with a dimer or trimer.
For V254R1, the reliability of the distance distribution crucially depends on the proper choice of background function. A single peak at 2.00 ± 0.04 nm with a width of 0.20 ± 0.10 nm is found under the assumption of a planar distribution of aggregates (Fig. 5 c), whereas a broad distribution covering the whole range between at least 1.5 and 5 nm is found when assuming a three-dimensional spatial distribution (Fig. 5 d). We have checked that the result for the planar distribution is self-consistent: For the distance distribution in Fig. 5 c the time-domain data at t > 600 ns are exclusively due to labels in neighboring nanoobjects. The dimensionality D can thus be determined independently by fitting a function exp(−ktD/3) to the decay at t > 600 ns. We find D = 2.009 in good agreement with the expectation of D = 2 for a planar distribution. Furthermore, as helix IX is thought to extend up to residue P257 (37), one would hardly expect a very broad distance distribution for residue V254R1 as seen in Fig. 5 d, even if one accounts for the conformational freedom of the MTSSL label. Finally the peak at 2 nm persists even with the three-dimensional background correction and is also found at pH 5.8 and 7.0 (data not shown). We thus assume that the distance distributions for H225R1 and V254R1 are both monomodal. In any case it is safe to conclude that in the observed oligomer of NhaA, labeled residues H225R1 are separated by more than 4 nm and there is at least a population of oligomers with a separation of <2.5 nm between labeled residues V254R1.
The probabilities to find a second labeled NhaA molecule at a distance within the DEER range from 1.75 to 6 nm can be compared for H225R1 and V254R1 by comparing the vertical scales of Fig. 5, a and c, or, more directly, the deviations from the smooth dashed line in Fig. 4. Clearly, this probability is lower for V254R1. This result could be explained by assuming that the mutation at position 225 and the attachment of the MTSSL label increase the tendency for oligomerization, or conversely, mutation and attachment at position 254 decrease this tendency. However, our experience with measurements of short distances in the Na+/proline transporter (10) and labeled peptides (J. Banham, G. Jeschke, and C. Timmel, unpublished) suggests that the low modulation depth for V254R1 could also be due to suppression of part of the distance distribution at the lower edge of the DEER distance range (see dotted line in Fig. 5 c), even if significant dipolar broadening cannot be detected in the CW EPR spectra. However, it cannot be excluded that the labeling efficiency at this position is less than that at position 225. Because of this ambiguity and the stronger dependency of data analysis on the choice of background function at position 254, we restrict further discussion of the pH dependence to the mutant H225R1.
Conformational changes that lead to a movement of the periplasmatic end of helix VIII should manifest in a peak shift in the distance distribution for residue H225R1, whereas changes in the degree of oligomerization should result in an amplitude scaling of P(r), as only labels within oligomers contribute to P(r). The data displayed in Fig. 5 a, which were obtained on the same sample with increasing pH, rule out any significant peak shift, but point to a sizeable amplitude change. Similar data were obtained on a second sample with decreasing pH (not shown). Quantitative analysis of the modulation depth can thus provide more information on the type and extent of oligomerization.
Dependence of the average degree of oligomerization on pH
As mentioned above, the average number 〈n〉 of labels per protein oligomer can be obtained from the modulation depth. To obtain precise values, the contribution due to labels in neighboring oligomers has to be removed by a background correction that is as accurate as possible. Ideally, the background is fitted at times where modulation due to labels within the same oligomer has decayed completely. For the distance of 4.36 nm in H225R1 oligomers of NhaA, such complete decay is not observed within experimentally feasible observation times. In this situation the background fit can still be optimized, since we know that for distances larger than the expected size of the nanoobject P(r) must approach zero. For a given background function, this optimization is done by varying the starting time t0 of the background fit and eliminating the background as well as computing P(r) by approximate Pake transformation (36) for each choice of t0. For all data sets of H225R1 samples, we find optimum values of t0 between 360 and 376 ns. Further analysis was thus performed with t0 = 368 ns as indicated by the dashed line in Fig. 6 a. The best planar background fit by a function exp(−kt2/3) is shown as a solid line. The dipolar evolution function corresponding to only labels within the same oligomer is now obtained by dividing the original data (dots) by the background function. The dipolar spectrum obtained by Fourier transformation of the corrected data (Fig. 6 b) has the expected shape of a broadened Pake pattern and is well fitted by the dipolar spectrum corresponding to the distance distribution in Fig. 5 a.
FIGURE 6.

Measurement of the pH dependence of the average oligomerization number 〈n〉 of the H225R1 spin-labeled mutant of NhaA. (a) Primary data (dots) at pH 8 normalized to the echo intensity at t = 0 with planar background fit (solid line). The fit range are times t > t0 with t0 indicated by the dashed line. (b) Dipolar spectrum obtained by Fourier transformation of background corrected data (dots) and fit by the spectrum corresponding to the distance distribution shown in Fig. 5 a. (c) Dipolar evolution function due to labels within the same oligomer (dots) obtained by the background correction and fit corresponding to the distance distribution shown in Fig. 5 a. Data are normalized to the background-corrected echo intensity at t = 0. The dashed lines and double arrow indicate the total modulation depth Δ. (d) pH dependences of 〈n〉 obtained in two series of measurements with increasing pH (▵, bold error bars) and decreasing pH (∇, slim lined error bars). Error bars are estimated from calibration experiments on a sample set of six chemically pure biradicals and one triradical.
This high fit quality indicates that we can confidently extract modulation depths Δ (Fig. 6 c) and thus reliable estimates of the average number 〈n〉 of labels per oligomer from our data. Indeed, the data in Fig. 6 d demonstrate that the dependence of 〈n〉 on pH for H225R1 samples of NhaA agrees within the expected error for two different sample preparations measured with increasing and decreasing pH, respectively. We find that 〈n〉 increases slightly but significantly upon shifting pH from 5.8 to 8. On the other hand, lowering pH from 8 to 5.8 decreases 〈n〉, indicating that the pH effect is completely reversible (Fig. 6 d).
To interpret 〈n〉 as an average number 〈nm〉 of NhaA molecules per oligomer, we have to make an assumption about the fraction f of labeled protein molecules. Incomplete labeling of the protein corresponds to f < 1. We find
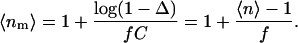 |
(3) |
Independently of the value of f, 〈n〉 > 1 implies 〈nm〉 > 1. Hence, our measurements show that even at the lowest pH value of 5.8, a significant amount of dimer is present. The value of 〈n〉 ≈ 1.6 at pH = 8 would correspond to complete dimerization for f = 0.6. Indeed, we found by determination of the spin concentration and protein concentration of a H225R1 sample after the DEER measurement that at least 60% of the protein molecules are labeled (f ≥ 0.6), hence 〈nm〉 ≤ 2. Within experimental error, our results are thus consistent with complete dimerization at pH = 8 but do not exclude incomplete dimerization at that pH value. Note that the relative change in 〈nm〉 − 1 is independent of the uncertainty in f and is nicely reproducible. We can thus safely conclude that for H225R1 mutants, a fraction of oligomers is already present at pH 5.8 (〈nm〉 − 1 > 0) and that the degree of oligomerization increases significantly when increasing pH form 5.8 to 8.
DISCUSSION
Comparison with structural models for NhaA dimers
For mutant H225R1, we find that the average number 〈n〉 of spin labels at distances in the DEER range from 1.75 to 6 nm depends on pH (Fig. 6 d). Clearly, a fraction of NhaA undergoes a change in the pH range between 5.8 and 8. Furthermore, distances H225R1/ H225R1′ shorter than 1.8 nm are inconsistent with the CW EPR spectra at any pH. We may thus safely conclude that for a fraction of NhaA, a decrease of pH from 8 to 5.8 causes an increase in the distance between H225R1 and H225R1′ from 4.36 nm to more than 6 nm. This observation is most easily explained by dissociation of a fraction of NhaA dimers at lower pH, i.e., by a pH-dependent dimerization equilibrium. In principle, our data could also be explained by pH-dependent dissociation of a symmetric trimer. However, as the low-resolution crystal structure (19,20) also suggests dimerization, we feel that the latter explanation is unlikely. A symmetric trimer could also hardly be reconciled with the cross-linking experiments (21), which show that dimers cross-linked between residues H254C are active and, for a long, flexible cross-linker, exhibit essentially the same pH profile of activity as NhaA that is not cross-linked. It seems implausible that such linked dimers could form a symmetric trimer in which residues H254R1 have a distance of only 2 nm. In the following, we therefore discuss our data in terms of a dimer structure.
Structural models of NhaA with different degrees of detail have been suggested by Williams (20) based on electron crystallography, by Ravna et al. (38) based on the model by Williams and an analogy to Lac permease, and by Rimon et al. (39) based on site-directed thiol cross-linking. The latter two models can essentially be considered tentative assignments of the helices detected in the crystal structure. For a discussion of such assignments, we may note that H225 is the last loop residue before the periplasmic end of helix VIII and V254 is separated by two residues from the cytoplasmic end of helix IX (18). We may thus relate our data and the helix assignments to the horizontal slices through the electron density of the putative dimer reported by Williams (20). The slices 0.8 nm ‘above’ and ‘below’ the membrane plane are shown in Fig. 7, a and b, respectively. It appears likely that the terms ‘above’ and ‘below’ the plane have to be assigned to the periplasmic and cytoplasmic side, respectively, as the cross-linking study (39) suggests that it is the periplasmic side where helices are more densely packed. Note that Ravna et al. (38) make the same assignment. In such projection maps, the locations of the spin labels that are consistent with our distance measurements can be visualized as circles centered at the twofold symmetry axis with a diameter corresponding to the measured distance. Given the size of the spin label and the distance of the labeled residues from the helix, the assignment of helix VIII by Ravna et al. might be consistent with our data (label R:VIII in Fig. 7 a) but the assignment of helix IX clearly is not (label R:IX in Fig. 7 b). Whatever the loop and helix conformations are, it is hardly possible for this assignment to lead to a distance H225R1/H225R1′ that is 2 nm longer than the distance V254R1/V254R1′.
FIGURE 7.
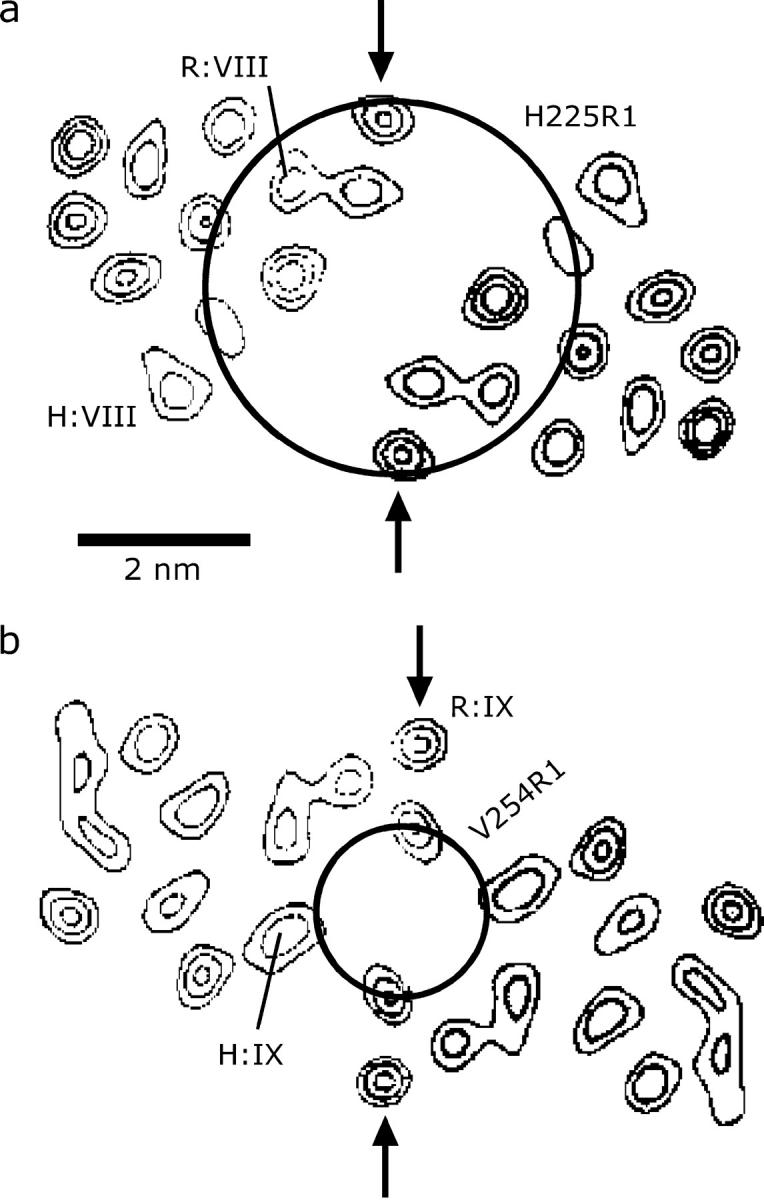
Comparison of intradimer distances between residues 225/225 and 254/254 to electron density maps of NhaA taken from Williams (20) and to assignments of transmembrane helices VIII and IX in these maps. Arrows pinpoint a helix-helix contact in the electron density maps that was assigned as an interdimer contact by Williams (20). Labels R:VIII and R:IX designate assignments made by Ravna et al. (38), whereas labels H:VIII and H:IX are assignments that are compatible with our measurements and conclusions by Rimon et al. (39). (a) Horizontal slice 0.8 nm above the center of the membrane, assigned here to the periplasmic side. (b) Horizontal slice 0.8 nm below the center of the membrane, assigned here to the cytoplasmic side.
Rimon et al. (39) did not explicitly map their helix assignment to the electron density map and made very clear that it is tentative. However, it seems to be clear from their results that helices VIII and IX reside on the outside rather than in the core of the helix bundle. If we assume that this is indeed the case, our data suggest a unique assignment of these two helices in the electron density map (labeled H:VIII and H:IX in Fig. 7).
We also checked whether the helix assignment of Ravna et al. (38) can be rescued by assuming a different choice of the dimer in the crystal structure. This idea is based on a close ‘interdimer’ contact between the helices labeled R:IX in Fig. 7 (see arrow) that was pointed out in the crystal structure by Williams (20). If this is interpreted as an intradimer contact, one obtains an alternative dimer that interacts only via helices R:IX. In this case, a distance V254R1/V254R1′ of 2 nm appears feasible, but the distance H225R1/H225R1′ would be expected to be significantly shorter than 4.3 nm. In principle, it might also be possible that the functional dimer existing in a biomembrane or a liposome is different from the dimer formed in crystals since crystallization is performed at pH 4, significantly below the range where NhaA is active. However, as our measurements show that the extent of dimerization depends only moderately on pH (Fig. 6 d) and distance H225R1/H225R1′ does not at all depend on pH, it appears likely that the dimer observed by us is also formed at pH 4 and sufficiently high concentrations of NhaA. Furthermore, it is possible that substitution of H225 or V254 by Cys and the subsequent labeling reaction affect the monomer-dimer equilibrium. Therefore, it cannot be excluded that the influence of pH on the oligomeric state of NhaA wild-type in the native membrane is much stronger than observed in our experiments. In any case, our assignment of helices VIII and IX in the electron density map is a prediction that can be tested when a crystal structure with atomistic resolution becomes available. Even if it turned out to be wrong, the two distances H225R1/H225R1′ and V254R1/V254R1′ determined by us should allow for identification of the functional dimer from such a structure.
Mechanistic implications of the observed pH dependence
Previous work has demonstrated that the activity of NhaA changes drastically in the studied range between pH 5.8 and 8, not only for the wild-type (23) but also for mutants H225R (24) and V254C (21). Considering this, the pH dependence of mobility, accessibility, and conformation close to residues H225R1 and V254R1 is surprisingly weak. The CW EPR spectra of mutant H225R1 indicate that residue 225 is slightly less mobile and resides in a slightly more polar environment in the dimer as compared to the monomer. These slight changes suggest that the basic mechanism of pH-dependent activation of NhaA does not depend on this residue. Rather, His-225 may serve only to fine tune the pH response of NhaA. In fact, it may not even be involved in the pH sensor but may belong to the part of the protein that conveys the signal from the site of the sensor to the site of the ion translocation pathway.
Note also that the low accessibility of position 225 in the mutant H225R1 to water-soluble CROX is in apparent contradiction to the at least moderate accessibility of this residue to the alkylation reagent N-ethylmaleimide in mutant H225C, as this reagent requires ionization of cystein (26). This contradiction could be resolved by assuming that the thiol group of Cys 225 is accessible, whereas the nitroxide group of the longer side chain of Cys-MTSSL 225 is buried. Alternatively, the residue might be buried in the protein but accessible to nearby water molecules that help to ionize the thiol group. We may not, however, exclude an alternative explanation based on the different timescales of the two experiments. In a dynamic environment, chemical experiments such as cross-linking or alkylation techniques sense proximity or exposure of residues, even if it is realized only during small fractions of the conformational trajectory, as reaction times are in the range of tens of minutes. EPR experiments, on the other hand, measure the time average of accessibility and, for ergodic systems, also the distribution of distances over the whole trajectory.
Just as for position 225, no significant changes of nitroxide accessibility were detected for V254R1. Furthermore, neither the mobility and polarity analyses nor the DEER measurements of distance V254R1/V254R1′ in dimers of NhaA revealed a significant conformational change close to this residue. For the DEER measurement, this failure to detect any change may be partially due to the small modulation depth and relatively poor signal/noise ratio in the measurements on mutant V254R1. We can thus neither exclude nor prove a small change of up to 0.3 nm in this distance in the range between pH 5.8 and 8. Much larger changes, however, can be excluded. On the other hand, a significant change at residue K249 is implied by the strong pH dependence of its accessibility to trypsin (25). Taken together, these results would suggest that loop VIII-IX moves only slightly or does not move at all close to its point of attachment to helix IX, where residue 254 is located, but undergoes a conformational change of its central part that exposes residue K249.
On the basis of the much better DEER data sets for mutant H225R1, we can safely exclude any significant change of distance H225R1/H225R1′. Taken together, these findings indicate that the monomer-monomer interface in the NhaA dimer does not undergo any large scale changes in the range between pH 5.8 and 8.
The increase in the degree of dimerization α of mutant H225R1 with increasing pH (Fig. 6 d) is the most significant change that we observed in this study. As 〈nm〉 = 1 + α, Eq. 3 yields
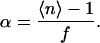 |
(4) |
If labeling is complete (f = 1), our data would correspond to a change from approximately α = 0.35 at pH 5.8 to α = 0.6 at pH = 8. With f = 0.6, which could be assumed as a lower limit of the degree of labeling, we find α ≈ 0.6 at pH 5.8 and α ≈ 1 at pH = 8. In any case, dimerization is substantial at pH 5.8 where antiporter activity of NhaA mutant H225C is almost absent. It may thus be safe to conclude that dimerization is not a sufficient condition for antiporter activity of NhaA. Dimerization may still be a necessary condition, as is indicated by the fact that coexpression of pairs of conditional lethal mutants may restore activity that is missing in the individual mutants (21).
The fact that the degree of dimerization and the structure at the monomer-monomer interface change only moderately with pH may suggest that the pH-dependent ion-translocation pathway is not located in this interface. This may also indirectly be deduced from the fact that many residues which are essential for NhaA function or selectivity are located in transmembrane helices IV, V, and XI (18,40,41), which appear to be remote from helix IX (39), and thus from residue 254 and the monomer-monomer interface. It thus appears to be likely that the pH-induced conformational change of loop VIII-IX translates the primary structural change at pH-sensing residues to the ion-translocation pathway through the membrane. In such a picture, dimerization serves to optimize the pH sensor. The pH dependence of the dimerization equilibrium would then be an indirect effect of the localization of pH sensing residues at or close to the monomer-monomer interface but not be directly related to control of the ion flow by pH.
Acknowledgments
We thank A. Godt and A. Koch for providing the biradicals and triradical for the calibration, and C. Bauer for technical assistance.
This work was financially supported by the Deutsche Forschungsgemeinschaft (SFB 431-P10 and 431-P18 (H.J. and H.J.S.)), Joint Lower Saxony, German-Israeli Research Projects (H.J. and E.P.), and the Israel Science Foundation (E.P.).
References
- 1.Veenhoff, L. M., E. H. M. L. Heuberger, and B. Poolman. 2002. Quaternary structure and function of transport proteins. Trends Biochem. Sci. 27:242–249. [DOI] [PubMed] [Google Scholar]
- 2.Hunte, C., and H. Michel. 2000. Membrane protein crystallization. In Membrane Protein Purification and Crystallization, 2nd ed. C. Hunte, G. Von Jagow, and H. Schaegger, editors. Elsevier Science, San Diego, CA. 143–160.
- 3.Hubbell, W. L., and C. Altenbach. 1994. Investigation of structure and dynamics in membrane proteins using site-directed spin labeling. Curr. Opin. Struct. Biol. 4:566–573. [Google Scholar]
- 4.Hubbell, W. L., D. S. Cafiso, and C. Altenbach. 2000. Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 7:735–739. [DOI] [PubMed] [Google Scholar]
- 5.Steinhoff, H.-J. 2002. Methods for study of protein dynamics and protein-protein interaction in protein-ubiquitination by electron paramagnetic resonance spectroscopy. Front. Biosci. 7:c97–110. [DOI] [PubMed] [Google Scholar]
- 6.Milov, A. D., A. G. Maryasov, and Y. D. Tsvetkov. 1998. Pulsed electron double resonance (PELDOR) and its application in free-radicals research. Appl. Magn. Reson. 15:107–143. [Google Scholar]
- 7.Borbat, P. P., and J. H. Freed. 2000. Double quantum ESR and distance measurements. In Distance Measurements in Biological Systems by EPR. L. Berliner, S. S. Eaton, and G. R. Eaton, editors. Kluwer, New York. 383–459.
- 8.Jeschke, G. 2002. Distance measurements in the nanometer range by pulse EPR. ChemPhysChem. 3:927–932. [DOI] [PubMed] [Google Scholar]
- 9.Pannier, M., S. Veit, A. Godt, G. Jeschke, and H. W. Spiess. 2000. Dead-time free measurement of dipole-dipole interactions between electron spins. J. Magn. Reson. 142:331–340. [DOI] [PubMed] [Google Scholar]
- 10.Jeschke, G., C. Wegener, M. Nietschke, H. Jung, and H.-J. Steinhoff. 2004. Inter-residual distance determination by four-pulse DEER in an integral membrane protein: the Na+/proline transporter PutP of Escherichia coli. Biophys. J. 86:2551–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borbat, P. P., H. S. Mchaourab, and J. H. Freed. 2002. Protein structure determination using long-distance constraints from double-quantum coherence ESR: study of T4 lysozyme. J. Am. Chem. Soc. 124:5304–5314. [DOI] [PubMed] [Google Scholar]
- 12.Jeschke, G., A. Bender, T. Schweikardt, G. Panek, H. Decker, and H. Paulsen. 2005. Localization of the N-terminal domain in light-harvesting chlorophyll a/b protein by EPR measurements. J. Biol. Chem. 280:18623–18630. [DOI] [PubMed] [Google Scholar]
- 13.Milov, A. D., A. B. Ponomarev, and Yu. D. Tsvetkov. 1984. Electron-electron double resonance in electron spin echo: model biradical systems and the sensitized photolysis of decalin. Chem. Phys. Lett. 110:67–72. [Google Scholar]
- 14.Jeschke, G., G. Panek, A. Godt, A. Bender, and H. Paulsen. 2004. Data analysis procedures for pulse ELDOR measurements of broad distance distributions. Appl. Magn. Reson. 26:223–244. [Google Scholar]
- 15.Godt, A., C. Franzen, S. Veit, V. Enkelmann, M. Pannier, and G. Jeschke. 2000. EPR probes with well defined, long distances between two or three unpaired electrons. J. Org. Chem. 65:7575–7582. [DOI] [PubMed] [Google Scholar]
- 16.Schuldiner, S., and E. Padan. 1993. Molecular analysis of the role of Na+/H+ antiporters in bacterial cell physiology. Int. Rev. Cytol. 137C:229–266. [PubMed] [Google Scholar]
- 17.Taglicht, D., E. Padan, and S. Schuldiner. 1993. Proton-sodium stoichiometry of NhaA, an electrogenic antiporter from Escherichia coli. J. Biol. Chem. 268:5382–5387. [PubMed] [Google Scholar]
- 18.Padan, E., M. Venturi, Y. Gerchman, and N. Dover. 2001. Na+/H+ antiporters. Biochim. Biophys. Acta. 1505:144–157. [DOI] [PubMed] [Google Scholar]
- 19.Williams, K. A., U. Geldmacher-Kaufer, E. Padan, S. Schuldiner, and W. Kühlbrandt. 1999. Projection structure of NhaA, a secondary transporter from Escherichia coli, at 4.0 A resolution. EMBO J. 18:3558–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams, K. A. 2000. Three-dimensional structure of the ion-coupled transport protein NhaA. Nature. 403:112–115. [DOI] [PubMed] [Google Scholar]
- 21.Gerchman, Y., A. Rimon, M. Venturi, and E. Padan. 2001. Oligomerization of NhaA, the Na+/H+ antiporter of Eschicheria coli in the membrane and its functional and structural consequences. Biochemistry. 40:3403–3412. [DOI] [PubMed] [Google Scholar]
- 22.Padan, E., T. Tzubery, K. Herz, L. Kozachkov, A. Rimon, and L. Galili. 2004. NhaA of Escherichia coli, as a model of a pH-regulated Na+/H+ antiporter. Biochim. Biophys. Acta. 1658:2–13. [DOI] [PubMed] [Google Scholar]
- 23.Taglicht, D., E. Padan, and S. Schuldiner. 1991. Overproduction and purification of a functional Na+/H+ antiporter coded by nhaA (ant) from Escherichia coli. J. Biol. Chem. 266:11289–11294. [PubMed] [Google Scholar]
- 24.Gerchman, Y., Y. Olami, A. Rimon, D. Taglicht, S. Schuldiner, and E. Padan. 1993. Histidine-226 is part of the pH sensor of NhaA, a Na+/H+ antiporter in Escherichia coli. Proc. Natl. Acad. Sci. USA. 90:1212–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerchman, Y., A. Rimon, and E. Padan. 1999. A pH-dependent conformational change of NhaA Na+/H+ antiporter of Escherichia coli involves loop VIII–IX, plays a role in the pH response of the protein, and is maintained by the pure protein in dodecyl maltoside. J. Biol. Chem. 274:24617–24624. [DOI] [PubMed] [Google Scholar]
- 26.Olami, Y., A. Rimon, Y. Gerchman, A. Rothman, and E. Padan. 1997. Histidine 225, a residue of the NhaA-Na+/H+ antiporter of Escherichia coli is exposed and faces the cell exterior. J. Biol. Chem. 272:1761–1768. [DOI] [PubMed] [Google Scholar]
- 27.Rimon, A., Y. Gerchman, Z. Kariv, and E. Padan. 1998. A point mutation (G338S) and its suppressor mutations affect both the pH response of the NhaA-Na+/H+ antiporter as well as the growth phenotype of Escherichia coli. J. Biol. Chem. 273:26470–26476. [DOI] [PubMed] [Google Scholar]
- 28.Jung, H., R. Rubenhagen, S. Tebbe, K. Leifker, N. Tholema, M. Quick, and R. Schmid. 1998. Topology of the Na+/proline transporter of Escherichia coli. J. Biol. Chem. 273:26400–26407. [DOI] [PubMed] [Google Scholar]
- 29.Jeschke, G., A. Bender, H. Paulsen, H. Zimmermann, and A. Godt. 2004. Sensitivity enhancement in pulse EPR distance measurements. J. Magn. Reson. 169:1–12. [DOI] [PubMed] [Google Scholar]
- 30.Weese, J. 1992. A reliable and fast method for the solution of Fredholm integral-equations of the 1st kind based on Tikhonov regularization. Comput. Phys. Commun. 69:99–111. [Google Scholar]
- 31.Altenbach, C., D. A. Greenhalgh, H. G. Khorana, and W. L. Hubbell. 1994. A collision gradient method to determine the immersion depth of nitroxides in lipid bilayers: application to spin-labeled mutants of bacteriorhodopsin. Proc. Natl. Acad. Sci. USA. 91:1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steinhoff, H.-J., N. Radzwill, W. Thevis, V. Lenz, D. Brandenburg, A. Antson, G. Dodson, and A. Wollmer. 1997. Determination of interspin distances between spin labels attached to insulin: comparison of electron paramagnetic resonance data with the x-ray structure. Biophys. J. 73:3287–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radzwill, N., K. Gerwert, and H.-J. Steinhoff. 2001. Time-resolved detection of transient movement of helices F and G in doubly spin-labeled bacteriorhodopsin. Biophys. J. 80:2856–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altenbach, C., T. Marti, H. G. Khorana, and W. L. Hubbell. 1990. Transmembrane protein structure: spin labeling of bacteriorhodopsin mutants. Science. 248:1088–1092. [DOI] [PubMed] [Google Scholar]
- 35.Anthony-Cahill, S. J., P. A. Benfield, R. Fairman, Z. R. Wasserman, S. L. Brenner, W. F. Stafford 3rd, C. Altenbach, W. L. Hubbell, and W. F. DeGrado. 1992. Molecular characterization of helix-loop-helix peptides. Science. 255:979–983. [DOI] [PubMed] [Google Scholar]
- 36.Jeschke, G., A. Koch, U. Jonas, and A. Godt. 2002. Direct conversion of EPR dipolar time evolution data to distance distributions. J. Magn. Reson. 155:72–82. [DOI] [PubMed] [Google Scholar]
- 37.Rothman, A., E. Padan, and S. Schuldiner. 1996. Topological analysis of NhaA, a Na+/H+ antiporter from Escherichia coli. J. Biol. Chem. 271:32288–32292. [DOI] [PubMed] [Google Scholar]
- 38.Ravna, A. W., I. Sylte, and S. G. Dahl. 2001. Molecular model of the Escherichia coli Na+/H+ antiporter NhaA. Receptors Channels. 7:319–328. [PubMed] [Google Scholar]
- 39.Rimon, A., T. Tzubery, L. Galili, and E. Padan. 2002. Proximity of cytoplasmic and periplasmic loops in NhaA Na+/H+ antiporter of Escherichia coli as determined by site-directed thiol cross-linking. Biochemistry. 41:14897–14905. [DOI] [PubMed] [Google Scholar]
- 40.Galili, L., A. Rothman, L. Kozachkov, A. Rimon, and E. Padan. 2002. Trans membrane domain IV is involved in ion transport activity and pH regulation of the NhaA-Na+/H+ antiporter of Escherichia coli. Biochemistry. 41:609–617. [DOI] [PubMed] [Google Scholar]
- 41.Galili, L., K. Herz, O. Dym, and E. Padan. 2004. Unraveling functional and structural interactions between transmembrane domains IV and XI of NhaA Na+/H+ antiporter of Escherichia coli. J. Biol. Chem. 279:23104–23113. [DOI] [PubMed] [Google Scholar]