

Abstract
Model systems mimicking the extracellular matrix (ECM) have greatly helped in quantifying cell migration in three dimensions and elucidated the molecular determinants of cellular motility in morphogenesis, regeneration, and disease progression. Here we tested the suitability of proteolytically degradable synthetic poly(ethylene glycol) (PEG)-based hydrogels as an ECM model system for cell migration research and compared this designer matrix with the two well-established ECM mimetics fibrin and collagen. Three-dimensional migration of dermal fibroblasts was quantified by time-lapse microscopy and automated single-cell tracking. A broadband matrix metalloproteinase (MMP) inhibitor and tumor necrosis factor-alpha, a potent MMP-inducer in fibroblasts, were used to alter MMP regulation. We demonstrate a high sensitivity of migration in synthetic networks to both MMP modulators: inhibition led to an almost complete suppression of migration in PEG hydrogels, whereas MMP upregulation increased the fraction of migrating cells significantly. Conversely, migration in collagen and fibrin proved to be less sensitive to the above MMP modulators, as their fibrillar architecture allowed for MMP-independent migration through preexisting pores. The possibility of molecularly recapitulating key functions of the natural extracellular microenvironment and the improved protease sensitivity makes PEG hydrogels an interesting model system that allows correlation between protease activity and cell migration.
INTRODUCTION
Cell migration through extracellular matrices (ECMs) is a key step in a variety of physiologic and pathophysiologic situations, ranging from morphogenesis and regeneration to tumor invasion and metastasis. The molecular mechanisms governing three-dimensional (3D) migration are highly complex, involving the coordination of biochemical as well as biophysical cell-matrix interactions (1–3). As the ECM often presents a barrier for migration, cells are forced to apply strategies to overcome the biophysical resistance of their surrounding matrix. Two main strategies for single-cell movement, namely proteolytic (or mesenchymal) and nonproteolytic (or amoeboid) migration (4–7), have been described for several cell types.
It is generally believed that tumor cells and most stromal cells such as fibroblasts or endothelial cells, apply proteolytic strategies for 3D migration (8,9). While migrating, these cells secrete soluble or cell-surface-associated proteases, including matrix metalloproteinases (MMPs) and serine proteases, enabling specific and localized matrix degradation (10–12). On the other hand, it has been shown by application of protease inhibitor cocktails that migrating leukocytes such as T lymphocytes and dendritic cells make use of nonproteolytic, path-finding migration strategies to overcome ECM barriers (4,5,13–16). In this case, 3D migration occurs independently of structural matrix remodeling as an amoeboid process driven by cell-shape adaptation, short-lived low-affinity interactions with the surroundings, propulsive squeezing through preexisting matrix pores, and elastic deformation of the ECM network.
Recent studies on neoplastic and nonneoplastic cells are now revealing that the use of one particular mode of 3D migration is not cell-type specific but rather dynamically and reversibly regulated by environmental cues (5,6,17). Consequently, cell migration has been extended by a new variable, plasticity in migration mode. Sahai and Marshall (18) recently connected this variable to specific intracellular signaling pathways. They observed that the two modes of migration are differentially regulated by Rho GTPases, establishing an important link between actin-determined cell morphology and migration strategy. To date, it has been possible to induce a mesenchymal-to-amoeboid transition in tumor cells by inhibiting protease activity (5,18) and integrin β1 function (17). This plasticity in migration strategy may have important implications, for example, on the efficiency of pharmacologic protease inhibitors. The disappointing outcome of clinical trials, testing MMP inhibitors as anticancer therapeutics, may be related to such plasticity allowing nonproteolytic tumor cell dissemination (19,20).
The use of native and engineered biopolymers as 3D model systems for invasion and migration assays has become indispensable in the development of migration-based therapies against cancer (e.g., agents inhibiting proteases or interfering with adhesion). Protein components of the ECM such as collagen, fibrin, laminin, or elastin (21–24), as well as mixtures of these materials (25,26) or other hybrid protein-based matrices such as Matrigel (27), are widely used as cell-culture substrates due to their inherent resemblance to the natural ECM and complex signaling capabilities. Most biopolymers form complex, 3D fibrillar matrices in a self-assembly and/or enzyme-catalyzed process. The matrix architecture can be altered by varying solid content or gelation conditions, or by adding molecules that induce fiber aggregation or interfere with the cross-linking mechanisms (e.g., Hayen et al. (23); and see Kuntz and Saltzman (51)). This simultaneously alters ligand densities and mechanical properties. The tertiary and quaternary structure of biopolymers features a wide distribution of pore sizes extending into the micrometer range, i.e., in the range of cellular processes involved in motility and the size of a cell body. It is thus to be expected that microscopic porosity in certain biopolymers could enable cell migration, in part or entirely, by nonproteolytic mechanisms as described for amoeboid migration. As a consequence, the microstructure of a model system may be a critical factor in a cell's selection of its migration paradigm, i.e., mesenchymal or amoeboid. To test or screen for candidate protease inhibitor and adhesion antagonists by measurement of migration characteristics, a model system is needed that does not allow the cell to use a mesenchymal-to-amoeboid transition as an escape mechanism. Such a matrix would need a microarchitecture that only allows migration by pericellular proteolysis and offers appropriate adhesion sites for mesenchymal migration.
In consideration of the above, we have explored a novel class of synthetic ECM analogs developed in our laboratory, which due to its homogeneous, nanoscale microstructure was anticipated to restrict cell migration to proteolytic remodeling. These molecularly engineered synthetic hydrogels are based on end-functionalized multiarm polyethylene glycol (PEG) macromers, reacted via Michael-type addition under mild conditions with cysteine-containing peptide sequences, resulting in a hybrid network that can be formed from aqueous precursors in the presence of cells (28,29). PEG acts as an inert structural platform due to its hydrophilicity and resistance to protein adsorption. Hence, it exhibits the desired biological signals uniquely from incorporated peptides or proteins, with minimal structural or chemical background. Similarly, as previously reported, we linked linear RGD-containing peptides as dangling ends (30) and protease-sensitive oligopeptides as elastically active network chains to the PEG macromers (31) to mimic the two essential biological functionalities of an ECM analog: cell adhesion and degradability by proteases. PEG hydrogels were cross-linked either with MMP- or plasmin-sensitive peptide sequences (M-PEG and P-PEG, respectively) to mimic specific degradability. Both sequences have been shown in animal studies to allow proteolytic remodeling and subsequent regeneration of critical-size bone defects (32,33).
We chose well-established standard formulation of collagen and fibrin gels as a benchmark to test the suitability of M-PEG and P-PEG gels as sensitive model systems for proteolytic cell migration. Physical network properties were characterized by confocal laser scanning microscopy (CLSM), perfusion measurements, and rheometry. Primary human neonatal foreskin fibroblasts (HFFs), nonneoplastic cells known to be conducive to tissue remodeling by secreting several classes of proteases, were incorporated into the different substrates and their 3D migration was quantitatively characterized by a persistent random walk. By abrogating MMP function with a broadband MMP inhibitor (GM6001) or stimulating MMP secretion with tumor necrosis factor-alpha (TNF-α) the proteolytic system was modulated and changes in migration were compared across all materials. TNF-α is a major inflammatory cytokine and induces MMP expression in several cell types, including fibroblasts (34). To detect contingently occurring mesenchymal-to-amoeboid transitions, morphometric parameters were recorded simultaneously.
We show that HFF migration in dense M-PEG gels is very sensitive to MMP modulation because it occurs solely by mesenchymal migration whereas in microporous collagen matrices migration occurs independent of MMPs. Furthermore, the pronounced differences in migration and morphology of HFF cultures in M-PEG gels as opposed to cultures in P-PEG gels emphasizes the potential of controlling matrix degradability in model systems for cell migration research. Combining our results from the structural analysis of the different materials and from migration experiments, we propose that the porosity of the matrix might be an important determinant for the sensitivity of 3D cell migration to protease modulation.
MATERIALS AND METHODS
Material and reagents
Branched 4arm PEG macromers, 20 kDa, were purchased from Shearwater Polymers (Huntsville, AL) and functionalized at the OH-termini. Divinyl sulfone was from Aldrich (Buchs, Switzerland). All standard peptide synthesis chemicals were analytical grade or better and were purchased from Novabiochem (Läufelfingen, Switzerland). Fibrinogen was obtained from Fluka (Buchs, Switzerland) and dialyzed as previously described (35). Thrombin and TNF-α were purchased from Sigma (St Louis, MO). Factor XIII was generously provided by Dr. A. Goessel (Baxter Biosciences, Vienna, Austria). Purified bovine dermal type I collagen solution (Vitrogen) was obtained from Cohesion (Palo Alto, CA). Broadband MMP inhibitor GM6001 was from Chemicon International (Temecula, CA). Branched PEG vinyl sulfones (PEG-VS) and peptides were synthesized and characterized as previously described (28). The degree of end-group functionalization of the PEG batch used for this work was ∼95%.
PEG hydrogel preparation
In a typical PEG gel preparation, 4arm-PEG-VS (20 kDa) was dissolved in triethanolamine (TEOA, 0.3 M, pH 8.0) to give a 10% (w/v) solution. The fibronectin derived, integrin-binding peptide Ac-GCGYGRGDSPG (adhesion domain in italic) (30,36) was added in TEOA to this solution. The density of integrin ligands in PEG was approximately matched to fibrin as follows: Human fibrinogen contains three potential integrin binding sites, two RGD sequences within the Aα-chain and a non-RGD sequence in the γ-chain (37). For a fibrin matrix containing 2 mg/ml fibrinogen the adhesion site density is thus ∼35 μM. The PEG-based system was correspondingly functionalized to contain the same density of pendant RGD peptide. After 15 min at room temperature, TEOA containing a bis-cysteine peptide with an MMP-sensitive (e.g., Ac-GCRD-GPQG↓IWGQ-DRCG) (28,38) or plasmin-sensitive (e.g., Ac-GC-YK↓NRD-CG) (33) sequence was added (↓ indicates the cleavage site). MMP-sensitive PEG hydrogels are herein referred to as M-PEG, whereas plasmin-sensitive PEG hydrogels are indicated as P-PEG. Gelation occurred within a few minutes; however, the cross-linking reaction was continued for ∼10 min at 37°C.
Pore-size determination
Pore diameters of 2 mg/ml fibrin and collagen gels were measured in a custom-made perfusion chamber by permeating PBS through the materials at constant pressure, monitoring the mass transport (39). Assuming simplified network architecture of irregularly packed long, straight rods, the pore diameter can be calculated from the Darcy constant (40). Perfusion measurements on PEG hydrogels were unfeasible with our low-pressure device due to the high density of the network. The mesh size, ξ, was therefore calculated on the basis of (41):
 |
(1) |
where 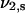 is the polymer volume fraction of the gel in the swollen state equal to the reciprocal value of the swelling ratio Q (
is the polymer volume fraction of the gel in the swollen state equal to the reciprocal value of the swelling ratio Q (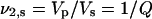 ; Vp, dry volume; Vs, volume in swollen state of gel), and
; Vp, dry volume; Vs, volume in swollen state of gel), and 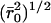 is the unperturbed mean-square end-to-end distance of the PEG:
is the unperturbed mean-square end-to-end distance of the PEG:
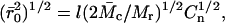 |
(2) |
where l is the average value of the bond length between C-C and C-O bonds in the repeat unit of PEG [-O-CH2-CH2-], taken as 1.46 Å, 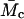 is the average molecular mass between cross-links in the network (11,800 g/mol), Mr is the molecular mass of the PEG repeat unit (44 g/mol) and Cn is the characteristic ratio for PEG, taken here as 4.
is the average molecular mass between cross-links in the network (11,800 g/mol), Mr is the molecular mass of the PEG repeat unit (44 g/mol) and Cn is the characteristic ratio for PEG, taken here as 4.
Rheological measurements
Storage and loss moduli (G′ and G″) on swollen gels were obtained by performing small-strain oscillatory shear experiments on a Bohlin CVO 120 high-resolution rheometer with plate-plate geometry at 25°C and under humidified atmosphere. PEG and fibrin gels were formed between sterile siliconized (Sigmacote; Fluka, Buchs, Switzerland) glass coverslips, detached after gelation and stored in PBS to swell to equilibrium. An amplitude sweep (G′ measured as a function of strain) was performed to confirm that subsequent measurements were conducted within the linear viscoelastic regime. Storage (G′) and loss (G″) moduli, as well as phase angle (δ), were measured as a function of frequency (from 0.01 to 10 Hz) in a constant-strain (0.05) mode to obtain mechanical spectra. Due to their fragility, collagen gels were polymerized between the plates of the rheometer using a gap size of 0.2 mm. After gelation for 1 h, viscoelasticity measurements were conducted as described for fibrin and PEG materials.
Cell culture
Human foreskin fibroblasts (neonatal normal human dermal fibroblasts; Clonetics, San Diego, CA) were cultured in fibroblast cell-culture medium (Dulbecco's Modified Eagle's Medium, with 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic, all Gibco BRL, Life Technologies, Grand Island, NY) at 37°C and 5% CO2. Cells were removed from culture substrates using 0.05% trypsin/0.02% EDTA (Gibco BRL), centrifuged at 400 × g for 5 min, and resuspended in culture medium. For all experiments, cells from subconfluent cultures at passages P9–P12 were used. All migration assays were carried out with medium containing 10% FBS. The same FBS batch was used throughout all experiments.
Confocal microscopy
The microarchitecture of the biopolymers and HFF cell morphology in PEG hydrogels were analyzed by CLSM using a Leica TCS SP2 confocal system (Leica Microsystems, Heidelberg, Germany) and an HCX PL APO 40×/0.75 NA oil immersion objective. Cell-free fibrin and collagen solutions were polymerized directly onto Lab-Tek chambered coverglass (Nalge Nunc International, Rochester, NY) and hydrated in phosphate-buffered saline (PBS) after polymerization. Visualization of fibrin fibrils was achieved by adding Oregon green-labeled fibrinogen (Juro-Chemicals, Lucerne, Switzerland) at a molar ratio of 1:50 to native fibrinogen before gel formation. Collagen fibers were imaged by confocal reflection microscopy at 488 nm (14,42). Both methods allow the assessment of unfixed specimens in the hydrated state. Z-series of 20 equidistant x-y scans at 200 nm intervals were acquired and projected onto a single plane using the Leica TCP-4D reconstruction software or the Imaris software package (Bitplane, Zurich, Switzerland). A preceding x-z scan assured a minimal distance of 25 μm between the imaged volume and the glass surface.
For nuclei and f-actin staining, samples were fixed and permeabilized in 3% formalin containing 0.2% Triton X-100 in PBS for 20 min at 4°C. Samples were incubated for 10 min in 0.1 M glycine followed by a wash step in PBS. For f-actin staining, the gels were incubated, protected from light, with 0.4 units/ml rhodamine-labeled phalloidin (R-415, Molecular Probes, Eugene, OR) in PBS with 1% BSA for 1 h at 4°C. After washing the samples in PBS three times for 5 min, cell nuclei were costained with 1 ng/μl DAPI (D-1306, Molecular Probes) in PBS for 10 min at 4°C.
Cell viability
Cell viability after incorporation into the different matrices was determined with a live/dead cytotoxicity kit (L-3224, Molecular Probes). HFFs were incorporated at a final density of 105 cells/ml in 135-μl gels. At 16 h after gelation the samples were washed twice in PBS and stained for 45 min (fibrin and collagen gels) or 60 min (M-PEG gels) with 200 μl calcein/ethidium homodimer-1 solution. Subsequently, samples were washed in PBS and fixed in 4% glutaraldehyde. Epifluorescence images were taken with a fluorescein and a rhodamine filter set at five randomly selected locations within the gels. The number of surviving cells per total number of cells was determined by standard digital image analysis with Object-Image 2.09 (Norbert Vischer, University of Amsterdam, http://simon.bio.uva.nl/object-image.html, based on NIH Image by Wayne Rasband).
Gel preparation for migration assay
We selected the collagen and fibrin concentration used herein according to widely used formulations in literature (5,23, and many others). In contrast to synthetic PEG hydrogels, collagen and fibrin gels are only subjected to negligible volume change upon polymerization and hydration (Table 1) and do not generate internal stresses without the active participation of cells when gelled in a confined state (e.g., adhered to a surface). To create the same stress-free initial condition, PEG hydrogels were hydrated under free-floating conditions, whereas the biopolymers were polymerized directly onto tissue culture polystyrene.
TABLE 1.
Physical and structural characteristics of PEG, collagen, and fibrin gels
| Material | Main cross-linking mechanism | Solid content (mg/ml) | Average pore diameter (μm) | G′ storage modulus (Pa) | G″ loss modulus (Pa) | δ phase angle (°) | Q swelling ratio | Volumetric swelling |
|---|---|---|---|---|---|---|---|---|
| PEG | covalent | 21.7 | 0.025 | 290 ± 18 | 2.6 | 2.3 | 42 ± 4 | 4.6 |
| Fibrin | covalent | 2 | 0.6 ± 0.1 | 26.8 ± 6.4 | 4.8 | 5.0 | — | 1 |
| Collagen | covalent and physical | 2 | 7.4 ± 1.1 | 6.8 ± 2.7 | 2.2 | 17.7 | — | 1 |
The average pore size and the swelling ratio Q for PEG was calculated as described in Materials and Methods. Pore size of fibrin and collagen gels is based on perfusion measurements (mean ± SD, n = 4) and calculated by Darcy's law (40). Both values are in good agreement with estimates from image analysis of CLSM projections. The values for storage and loss modulus, G′ and G″, as well as the phase angle, δ, were derived as means from rheological measurements between 0.5 and 1 Hz. For G′, SD for n = 4 is given. Volumetric swelling was measured on a buoyancy balance.
M-PEG and P-PEG gels for 3D experiments were polymerized in the presence of cells as described above between glass slides for 10 min at 37°C. Subsequently, samples were detached and transferred into normal cell-culture medium to allow swelling under unconfined conditions.
Fibrin gels were made in 24-well flat-bottomed culture plates by mixing 6.8 mg/ml fibrinogen solution, Tris-buffered saline (10 mM Tris/HCl, pH 7.4, 150 mM NaCl), cell suspension, 1 NIH units/ml thrombin and 10 NIH units/ml factor XIII to give a final concentration of 2 mg/ml. At 10 min after inducing gelation by added thrombin, the samples were overlaid with cell-culture medium and allowed to continue gelation for at least 1 h at 37°C.
Collagen gels were reconstituted from purified bovine dermal type I collagen solution (Vitrogen Cohesion Technologies, Palo Alto, CA) to form 2-mg/ml collagen matrices. Physiological pH and ionic concentration were adjusted with 10× PBS and 0.1 M NaOH. The collagen cell suspension was pipetted into 24-well plates and allowed to polymerize at 37°C for 60 min in a 5% CO2 atmosphere before addition of cell-culture medium. Due to the slow polymerization compared to the PEG or fibrin system, part of the cell population sank to the bottom of the gels during fibrillogenesis. To prevent cell contact with tissue-culture polystyrene, the wells were prelayered with a thin film of hydrated bovine dermal type I collagen. This procedure led to three-dimensionally incorporated cells and attached gels firmly to the well bottom, preventing gel compaction. All gels reached a final volume of 135 μl in the equilibrium-swollen state and had a typical cell density of 105 cells/ml gel.
Unless indicated otherwise, cells were incubated for 12 h after gelation before exogenous factors were added. The imaging sequence followed a preincubation period of 12 h after addition of factors to exclude transient effects. A relatively large volume of culture volume per cell (1 ml/104 cells) and saturating concentrations of exogenous factors (GM6001: 10 μM; TNF-α: 5 ng/ml) prevented depletion of factors over the course of the experiment. To minimize evaporation of the medium during the imaging period, a sterile, light, gas-permeable silicon oil (Silfar S184, Wacker, Munich, Germany) was overlaid on the medium.
PEG gels were not adherent to the well bottom. To minimize dislocation of the samples during stage movements, a 0.8-mm-thick Teflon annulus was placed on top of the gel, touching only at the outer rim but stabilizing its position. The biopolymers were adherent to the polystyrene of the 24-well plates.
Migration assay
Multiple cells were tracked at low magnification by their 2D projections of the real 3D tracks, similarly to an approach described by Dickinson and coworkers (43). This method has the advantage of a high tracking rate, and we were thus able to simultaneously track up to 2000 cells with an interval Δt = 15 min. Standard sized gels containing 105 cells/ml were placed in a 24-well plate on a Leica DM-IRBE inverted optical microscope equipped with a software-controlled motorized stage and focus for automated x, y, and z positioning. A stage incubation system (Tempcontrol 37-2 and CTI-Controller 3700; PeCon, GmbH, Erbach Bach, Germany) maintained the cells at physiological conditions (37°C, 5% CO2 and high relative humidity). Bright-field images were acquired through a 10 × 0.25 NA objective with a charge-coupled device camera (XC-75CE, Sony) mounted on a 0.55× zoom lens yielding a measurement window of 1138 × 887 μm. Each gel was repeatedly imaged in 15 min intervals at 4–6 randomly chosen locations over 50–100 time steps (12.5–25 h). Attention was paid to assure that the imaging volume did not comprise any sample boundaries. To allow high tracking rates, the image time stacks from each location were analyzed off-line by custom software macros written for a modified version of Object-Image 2.09 (Norbert Vischer, based on NIH Image by Wayne Rasband) and yielded cell centroid coordinates and morphometric parameters assigned to a cell track.
Statistical analysis of migration parameters
A common method of cell migration analysis is based on the correlated random walk model, assuming that the correlation of the cell velocity decays exponentially in time with the characteristic time constant P, called directional persistence time (44,45). The mean-squared displacement of a cell, 〈d2(t)〉 during a time interval t is related to P and the root-mean-squared speed S by the relation
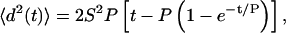 |
(3) |
which applies for cells migrating on planar substrates as wells as in 3D. In a conventional-microscopy setup for cell-migration analysis, only 2D projections of the actual 3D migration path can be recorded. By assuming isotropic material properties, and therefore isotropic migration, the measured, projected cell speed S2D was corrected by a geometry factor of sqrt(3/2) to give an estimate of S. To validate the assumption of isotropic cell movement, the migrated distance in 1 h was plotted against the migration direction in a polar plot (see Fig. 5 A). No preferential direction was detected. The mean-squared displacements 〈d2(t)〉 from experimental cell-track data were calculated by the method of overlapping intervals (46) and corrected as well to predict the 3D displacement from the 2D projections. Using the dimension-corrected cell speed S as an input parameter and fitting Eq. 3 to the dimension-corrected displacement data by a nonlinear least-square regression analysis, an estimate of the directional persistence time P was obtained. The persistence length L was defined as the product L = P × S.
FIGURE 5.

Polar plots of migrated distance and histograms of migration parameters for the four matrices. Each track is assigned either to a migrating (black) or a nonmigrating (shaded) group based on the persistence length criterion Lcrit ≥ 3 μm. (A) Cell tracks were segmented into 60-min parts. Distance and direction migrated during that time is displayed in polar coordinates (r, φ), where r is the distance in μm and φ the angle from start to end-point. Nonmigrating cells are confined to the center. For P-PEG, very few cells were detected as migrating. With increasing pore size, migrated distances increase noticeably. No preferential direction was apparent. (B) Histograms show the pooled data of three independent experiments consolidating n cell tracks with a minimal length of 150 min. Depicted are mean cell speed (left column) and persistence time (right column) of individual tracks from migrating (black) or nonmigrating (shaded) cells, including a nonparametric density curve for the whole population. The number of cells having a mean speed or persistence time within a certain bin of the histogram is divided by the total number of cells to give the normalized number indicated on the probability axis. Tracks with persistence time values derived from regression analysis with R2 < 0.75 were excluded from histograms.
The S and P values of individual cell tracks were pooled from all locations in all samples with the same treatment to generate cell-population histograms and box plots extending from the 25th to the 75th percentile, including the median, whiskers from the 10th to the 90th percentile, and a symbol corresponding to the arithmetic mean. Tracks with persistence time values derived from regression analysis with R2 < 0.75 were excluded from the study, disqualifying 3–14% of all cell tracks. Confidence intervals of the mean for an α-level of 0.05, as well as SE, were calculated for each group to make an initial assessment of the differences between means. Homogeneity of variances and normality of errors was not given for migration parameters. For statistical analysis, multiple comparisons were therefore conducted with the Dunn's test, a nonparametric rank sum test suitable for unequal sample sizes.
In addition to the above-mentioned analysis of whole-cell populations, it proved to be useful to define a criterion for dividing the cell ensemble into a motile and a nonmotile fraction. We used a persistence length criterion Lcrit = 3 μm as a threshold: only those cells with a persistence length L ≥ Lcrit were defined as motile. Since we were interested in the proteolytic migration activity of cells, this criterion allowed us to exclude cells from the motile population, which were either dead or only migrating back and forth in preformed cavities developed during the gelation process. This separation was especially useful for M-PEG gels, since volume, occupied by the cell during gelation increased nearly by a factor of 5 during hydration, creating a small cavity in which the cell could freely migrate without proteolytic effort. The threshold level of Lcrit = 3 μm was estimated by the mean cell size and validated experimentally.
Cell morphometric analysis
During automated cell tracking, simple morphometric parameters were recorded for each cell over time. Cell shapes were transformed into ellipses of equal area and orthogonal moments of inertia, allowing the definition of a cell orientation and compactness. From the ellipse area A and the perimeter U, the normalized compactness C was computed by C = 4π × A/U2 (C = 1 for a circle, C < 1 for an ellipse). These parameters were averaged for each cell over the entire tracking time to give mean area and mean compactness values before being pooled for each condition; ∼400 cell tracks from four samples per material and condition were analyzed. The distribution of the mean cell area and the mean ellipse compactness were displayed as box plots as described for migration parameters. Statistical analysis was performed by the nonparametric Dunn's test.
Gelatin zymography
Supernatants of TNF-α-treated and untreated HFF cultures in different matrices were analyzed for gelatinolytic activity by SDS-PAGE under nonreducing conditions. Fibroblasts were cultured in 3D for 2 days before 5 ng/ml TNF-α was added for 24 h, unless indicated otherwise. Supernatants were collected and total protein concentrations were determined using the Pierce BCA protein assay (Pierce, Rockford, IL) with bovine serum albumin standards. Equal amounts of protein (3.5 μg) were loaded on acrylamide gels containing 1 mg/ml gelatin and 8% acrylamide. Electrophoresis was carried out for 45 min at room temperature. After washing in water, SDS was extracted from acrylamide gels with 2.5% Triton X-100 for 2 × 30 min. Gelatinolytic activities were developed in a buffer, pH 8.0, containing 50 mM Tris-HCl, 5 mM CaCl2 and 0.02% NaN3 at 37°C for ∼24 h and visualized by staining with Coomassie Blue R-250.
RESULTS
Biopolymer microarchitecture
CLSM images of the 3D structure of fibrin and reconstituted collagen gels (both at 2 mg/ml solid content) demonstrate the microarchitectural disparity between the two (Fig. 1). Collagen consists of long, seldom end-linked fibers with very low curvature. The pore diameter was in the range of 1–10 μm. Fiber diameter was uniform and ∼500 nm, which is in accordance with the literature (47). The fibril density was much higher for fibrin, the distance between apparent cross-links being accordingly smaller. The pore size of these matrices was in the range of 0.1–1 μm. Since pore size estimations from CLSM projections can be afflicted with errors, additional perfusion measurements were conducted on the biopolymers. The resulting pore diameter of ∼0.6 μm for fibrin and ∼7 μm for collagen confirm our visual estimations as well as values from literature (22,23,42,48) (Table 1).
FIGURE 1.

Representative CLSM images of 2 mg/ml fibrin (A) and 2 mg/ml reconstituted type I collagen (B) demonstrate considerable architectural discrepancy between the two biopolymers. Both matrices show a fibrillar structure, but with pore size differing by 1–2 orders of magnitude. The pore structures were quantified as reported in Table 1. Scale bar, 20 μm.
PEG hydrogels, on the other hand, consist of a dense network of macromolecular chains, which can neither be investigated by perfusion measurements nor visualized by microscopy. As an approximation of the molecular gel porosity, the mesh size of the network was calculated and is indicative of the maximum diameter of a molecule that can still diffuse through the network. Using the theoretical molecular weight between cross-links of 11,800 g/mol and an experimentally determined swelling ratio Q (swollen gel volume/dry gel volume) of 49 ± 2, a value of ∼25 nm was calculated. The mesh size is a crude estimation of the average porosity in the synthetic network presuming ideal cross-linking. In reality, molecular defects such as unreacted groups leading to dangling chains and macroscopic inhomogeneities will be present that substantially increase the effective mesh size (28). Physical entanglements, on the other hand, which are not accounted for in the estimation, will decrease the mesh size. Nevertheless, the value estimated from swelling measurements gives a correct order of magnitude, and the pore diameter of the PEG hydrogels is far smaller than that of either the fibrin or collagen matrices and also far smaller than that of cellular processes involved in migration.
Mechanical characteristics
As a further indicator of physical network structure, storage and loss moduli (G′ and G″) were evaluated as a function of frequency by conducting small strain oscillatory shear experiments (Fig. 2). In the frequency range 0.1–1 Hz, the three matrices exhibited storage moduli G′ at low levels ranging from 7 Pa for collagen to 290 Pa for PEG hydrogels (Table 1). As G′ in the plateau range can be correlated with the concentration of elastically active network chains or cross-link density (including both covalent and physical cross-links), the mechanical characteristic of three materials seem to correlate with their previously studied microarchitecture. The phase angle δ, characterizing the ratio of the two moduli (tanδ = G″/G′) was highest for collagen, followed by fibrin and PEG, reflecting the difference of the networks with respect to their noncovalently cross-linked nature. These data demonstrate that PEG and fibrin gels are almost ideal elastic materials at low frequencies (δPEG = 2.3°, δFib = 2.3°), whereas the viscous component in collagen cannot be neglected (δCol = 17.7°). A dramatic increase of the moduli at higher frequencies was observed for the biopolymer gels, which is an additional indication of noncovalently bound network structures relaxing at higher frequencies.
FIGURE 2.

Typical mechanical spectra for PEG (A), fibrin (B), and collagen (C). G′ exhibited a plateau at lower frequencies and G″ was typically 1–3 orders of magnitude smaller than G′, representing the predominantly elastic behavior of all gels. The difference in cross-linking density resulting from distinct gel formation mechanisms (covalent bond formation in PEG and partially in fibrin; physical cross-linking in collagen and partly in fibrin) is visible by the magnitude of the phase angle and the increase of the moduli at higher frequencies.
Cell viability
Cell viability for all three materials was ∼90–95% (Fig. 3). In M-PEG gels it was observed that the survival rate critically depended on the gelation duration. As a consequence, gelation condition and duration for synthetic P- and M-PEG matrices was kept strictly constant.
FIGURE 3.

Mean cell viability (shaded) from five fields of three gels per condition are shown. Error bars represent SD of the mean sample values. Dark shaded portions of the bars above the 100% limit indicate dead cells after harvesting, before gelation (∼3.5%).
Cell morphology in MMP-sensitive PEG hydrogels is comparable to biopolymers
To determine if a preferential HFF morphology exists for a certain ECM structure, mean cell area and mean compactness recorded during time-lapse experiments were analyzed. By subjecting the cultures to the MMP inhibitor GM6001 and TNF-α treatment, we investigated the sensitivity of cell morphology to modulations of the MMP system (Fig. 4, A–D).
FIGURE 4.

Cell morphology was analyzed by the projected cell area and the normalized ellipse compactness as a function of the substrate. (A and D) The MMP-sensitive cross-linker allows HFFs to spread and attain cell shapes in synthetic M-PEG gels (M-PEG) very similarly to HFFs in biopolymers (FIB, COL). In contrast, HFFs are not able to form a spindle-shaped morphology in plasmin-sensitive PEG hydrogels (P-PEG) as seen by the increased compactness and a decrease in projected cell area. (B) For TNF-α-treated cultures, no differences in morphometric parameters were detected. (C) MMP-inhibited cultures (GM6001) in fibrin showed a significantly increased mean compactness (n = 4; ∼400 cell tracks; (*) p < 0.05; (**) p < 0.01; (***) p < 0.001 for difference to M-PEG or between groups if indicated with brackets; nonparametric multiple comparison with Dunn's test, α-level 0.01). (E) Representative brightfield images of HFFs within the four materials. (F) Representative reconstructed CLSM images of HFFs in MMP-sensitive PEG hydrogels (M-PEG), 2 mg/ml fibrin (FIB), and 2 mg/ml reconstituted type I collagen (COL) stained for f-actin (rhodamine-phalloidin, green) and nuclei (DAPI, red). Scale bars, 100 μm (E) and 30 μm (F).
No significant difference in compactness or projected area was observed in M-PEG gels compared to either of the two biopolymers, indicating the development of a natural mesenchymal morphology in the MMP-degradable synthetic matrix. On the other hand, a pronounced difference in HFF morphology was found between M-PEG and P-PEG gels with identical adhesive properties. Cells cultured in P-PEG gels kept a spherical geometry after gelation, did not extend processes, and stayed smaller (Fig. 4). A similar morphology was observed in synthetic hydrogels lacking adhesion sites or cross-linked with a nondegradable peptide sequence (data not shown).
Antagonizing MMP function by a broadband inhibitor led to a significant increase in cell compactness in fibrin but not in M-PEG gels or collagen (Fig. 4 C). With TNF-α stimulation, no measurable effect on cell morphology was seen (Fig. 4 B). HFFs were cultured for 12 h before GM6001 or TNF-α was added. As a consequence, cells had already migrated and attained a spindle-shaped morphology before MMP inhibition was induced. However, for cultures in M-PEG gels subjected to GM6001 early on, cell morphology stayed spherical as in P-PEG gels (data not shown).
To establish whether HFFs in M-PEG gels were able to build a structured and functional cytoskeleton comparable to cells in biopolymers, we stained for f-actin and imaged by CLSM. Images revealed spindle-shaped cells with very similar f-actin distribution, i.e., cytoskeleta organized into stress fibers, featuring spiky endings in all three matrices (Fig. 4 F). The f-actin was distributed throughout the entire cell body and not confined to the cortex underneath the plasma membrane. This cytoskeletal morphology indirectly suggests firm, integrin-mediated and traction-transducing cell-matrix binding in M-PEG gels, fibrin, and collagen, characteristic for mesenchymal migration.
Cell migration behavior
Single-cell migration parameters were quantified as a function of the surrounding matrix from time-lapse movies (see Supplementary Material, time-lapse sequences). To this end, we calculated normalized speed and persistence time histograms by analyzing the displacement data and created polar plots from 60-min segments of migration paths to verify directional independence of cell movement. Each cell track was assigned to the migrating (black) or the nonmigrating (shaded) group according to the criterion of Lcrit ≥ 3 μm (Fig. 5). With increasing pore size, HFFs migrated longer distances but showed no preferential direction (Fig. 5 A), typical for an unbiased random walk. Therefore, the assumption of an isotropic cell movement, made for estimating 3D parameters from 2D projections of cell tracks, held true. Speed histograms of HFFs migrating in M-PEG gels showed a high fraction of very slow migrating cells (Fig. 5 B, left column). The pronounced histogram peak was located at 0.2 μm/min and <10% of the cells migrated faster than 0.45 μm/min. Cells in collagen migrated on average with a much higher velocity (0.55 μm/min). The cell speed of highest probability in fibrin was similar to that in M-PEG; however, the distribution was broader and the peak of the unimodal speed distribution shifted to higher values, indicating an increased migratory behavior in fibrin compared to M-PEG; >25% of the cells in fibrin migrated at velocities >0.4 μm/min. In collagen, the speed distribution was again broader, changing from a positively skewed to a symmetrical distribution. The distribution of persistence time from cells migrating in M-PEG and fibrin was very similar, with peak values between 4 and 6 min. For collagen, the distribution was only slightly broader and shifted to higher values (Fig. 5 B, right column). The persistence time data of the nonmigrating group showed very low values, whereas the separation of the mean speed for nonmigrating cells was less pronounced. This indicates that cells moving back and forth without migrating (having a considerable speed but no persistence) were assigned as intended to the nonmigrating group. This was particularly the case in P-PEG, where almost the entire fibroblast population was allocated to the nonmigrating group (Fig. 5 B, top row).
TNF-α induces pro-MMP-9 production in human foreskin fibroblasts
To identify the important MMPs involved in cell migration within the three matrices and to elucidate the mechanism by which TNF-α induced MMP-dependent migration, supernatants from HFF cultures were assayed by gelatin zymography (Fig. 6). Zymograms for fibroblasts cultured in M-PEG gels, fibrin, and collagen for 2 days revealed the presence of gelatinolytic activity at 72 kDa and 62 kDa. The identity of these bands was confirmed by Western blot to be proMMP-2 and active MMP-2, respectively. Both signals increased over a 5-day culture period (data not shown). TNF-α did not induce proMMP-2 or MMP-2, but led to a significant increase of the 92 kDa activity, which was shown by Western blot to correspond to proMMP-9. Cells within collagen showed considerably higher signals for active MMP-2 (62 kDa) whereas supernatants from fibrin and M-PEG cultures had indistinguishable gelatinolytic activity.
FIGURE 6.

Typical gelatin zymogram showing TNF-α-induced production of proMMP-9 (92 kDa) by fibroblasts within M-PEG gels (M-PEG), fibrin (FIB), and collagen (COL). Fibroblasts were cultured for 2 days before TNF-α was added at a concentration of 5 ng/mL for 25 h. Cell culture in collagen matrices led to increased gelatinolytic activity at 62 kDa, corresponding to active MMP-2. S, MMP-2 and -9 standard.
MMP inhibition or stimulation has a pronounced effect on single-cell migration in M-PEG gels, but not in fibrin and collagen
To establish whether HFFs were using MMP-dependent migration strategies, we investigated single-cell migration by measurements of mean cell speed (Fig. 7 A) and persistence time in response to MMP stimulation by TNF-α treatment and MMP inhibition by GM6001. As a control, migration on the surface of the same matrices was quantified (Fig. 7 B). P-PEG was not used in the gel surface assay since the same results as for M-PEG were anticipated. The percentage of migrating cells on the gel's surface and in 3D was determined based on the persistence length criterion Lcrit ≥ 3 μm (Fig. 7 C).
FIGURE 7.

Single-cell behavior as a function of MMP stimulation or inhibition with 5 ng/ml TNF-α (TNF-a) or 10 μM GM6001 (GM6001) in MMP-degradable PEG gels (M-PEG), plasmin-degradable PEG gels (P-PEG), 2-mg/ml fibrin gels (FIB), and 2-mg/ml collagen gels (COL). Mean cell speed (A) was analyzed from tracks longer than 10 time intervals and displayed as box plots ranging from the 25th to 75th percentile including the median and whiskers from the 10th to 90th percentile. Diamonds and error bars indicate the group mean ± SE. Additionally, cell migration on the surface of identical materials was quantified (B). The cell populations were split into a migrating and a nonmigrating group, based on the persistence distance criterion L ≥ 3 μm, to visualize differences between materials, migration on the gel surface and within the materials, and the sensitivity to MMP modulation (C), mean ± SD between different samples, n ≥ 4). Migration on the surface of P-PEG was not investigated. Statistical analysis was performed by multiple comparisons with a nonparametric Dunn's rank sum test. For each material, the MMP inhibited and TNF-α-stimulated samples were compared to the control and the controls were compared across all materials with M-PEG ((*) p < 0.05; (**) p < 0.01; (***) p < 0.001).
Between the different matrices tested, an increasing 3D cell speed with increasing pore size was apparent. Cells were able to migrate in collagen more than twice as fast as in M-PEG gels. The main finding, however, was that the sensitivity of cell speed to MMP modulation was only significant between conditions on stimulation versus inhibition in M-PEG, partly in fibrin but not in collagen. In other words, sensitivity increased with increasing density of the matrix. The persistence time had a similar, but somewhat different, trend. Analogous to what was seen for cell speed, a significant difference in persistence time was only observed in M-PEG gels (data not shown).
As anticipated, cell speed disparity on the gel's surface was much smaller than in 3D (mean speed 0.36–0.44 μm/min). It is noteworthy that cell tracks were much more tortuous on collagen than on M-PEG or fibrin, resembling 3D cell tracks within collagen (Fig. 7 B). In fact, when comparing the results for surface and 3D migration, cell speed and persistence time are very similar for collagen, but not for PEG or fibrin, as P was several fold higher on the gel's surface, indicating straighter cell tracks. This illustrates that cell migration within compared to upon M-PEG and fibrin is clearly dissimilar, but migration on the surface of a material with a pore size like collagen resembles 3D migration.
The most substantial differences between materials and MMP modulation conditions were found in the data of relative migrating cell number (Fig. 7 C). In M-PEG gels, the number of migrating cells increased by a factor of 2 with TNF-α stimulation to levels comparable with collagen. MMP inhibition had a pronounced opposing effect and suppressed migration almost completely. On the other hand, the number of migrating cells in collagen remained at the same high levels, irrespective of MMP stimulation or inhibition. The percentage of migrating cells in fibrin was markedly reduced by MMP inhibition. However, inhibition was less efficient than in M-PEG gels. The response of fibrin-hosted cells to TNF-α was surprisingly inhibitory rather than stimulatory, but statistically not significant (p > 0.2). Strikingly, irrespective of MMP inhibition, <1% of the HFFs were able to migrate in P-PEG gels. The fraction of migrating cells on the surface of the matrices was ∼85% and similar for all materials.
DISCUSSION
Recent findings have added a new variable to the process of migration, plasticity in migration mode (5,6,17). In this report, we introduce synthetic PEG hydrogels as a novel model system for quantifying 3D cell migration and provide evidence that this matrix restricts migration to adhesion-dependent proteolytic matrix remodeling due to its nanoscale microarchitecture. The variable of migration mode is thereby eliminated and the correlation between proteolysis and migration enabled. This material is thoroughly compared on a biophysical and a biological level with standard formulations of two widely used biopolymers in cell migration research, namely collagen and fibrin.
Biophysical comparison of model matrices
Biophysical properties of collagen, fibrin, and PEG hydrogels were investigated by perfusion measurements, rheology, CLSM, and, in the case of PEG hydrogels, estimation of pore sizes by a theoretical approach. Our structural analysis of fibrin and collagen gels revealed pore sizes in the same range as cellular processes involved in motility. Perfusion measurements are in line with CLSM images and similar to literature values (23,39) indicating pore diameters of 7 μm and 0.6 μm for collagen and fibrin, respectively (Table 1, Fig. 1). It has to be noted, however, that these values are means of a broad distribution of sizes that cannot be assessed by perfusion measurements. Collagen fibril-fibril associations are to a high degree noncovalent in nature (entanglements), which was substantiated by rheology measurements and CLSM of living cells, demonstrating that cells were able to dislocate single collagen fibers (data not shown). In entangled networks, viscoelastic relaxation mechanisms can take place and are responsible for a higher phase angle in rheological measurements of collagen. Indeed, at long times (hours), collagen gels have been demonstrated to exhibit the characteristics of a linear viscoelastic fluid (48,49). This implies that the matrix resistance against a slowly invading cell is reduced due to creep mechanisms and that cells might be able to migrate through the entangled network only by fiber dislocation. Therefore, cell-mediated local fiber rearrangement necessary for migration can be expected to occur to a certain degree by force application and not purely by proteolytic remodeling. The measured storage modulus of collagen, G′Col ≈ 7 Pa, is in good agreement with literature (15.5 Pa with 2.1 mg/ml reconstituted type I collagen (50,51)), as well as with the theoretical estimate calculated from the Stokes-Einstein equation (G0 = c × k × T/Me = 12.5 Pa, where Me = 407 kDa is the molecular weight between entanglements, estimated from a triple-helical type I collagen molecule). Fibrin monomers associate spontaneously to fibrils, which undergo covalent interfibril γ-chain cross-linking to form a stable fibrin network (22). The elastic behavior of fibrin at low frequencies and the intermediate storage modulus, G′Fib ≈ 30 Pa, mainly originate from these covalent cross-links (Fig. 2 B). In this case, it is probable that matrix remodeling must be accomplished to a substantial part by cell-secreted enzymes during migration. With a typical pore being <1 μm in diameter, and with the fibrils being more or less rigidly fixed due to interfibril cross-links, it is only through the occasional large pore that a cell may migrate nonproteolytically. In marked contrast, the investigated synthetic hydrogels are block copolymers with a mean distance between cross-links in the nanometer range, and therefore, at low frequencies they have a negligible viscous component and possess the highest storage modulus, G′PEG ≈ 300 Pa, of the three materials (Fig. 2 A). PEG hydrogels present themselves to incorporated cells as an isotropic continuum without sensible physical architecture. Consequently, embedded cells should not be able to migrate without proteolytic effort.
In summary, it can be stated that the three different ECM model systems (PEG hydrogels, fibrin, and collagen) exhibit extensive microarchitectural differences (pore sizes < 0.1 μm, 1 μm, and 10 μm, respectively) and, to a lesser extent, also mechanical diversity (storage moduli 300 Pa, 30 Pa, and 10 Pa, respectively). The adhesiveness of the materials was not assessed experimentally but RGD density in PEG hydrogels was adjusted to the adhesion-site density in fibrin.
Cell migration in PEG hydrogels is restricted to proteolysis
It has been shown for collagen (52) and fibrin (23) that the microarchitecture of biopolymers is a major determinant of 3D cell migration. Hayen and coworkers proposed that highly porous biopolymers reduce the need for proteolysis, whereas in compact fine fiber gels pericellular proteolysis may be the rate-limiting step in cell migration (23). The observation that neoplastic cells can undergo mesenchymal-to-amoeboid transition as an escape mechanism to retain their migration capacity when deprived of their matrix-degrading (5,6,17) or matrix-binding (18) machinery can further complicate the interpretation of experimental migration results in terms of pericellular proteolysis. Although the interrelation between matrix microarchitecture and the ability of a cell to choose between migration modes have not been studied systematically yet, it is very likely that below a critical porosity, amoeboid migration is not possible. We hypothesized that synthetic protease-degradable PEG hydrogels, although providing the essential signals for cell survival, features pore sizes far below that critical limit and that pericellular proteolysis is the rate-limiting step in migration. To test this hypothesis we investigated viability, morphology, organization of the f-actin cytoskeleton, and migration of HFFs in the different materials.
Cell viability for collagen, fibrin, and M-PEG was high and very similar (Fig. 3). Any intermaterial differences in other parameters investigated in this study are therefore unlikely to be related to viability. Projected mean cell area and mean compactness were used as simple morphometric characteristics for cell-matrix interactions and as indicators for a mesenchymal or amoeboid migration mode. Morphometric parameters of HFFs in biopolymers did not differ significantly from those in M-PEG gels and indicated a spindle-shaped, mesenchymal HFF morphology. However, a pronounced dissimilarity was seen between M-PEG and P-PEG gels. HFFs remained rounded and did not spread or migrate in P-PEG gels, whereas only the protease sensitivity of the material was different from M-PEG gels. This is likely due to a proteolytic deficit within the cellular system in culture, described as follows. Plasminogen is produced as an inactive precursor mainly in the liver and activated by a number of enzymes, including tissue- and urokinase-type plasminogen activators (53) and MMPs (54). Plasminogen is also present in the extravascular space of most tissues, and some cells in the connective tissues are capable of expressing it. Our results suggest that HFFs are missing one or more components of the serine protease system to secrete, bind, and activate plasminogen for the degradation of P-PEG gel. Consequently, they are unable to acquire normal mesenchymal morphology, and migration is suppressed. Forcing cells into a spherical shape by culturing them in PEG matrices that do not match their proteolytic machinery (e.g., HFFs in P-PEG) might show unexpected differences in cytoskeletal arrangement, integrin usage, enzyme production, or apoptosis when compared to cultures in degradable PEG matrices (e.g., HFFs in M-PEG) despite very similar adhesiveness and physical matrix properties. In M-PEG gels, embedded cells changed within a couple of hours from spherical into polarized, often spindle-shaped morphology, requiring the development of a tensile force between front and rear. F-actin was organized into stress fibers, indicating load-bearing cell-matrix binding. In the case of M-PEG gels, in which macroscopic pores do not exist, these findings emphasize the ability of HFFs to induce highly localized, protease-mediated material remodeling without losing integrin-mediated traction to the surrounding matrix.
One of the main differences between migration on a surface and in 3D is the biophysical resistance imposed on a moving cell (55). By comparing migration on the surface with 3D migration in the same material, predictions about the influence of matrix microarchitecture and degradability on migration can be made. We found the most pronounced dissimilarities in M-PEG gels. Contrarily, migration parameters in collagen were very similar on the gel's surface and in 3D (Fig. 7). This indicates that the biophysical resistance against 3D migration is higher in M-PEG than in collagen. Surprisingly, migration parameters of fibroblasts migrating on the substrates were similar despite the vastly different materials (Fig. 7, B and C). Especially between M-PEG and fibrin, no significant dissimilarity was measured. Combining these results, it becomes likely that it is not intermaterial disparity in adhesion or mechanical properties but the microarchitectural differences that are a major determinant of 3D cell-migration dissimilarity in the three model systems.
We modulated the MMP system to further investigate the importance of MMP-derived proteolysis for 3D migration. Inhibition of active MMPs resulted in almost complete suppression of 3D cell migration in M-PEG matrices, whereas TNF-α simulation increased the number of migrating cells by a factor of 2. These findings show that synthetic M-PEG gels have indeed an almost defect-free microstructure, allowing only marginal nonproteolytic migration, and are highly MMP-sensitive. In fibrin, a partial inhibition was observed by blocking active MMPs. Plasmin inhibition by aprotinin did not significantly influence HFF migration (data not shown). Although plasmin plays a key role in fibrinolysis, there is evidence that pericellular fibrinolysis may also occur independently of the plasminogen cascade, through MMPs (membrane-type MMP and stromelysin) (56). Combined with our observations that HFFs were not able to migrate in the plasmin-sensitive P-PEG networks, this demonstrates that fibrin allows substantial nonproteolytic migration and is sensitive to MMPs, and that HFFs possess only an incomplete serine protease system. On the other hand, within highly porous collagen, cell migration was able to occur independent of MMPs. MMP inhibition did not compromise cell dispersion, and we did not detect a cell-shape change toward a rounded, amoeboid morphology. HFFs kept a spindlelike shape, characteristic for adhesion-dependent interaction with the substrate. Consequently, there was no evidence for a mesenchymal-to-amoeboid transition for HFFs in collagen after abrogating MMP function, unlike what was reported for neoplastic cells in a similar matrix (5). Likewise, GM6001 did not induce a mesenchymal-to-amoeboid transition in M-PEG gels.
We used gelatin zymography of supernatants as a screening method to analyze gelatinolytic enzymes enabling proteolytic migration in the three matrices. ProMMP-2 was secreted in comparable levels in all materials, whereas TNF-α strongly induced proMMP-9, which is in line with an earlier study by Sato et al. (57). Activation of MMP-2 in fibroblasts requires binding to fibrillar collagen (58) via integrin α2β1 (59) and MT1-MMP (60). This may explain the low levels of active MMP-2 in the other materials. Our results show that protease expression is not correlated with migration in collagen gels, as increased levels of active MMP-2 did not enhance cell migration, thereby giving further evidence that proteolysis is not a limiting factor in this case. In M-PEG matrices, increased MMP-9 levels after stimulation coincided with elevated migration. However, TNF-α has been shown to possess promigratory potential by regulating expression and activation of certain integrins (61,62). Hence, increased cell dispersion may not be attributed solely to augmented MMP levels. Except for MMP-2 in collagen matrices, only minor quantities of active MMPs were detected by gelatin zymography. Membrane-anchored MMPs (MT-MMPs) and membrane associated MMPs will not be detected in the supernatant, but might play a pivotal role in localized pericellular proteolysis. MMP-2 and MMP-9 can both associate with membrane proteins. MMP-2 binds via TIMP2 to MT1-MMP (63) or interacts with integrin αvβ3 (64). Active MMP-9 binds to CD44 (65) or, in latent form, to its own substrate type IV collagen on the cell surface (66). These mechanisms are important regulators of protease dynamics in a natural ECM (67) and are likely to enable localized proteolysis in our purely synthetic material as well. Further evidence for highly localized degradation by bound proteases can be found in long-term experiments in M-PEG gels (29), in which an interconnected cellular meshwork developed within an otherwise intact hydrogel.
PEG hydrogels as novel 3D model matrix
The benefits of 3D compared to conventional 2D cell-culture systems have lately earned much recognition (68), as 3D matrices allow investigation of cell signaling (69) and cell-matrix interactions (70) in a much more in vivo-like context. These ECM mimetics are currently establishing their rank between classical 2D in vitro and resource-intensive in vivo experiments. The matrix materials used for this task have mainly been biopolymers like collagen, fibrin, or Matrigel (71), with limited control over physical and chemical properties. The class of synthetic PEG hydrogels used herein offers the possibility of systematically reconstituting a specific extracellular environment on a molecular level, allowing a rigorous investigation of individual physical and chemical factors on cellular processes. Thus, the complex array of properties and functions exhibited in vivo by natural ECMs can be dissected into single independent modules that can then be reassembled to obtain a well-defined matrix for cell-culture experiments or in vivo applications in regenerative medicine. Our expanding toolbox of building blocks currently contains different adhesion ligands, cross-linkers that are sensitive to specific classes of proteases, PEG macromers with various molecular weights and number of arms, growth factors, and fluorogenic protease substrates. Therefore, engineered model matrices will ultimately allow investigation of specific biological questions. In clear contrast to biopolymers, PEG hydrogels do not possess a fibrillar microarchitecture. This might be a shortcoming for certain cell types, especially sensitive progenitor or stem cells. In this case, a less dense environment that allows cellular spatial organization without proteolytic matrix remodeling may be favorable. Under these circumstances, porous matrix architectures may be a necessity for synthetic ECM model systems. Such materials are the subject of further biomaterial development (M. P. Lutolf and J. A. Hubbell, unpublished). Furthermore, specialized cells will demand additional biological signals for survival, proliferation, or migration. For example, human umbilical vein endothelial cells were able to invade identical MMP-degradable PEG hydrogels when supplied with vascular endothelial growth factor (72). The investigated material system allows the covalent incorporation of such additional factors.
We applied PEG matrices in this report for cell migration studies with proteolytically competent dermal fibroblasts. Despite very limited biological signaling capabilities and the lack of a fibrillar microstructure of the synthetic PEG matrix as compared to biopolymers, cell viability, morphology, and migration were surprisingly similar when the material's protease sensitivity overlapped with the protease profile of the residing cells. Cell migration in MMP-sensitive gels was fully competent in culture, whereas the cells in culture seemed to lack a key component of the plasminogen activation cascade and remained nonmigratory. The use of this synthetic ECM model system thus permitted the restriction of cell migration to a proteolytic strategy, a characteristic that is indispensable for the development of migration-impeding anticancer therapeutics, for example. Using this approach, we believe that PEG model matrices, in combination with elaborate methods for measuring and especially localizing enzyme action, might help to further investigate the molecular steps involved in proteolytic migration.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank Prof. Thomas Baechi and Dr. Matthias Hoechli from the Electron Microscopy Central Laboratory of the University of Zurich for their competent help with the confocal laser scanning microscopy.
M. P. Lutolf's present address is Baxter Laboratory in Genetic Pharmacology, Stanford University School of Medicine, 269 Campus Drive, CCSR 3200, Stanford, CA 94305-5175.
References
- 1.Ridley, A. J., M. A. Schwartz, K. Burridge, R. A. Firtel, M. H. Ginsberg, G. Borisy, J. T. Parsons, and A. R. Horwitz. 2003. Cell migration: integrating signals from front to back. Science. 302:1704–1709. [DOI] [PubMed] [Google Scholar]
- 2.Lauffenburger, D. A., and A. F. Horwitz. 1996. Cell migration: a physically integrated molecular process. Cell. 84:359–369. [DOI] [PubMed] [Google Scholar]
- 3.Friedl, P., and K. Wolf. 2003. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer. 3:362–374. [DOI] [PubMed] [Google Scholar]
- 4.Wolf, K., R. Muller, S. Borgmann, E. B. Brocker, and P. Friedl. 2003. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood. 102:3262–3269. [DOI] [PubMed] [Google Scholar]
- 5.Wolf, K., I. Mazo, H. Leung, K. Engelke, U. H. von Andrian, E. I. Deryugina, A. Y. Strongin, E. B. Brocker, and P. Friedl. 2003. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 160:267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedl, P. 2004. Prespecification and plasticity: shifting mechanisms of cell migration. Curr. Opin. Cell Biol. 16:14–23. [DOI] [PubMed] [Google Scholar]
- 7.Wang, W., J. B. Wyckoff, V. C. Frohlich, Y. Oleynikov, S. Huttelmaier, J. Zavadil, L. Cermak, E. P. Bottinger, R. H. Singer, J. G. White, J. E. Segall, and J. S. Condeelis. 2002. Single cell behavior in metastatic primary mammary tumors correlated with gene expression patterns revealed by molecular profiling. Cancer Res. 62:6278–6288. [PubMed] [Google Scholar]
- 8.Murphy, G., and J. Gavrilovic. 1999. Proteolysis and cell migration: creating a path? Curr. Opin. Cell Biol. 11:614–621. [DOI] [PubMed] [Google Scholar]
- 9.Nabeshima, K., T. Inoue, Y. Shimao, and T. Sameshima. 2002. Matrix metalloproteinases in tumor invasion: Role for cell migration. Pathol. Int. 52:255–264. [DOI] [PubMed] [Google Scholar]
- 10.Woessner, J. F., and H. Nagase. 2000. Matrix Metalloproteinases and TIMPs. Oxford University Press, New York.
- 11.Sternlicht, M. D., and Z. Werb. 2001. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 17:463–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stefansson, S., and D. A. Lawrence. 2003. Old dogs and new tricks: proteases, inhibitors, and cell migration. Sci STKE 2003. 189:pe24. [DOI] [PubMed]
- 13.Friedl, P., F. Entschladen, C. Conrad, B. Niggemann, and K. S. Zaenker. 1998. T lymphocytes migrating in 3-D collagen lattices lack focal adhesions and utilize β1 integrin-independent strategies for polarization, interaction with collagen fibres, and migration. Eur. J. Immunol. 28:2331–2343. [DOI] [PubMed] [Google Scholar]
- 14.Friedl, P., K. Maaser, C. E. Klein, B. Niggemann, G. Krohne, and K. S. Zanker. 1997. Migration of highly aggressive MV3 melanoma cells in 3-dimensional collagen lattices results in local matrix reorganization and shedding of alpha2 and beta1 integrins and CD44. Cancer Res. 57:2061–2070. [PubMed] [Google Scholar]
- 15.Gunzer, M., E. Kampgen, E. B. Brocker, K. S. Zanker, and P. Friedl. 1997. Migration of dendritic cells in 3D-collagen lattices. Visualisation of dynamic interactions with the substratum and the distribution of surface structures via a novel confocal reflection imaging technique. Adv. Exp. Med. Biol. 417:97–103. [PubMed] [Google Scholar]
- 16.Schor, S. L., T. D. Allen, and B. Winn. 1983. Lymphocyte migration into three-dimensional collagen matrices: a quantitative study. J. Cell Biol. 96:1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hegerfeldt, Y., M. Tusch, E. B. Brocker, and P. Friedl. 2002. Collective cell movement in primary melanoma explants: plasticity of cell-cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 62:2125–2130. [PubMed] [Google Scholar]
- 18.Sahai, E., and C. J. Marshall. 2003. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 5:711–719. [DOI] [PubMed] [Google Scholar]
- 19.Coussens, L. M., B. Fingleton, and L. M. Matrisian. 2002. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 295:2387–2392. [DOI] [PubMed] [Google Scholar]
- 20.Overall, C. M., and C. Lopez-Otin. 2002. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat. Rev. Cancer. 2:657–672. [DOI] [PubMed] [Google Scholar]
- 21.Silver, F. H., and G. Pins. 1992. Cell growth on collagen: a review of tissue engineering using scaffolds containing extracellular matrix. J. Long Term Eff. Med. Implants. 2:67–80. [PubMed] [Google Scholar]
- 22.Mosesson, M. W., K. R. Siebenlist, and D. A. Meh. 2001. The structure and biological features of fibrinogen and fibrin. Ann. N. Y. Acad. Sci. 936:11–30. [DOI] [PubMed] [Google Scholar]
- 23.Hayen, W., M. Goebeler, S. Kumar, R. Riessen, and V. Nehls. 1999. Hyaluronan stimulates tumor cell migration by modulating the fibrin fiber architecture. J. Cell Sci. 112:2241–2251. [DOI] [PubMed] [Google Scholar]
- 24.Lee, C. H., A. Singla, and Y. Lee. 2001. Biomedical applications of collagen. Int. J. Pharm. 221:1–22. [DOI] [PubMed] [Google Scholar]
- 25.Hafemann, B., S. Ensslen, C. Erdmann, R. Niedballa, A. Zuhlke, K. Ghofrani, and C. J. Kirkpatrick. 1999. Use of a collagen/elastin-membrane for the tissue engineering of dermis. Burns. 25:373–384. [DOI] [PubMed] [Google Scholar]
- 26.Daamen, W. F., H. T. van Moerkerk, T. Hafmans, L. Buttafoco, A. A. Poot, J. H. Veerkamp, and T. H. van Kuppevelt. 2003. Preparation and evaluation of molecularly-defined collagen-elastin-glycosaminoglycan scaffolds for tissue engineering. Biomaterials. 24:4001–4009. [DOI] [PubMed] [Google Scholar]
- 27.Benelli, R., and A. Albini. 1999. In vitro models of angiogenesis: the use of Matrigel. Int. J. Biol. Markers. 14:243–246. [DOI] [PubMed] [Google Scholar]
- 28.Lutolf, M. P., and J. A. Hubbell. 2003. Synthesis and physicochemical characterization of end-linked poly(ethylene glycol)-co-peptide hydrogels formed by Michael-type addition. Biomacromolecules. 4:713–722. [DOI] [PubMed] [Google Scholar]
- 29.Lutolf, M. P., G. P. Raeber, A. H. Zisch, N. Tirelli, and J. A. Hubbell. 2003. Cell-responsive synthetic hydrogels. Adv. Mater. 15:888–892. [Google Scholar]
- 30.Hern, D. L., and J. A. Hubbell. 1998. Incorporation of adhesion peptides into nonadhesive hydrogels useful for tissue resurfacing. J. Biomed. Mater. Res. 39:266–276. [DOI] [PubMed] [Google Scholar]
- 31.West, J. L., and J. A. Hubbell. 1999. Polymeric biomaterials with degradation sites for proteases involved in cell migration. Macromolecules. 32:241–244. [Google Scholar]
- 32.Lutolf, M. P., J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields, and J. A. Hubbell. 2003. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: engineering cell-invasion characteristics. Proc. Natl. Acad. Sci. USA. 100:5413–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pratt, A. B., F. E. Weber, H. G. Schmoekel, R. Muller, and J. A. Hubbell. 2004. Synthetic extracellular matrices for in situ tissue engineering. Biotechnol. Bioeng. 86:27–36. [DOI] [PubMed] [Google Scholar]
- 34.Reference deleted in proof.
- 35.Hall, H., T. Baechi, and J. A. Hubbell. 2001. Molecular properties of fibrin-based matrices for promotion of angiogenesis in vitro. Microvasc. Res. 62:315–326. [DOI] [PubMed] [Google Scholar]
- 36.Pierschbacher, M. D., and E. Ruoslahti. 1984. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 309:30–33. [DOI] [PubMed] [Google Scholar]
- 37.Herrick, S., O. Blanc-Brude, A. Gray, and G. Laurent. 1999. Fibrinogen. Int. J. Biochem. Cell Biol. 31:741–746. [DOI] [PubMed] [Google Scholar]
- 38.Lutolf, M. P., N. Tirelli, S. Cerritelli, L. Cavalli, and J. A. Hubbell. 2001. Systematic modulation of Michael-type reactivity of thiols through the use of charged amino acids. Bioconjug. Chem. 12:1051–1056. [DOI] [PubMed] [Google Scholar]
- 39.Carr, M. E., Jr., and C. L. Hardin. 1987. Fibrin has larger pores when formed in the presence of erythrocytes. Am. J. Physiol. 253:H1069–H1073. [DOI] [PubMed] [Google Scholar]
- 40.Niven, R. K. 2002. A derivation of Darcy's law. Ground Water. 40:670–671; discussion 671–672. [DOI] [PubMed] [Google Scholar]
- 41.Canal, T., and N. A. Peppas. 1989. Correlation between mesh size and equilibrium degree of swelling of polymeric networks. J. Biomed. Mater. Res. 23:1183–1193. [DOI] [PubMed] [Google Scholar]
- 42.Brightman, A. O., B. P. Rajwa, J. E. Sturgis, M. E. McCallister, J. P. Robinson, and S. L. Voytik-Harbin. 2000. Time-lapse confocal reflection microscopy of collagen fibrillogenesis and extracellular matrix assembly in vitro. Biopolymers. 54:222–234. [DOI] [PubMed] [Google Scholar]
- 43.Dickinson, R. B., S. Guido, and R. T. Tranquillo. 1994. Biased cell migration of fibroblasts exhibiting contact guidance in oriented collagen gels. Ann. Biomed. Eng. 22:342–356. [DOI] [PubMed] [Google Scholar]
- 44.Dunn, G. A. 1983. Characterising a kinesis response: time averaged measures of cell speed and directional persistence. Agents Actions Suppl. 12:14–33. [DOI] [PubMed] [Google Scholar]
- 45.Othmer, H. G., S. R. Dunbar, and W. Alt. 1988. Models of dispersal in biological systems. J. Math. Biol. 26:263–298. [DOI] [PubMed] [Google Scholar]
- 46.Dickinson, R. B., and R. T. Tranquillo. 1993. Optimal estimation of cell movement indices from the statistical analysis of cell tracking data. AIChE J. 39:1995–2010. [Google Scholar]
- 47.Roeder, B. A., K. Kokini, J. E. Sturgis, J. P. Robinson, and S. L. Voytik-Harbin. 2002. Tensile mechanical properties of three-dimensional type I collagen extracellular matrices with varied microstructure. J. Biomech. Eng. 124:214–222. [DOI] [PubMed] [Google Scholar]
- 48.Knapp, D. M., E. F. Helou, and R. T. Tranquillo. 1999. A fibrin or collagen gel assay for tissue cell chemotaxis: assessment of fibroblast chemotaxis to GRGDSP. Exp. Cell Res. 247:543–553. [DOI] [PubMed] [Google Scholar]
- 49.Barocas, V. H., and R. T. Tranquillo. 1994. Biphasic theorie and in vitro assay of cell-fibril mechanical interactions in tissue-equivalent gels. In Cell Mechanics and Cellular Engineering. V. C. Mow, editor. Springer, New York. 185–209.
- 50.Barocas, V. H., A. G. Moon, and R. T. Tranquillo. 1995. The fibroblast-populated collagen microsphere assay of cell traction force–Part 2: Measurement of the cell traction parameter. J. Biomech. Eng. 117:161–170. [DOI] [PubMed] [Google Scholar]
- 51.Knapp, D. M., V. H. Barocas, A. G. Moon, K. Yoo, L. R. Petzold, and R. T. Tranquillo. 1997. Rheology of reconstituted type I collagen gel in confined compression. J Rheol. 41:971–993. [Google Scholar]
- 52.Kuntz, R. M., and W. M. Saltzman. 1997. Neutrophil motility in extracellular matrix gels: mesh size and adhesion affect speed of migration. Biophys. J. 72:1472–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Colman, R. W., J. Hirsch, V. J. Marder, A. W. Clowes, and J. N. George 2001. Hemostasis and Thrombosis. Basic Principles and Clinical Practice. Lippincott Williams & Wilkins, Philadelphia.
- 54.Visse, R., and H. Nagase. 2003. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 92:827–839. [DOI] [PubMed] [Google Scholar]
- 55.Friedl, P., and E. B. Brocker. 2000. The biology of cell locomotion within three-dimensional extracellular matrix. Cell. Mol. Life Sci. 57:41–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hiraoka, N., E. Allen, I. J. Apel, M. R. Gyetko, and S. J. Weiss. 1998. Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell. 95:365–377. [DOI] [PubMed] [Google Scholar]
- 57.Sato, T., A. Ito, Y. Ogata, H. Nagase, and Y. Mori. 1996. Tumor necrosis factor alpha (TNF-alpha) induces pro-matrix metalloproteinase 9 production in human uterine cervical fibroblasts but interleukin 1alpha antagonizes the inductive effect of TNF-alpha. FEBS Lett. 392:175–178. [DOI] [PubMed] [Google Scholar]
- 58.Ruangpanit, N., D. Chan, K. Holmbeck, H. Birkedal-Hansen, J. Polarek, C. Yang, J. F. Bateman, and E. W. Thompson. 2001. Gelatinase A (MMP-2) activation by skin fibroblasts: dependence on MT1-MMP expression and fibrillar collagen form. Matrix Biol. 20:193–203. [DOI] [PubMed] [Google Scholar]
- 59.Zigrino, P., C. Drescher, and C. Mauch. 2001. Collagen-induced proMMP-2 activation by MT1-MMP in human dermal fibroblasts and the possible role of alpha2beta1 integrins. Eur. J. Cell Biol. 80:68–77. [DOI] [PubMed] [Google Scholar]
- 60.Murphy, G., H. Stanton, S. Cowell, G. Butler, V. Knauper, S. Atkinson, and J. Gavrilovic. 1999. Mechanisms for pro matrix metalloproteinase activation. APMIS. 107:38–44. [DOI] [PubMed] [Google Scholar]
- 61.Scott, K. A., C. H. Arnott, S. C. Robinson, R. J. Moore, R. G. Thompson, J. F. Marshall, and F. R. Balkwill. 2004. TnF-alpha regulates epithelial expression of MMP-9 and integrin alpha v beta 6 during tumour progression. Oncogene. 23:6954–6966. [DOI] [PubMed] [Google Scholar]
- 62.Gao, B., T. M. Saba, and M. F. Tsan. 2002. Role of alpha(v)beta(3)-integrin in TNF-alpha-induced endothelial cell migration. Am. J. Physiol. Cell Physiol. 283:C1196–1205. [DOI] [PubMed] [Google Scholar]
- 63.Strongin, A. Y., I. Collier, G. Bannikov, B. L. Marmer, G. A. Grant, and G. I. Goldberg. 1995. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J. Biol. Chem. 270:5331–5338. [DOI] [PubMed] [Google Scholar]
- 64.Brooks, P. C., S. Stromblad, L. C. Sanders, T. L. von Schalscha, R. T. Aimes, W. G. Stetler-Stevenson, J. P. Quigley, and D. A. Cheresh. 1996. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell. 85:683–693. [DOI] [PubMed] [Google Scholar]
- 65.Yu, Q., and I. Stamenkovic. 1999. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 13:35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Olson, M. W., M. Toth, D. C. Gervasi, Y. Sado, Y. Ninomiya, and R. Fridman. 1998. High affinity binding of latent matrix metalloproteinase-9 to the alpha2(IV) chain of collagen IV. J. Biol. Chem. 273:10672–10681. [DOI] [PubMed] [Google Scholar]
- 67.Werb, Z. 1997. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 91:439–442. [DOI] [PubMed] [Google Scholar]
- 68.Abbott, A. 2003. Cell culture: biology's new dimension. Nature. 424:870–872. [DOI] [PubMed] [Google Scholar]
- 69.Schmeichel, K. L., and M. J. Bissell. 2003. Modeling tissue-specific signaling and organ function in three dimensions. J. Cell Sci. 116:2377–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cukierman, E., R. Pankov, and K. M. Yamada. 2002. Cell interactions with three-dimensional matrices. Curr. Opin. Cell Biol. 14:633–639. [DOI] [PubMed] [Google Scholar]
- 71.Kleinman, H. K., M. L. McGarvey, L. A. Liotta, P. G. Robey, K. Tryggvason, and G. R. Martin. 1982. Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry. 21:6188–6193. [DOI] [PubMed] [Google Scholar]
- 72.Zisch, A. H., M. P. Lutolf, M. Ehrbar, G. P. Raeber, S. C. Rizzi, N. Davies, H. Schmokel, D. Bezuidenhout, V. Djonov, P. Zilla, and J. A. Hubbell. 2003. Cell-demanded release of VEGF from synthetic, biointeractive cell-ingrowth matrices for vascularized tissue growth. FASEB J. 17:2260–2262. [DOI] [PubMed] [Google Scholar]