

Abstract
In biology experiments, oligonucleotide microarrays are contacted with a solution of long nucleic acid targets. The hybridized probes thus carry long tails. When the surface density of the oligonucleotide probes is high enough, the progress of hybridization gives rise to a polyelectrolyte brush due to mutual crowding of the nucleic acid tails. The free-energy penalty associated with the brush modifies both the hybridization isotherms and the rate equations: the attainable hybridization is lowered significantly as is the hybridization rate. When the equilibrium hybridization fraction, xeq, is low, the hybridization follows a Langmuir type isotherm, xeq/(1 − xeq) = ctK where ct is the target concentration and K is the equilibrium constant. K is smaller than its bulk value by a factor (n/N)2/5 due to wall effects where n and N denote the number of bases in the probe and the target. At higher xeq, when the brush is formed, the leading correction is 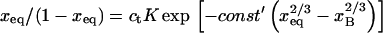 where xB corresponds to the onset of the brush regime. The denaturation rate constant in the two regimes is identical. However, the hybridization rate constant in the brush regime is lower, the leading correction being
where xB corresponds to the onset of the brush regime. The denaturation rate constant in the two regimes is identical. However, the hybridization rate constant in the brush regime is lower, the leading correction being 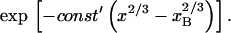
INTRODUCTION
The growing availability of genomic DNA sequences enables research on profiles of gene expression, single nucleotide polymorphism and their role, molecular diagnostics for cancer, etc. In turn, these activities require simultaneous interrogation of a given sample for the presence of numerous different nucleic acid sequences. DNA microarrays, “DNA chips,” emerged as an important method for such parallel analysis (1–4). DNA chips function by parallel hybridization of labeled nucleic acid sequences in the solution, known as targets, to an array of nucleic acid probes bound to a surface. Numerous identical probes are localized at a small area known as “spot” or “probe cell.” The composition of the sample is deduced from the label intensities of the different spots after the hybridization. DNA chips are produced in one of two main formats. In cDNA microarrays, long cDNA targets are physisorbed onto the substrate whereas in oligonucleotide chips short oligonucleotides are chemically bound to the surface via their terminal groups. Our theoretical considerations address the hybridization behavior, kinetics and thermodynamics, of oligonucleotide microarrays when the targets are much longer than the probes as is typically the case in biology experiments (see, for examples, Guo et al. and others (5,6)). In particular, we analyze the consequences of the interactions between the long hybridized targets at the surface (Fig. 1).
FIGURE 1.

A schematic picture of the hybridization of long targets at a layer of short probes. For simplicity we depict the case of targets with a terminal hybridization site, when each hybridized probe carries a long ssDNA tail. Three regimes occur: (a) in the 1:1 regime the distance between the probes, 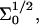 is large and each hybridized target can only interact with its own probe. There is no crowding of the tails. (b) In the 1:q regime the probe density is higher. At low hybridization fraction each target interacts with
is large and each hybridized target can only interact with its own probe. There is no crowding of the tails. (b) In the 1:q regime the probe density is higher. At low hybridization fraction each target interacts with  probes. (c) As the hybridization fraction increases the hybridized targets begin to crowd each other thus forming a brush with an area per chain
probes. (c) As the hybridization fraction increases the hybridized targets begin to crowd each other thus forming a brush with an area per chain 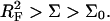 Note that in the general case the hybridization site is situated roughly in the middle of the target and each hybridized probe carries two tails (d).
Note that in the general case the hybridization site is situated roughly in the middle of the target and each hybridized probe carries two tails (d).
A growing theory effort aims to clarify the underlying physics of DNA chips with view of assisting in their design and in the analysis of the results. The Langmuir isotherm and the corresponding kinetic scheme provide a natural starting point for the modeling (7–14) as well as the analysis of the experimental results (15–23). Within this model, the probes, irrespective of their hybridization state, do not interact. This assumption is justified when the probe density in the spots is sufficiently low. At higher probe densities interactions are no longer negligible and the Langmuir model requires modifications. As we shall discuss, the necessary modifications depend crucially on the length of the targets as characterized by N, the number of bases or monomers. Importantly, in biology experiments the targets are usually significantly longer than the probes. As a result, each hybridized probe binds a long segment of single-stranded nucleic acid formed by the unhybridized part of the target (Fig. 1). This leads to two effects. First, when the tails do not overlap the hybridization at an impenetrable surface incurs an entropic penalty. This reduces the equilibrium constant of hybridization with respect to its bulk value. Second, it is necessary to allow for the crowding of these unhybridized “tails” as the fraction of hybridized probes grows. This crowding gives rise to a polymer brush, a phenomenon that was extensively studied in polymer physics (24–26). The theory of polyelectrolyte brushes (26–28), as modified to allow for target-probe interactions and wall effects, enables us to analyze the effects of the crowding on the thermodynamics and kinetics of hybridization on DNA chips. In particular, we obtain the hybridization isotherm and the rate equation in the brush regime when the unhybridized tails overlap. As we shall see, the free-energy penalty associated with the brush gives rise to distinctive modification of the Langmuir isotherm and kinetics. Importantly, the brush penalty reflects both the electrostatic interactions within the probe layer and the entropic price due to the extension of the crowded chains. It results in slower hybridization and lower attainable hybridization.
Our analysis focuses on oligonucleotide microarrays hybridizing with long targets of single-stranded (ss) DNA. For simplicity we limit the discussion to the experimentally attainable case of monodispersed targets and probes, a passivated surface that eliminates physical adsorption of DNA and probes anchored to the surface via short spacer chains. The qualitative features of our results apply, however, to a wider range of systems. Two hybridization regimes appear, depending on the equilibrium hybridization fraction, xeq, as set by the bulk concentration of the target, ct. For low xeq, the hybridization isotherm is of the Langmuir form, xeq/(1 − xeq) = ctK, where K is the equilibrium constant of the hybridization reaction at the surface. For probes comprising 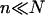 bases, K at an impenetrable surface is reduced by a factor of (n/N)2/5 with respect to the equilibrium constant of the free chains in solution. At higher xeq, obtained at higher ct, the effective equilibrium constant is modified because of the brush penalty. The leading correction to the hybridization isotherm is
bases, K at an impenetrable surface is reduced by a factor of (n/N)2/5 with respect to the equilibrium constant of the free chains in solution. At higher xeq, obtained at higher ct, the effective equilibrium constant is modified because of the brush penalty. The leading correction to the hybridization isotherm is  where xB corresponds to the onset of brush formation. The formation of the brush does not affect the denaturation rate constant of the hybridized probe. However, it does lower the hybridization rate constant by a factor of
where xB corresponds to the onset of brush formation. The formation of the brush does not affect the denaturation rate constant of the hybridized probe. However, it does lower the hybridization rate constant by a factor of  where x is the instantaneous hybridization fraction. The proportionality constant scales with
where x is the instantaneous hybridization fraction. The proportionality constant scales with 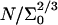 where Σ0 is the area per probe.
where Σ0 is the area per probe.
To our knowledge, there has been no direct experimental study of the effects of brush formation on the hybridization isotherms and the hybridization rates. Yet, experimental evidence of brush effects has been reported. Guo et al. (5) observed that the maximum attainable hybridization fraction is reached at higher Σ0 when N increases. Su et al. (29) reported slower hybridization as N increases at fixed Σ0. A similar effect was reported for RNA targets by Dai et al. (20). Further support for the existence of the brush effect is lent by the widespread use of sample fragmentation to achieve a lower average N (see, for example, Rosenow et al. and others (30,31)).
The practical implications of our analysis concern three issues: the design of DNA chips, the sample preparation, and the analysis of the data. The design of DNA chips currently reflects the view that an increase in the oligonucleotide density in a spot should increase the signal intensity and therefore the sensitivity (32). Certain limitations of this strategy, due to the increase of the DNA diameter upon hybridization and the resulting steric hindrance, has been long recognized (33). In marked distinction, our analysis highlights limitations due to the crowding of the long nonhybridized tails of the targets. Thus, in choosing Σ0 it is useful to bear in mind the anticipated N of the sample and its effect on the attainable hybridization. When Σ0 is fixed, our analysis provides guidelines for the sample preparation. In particular, the choice of N as determined by the polymerase chain reaction (PCR) primers or the fragmentation procedure. Concerning data analysis, our discussion identifies possible sources of error when comparing spot intensities of samples with different N. These may occur because both the onset of saturation and the hybridization rate vary with N. In quantitative terms, our analysis yields two guidelines: Concerning equilibrium hybridization, it leads to a simple relationship between the area per probe, Σ0, the number of bases in the probe, n, the number of bases in the target, N, and the attainable sensitivity as measured by c50, i.e., the target concentration resulting in 50% hybridization at the spot. Regarding the kinetics, it yields a simple criterion for the onset of slowdown due to the brush formation.
Experiments using DNA chips involve many control parameters concerning the chip design, the sample preparation, and the hybridization conditions. These are outlined in “Design of oligonucleotide microarray experiments” together with a discussion of the resulting hybridization regimes and the choice of parameters used in our numerical calculations. Our analysis incorporates ingredients from the theory of polymer brushes. These are summarized in “Background on polymer brushes”. This section describes the Flory version of the Alexander model of brushes as applied to terminally anchored polyelectrolytes in aqueous solution of high ionic strength. The model is modified to incorporate the effect of an impenetrable grafting surface. This is important to ensure crossover to the mushroom regime of nonoverlapping tails, and to enable comparison of the hybridization constants at the surface and in the bulk. Because the hybridization site is typically situated within the target, each hybridized probe carries two unhybridized tails. The necessary modifications are also discussed. When brush formation is possible, the hybridized targets also interact with neighboring probes. The resulting free-energy penalty, within the Flory approximation, is described in “Target-Probe Interactions”. The free energies associated with the brush and with the target-probe interactions enable us to obtain the equilibrium hybridization isotherms. The derivation is discussed in “Brush effects: thermodynamics of hybridization”. The hybridization isotherms allow us to quantify the sensitivity in terms of the corresponding c50's. In turn, these yield design guidelines relating the sensitivity to n, N, and Σ0. Assuming, and later checking, that the hybridization rate at the surface is reaction controlled enables us to specify the hybridization and denaturation rate constants in the different regimes. The necessary background, on the hybridization kinetics in the bulk and the desorption dynamics out of a brush, as well as the resulting rate equations are discussed in "Brush effects: kinetics of hybridization". The second virial coefficient, v, specifying the interactions between the charged monomers of polyelectrolytes in the high-salt regime is discussed in Appendix A. Using this v, we recover our earlier results of the n = N case and discuss the comparison to the 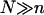 scenario. In Appendix B we present an alternative derivation of our result for the hybridization constant in the low surface density regime. This utilizes exact results, thus avoiding the approximations inherent in the “Alexander-Flory” approximation.
scenario. In Appendix B we present an alternative derivation of our result for the hybridization constant in the low surface density regime. This utilizes exact results, thus avoiding the approximations inherent in the “Alexander-Flory” approximation.
DESIGN OF OLIGONUCLEOTIDE MICROARRAY EXPERIMENTS
Oligonucleotide chip experiments vary widely in their design. A brief summary of the possible designs is necessary to delineate the range of applicability of our analysis and the different possible regimes. To this end it is helpful to distinguish between three groups of design parameters: the chip design, the sample characteristics, and the hybridization conditions. The primary parameters in the chip design are the area per probe, Σ0, and the number of bases in the probe, n (32); n values in the range 10–60 are typical. In this context one should discriminate two approaches to the production of oligonucleotide chips. In one, the probes are synthesized in situ using photolithography or ink jet technology. In the other, presynthesized oligonucleotides with functionalized end groups are delivered to the spot. In the first approach it is necessary to allow for the production of incomplete sequences leading to polydispersity in n (15). The reported probe densities within spots vary between 1.2 × 1010 and 4 × 1013 probes per cm2 corresponding to 2.5 × 102Å2 ≤ Σ0 ≤ 8.3 × 105Å2. The chip characteristics also include the nature of the surface treatment used to minimize nonspecific adsorption and of the spacer chains joining the probe to the anchoring functionality (length, charge, hydrophobicity, etc.).
A key qualitative characteristic of the sample is the chemical nature of the targets (1,3,4). To begin, it is necessary to distinguish between DNA and RNA targets, which differ in two respects: first, single-stranded (ss) RNA exhibits a pronounced secondary structure (loops, hairpins, etc.) that is largely absent in ssDNA. Second, the hybridization free energy of RNA-DNA complexes is higher than that of DNA-DNA ones. For DNA samples, it is further necessary to distinguish between samples of double-stranded (ds) DNA, as obtained from symmetric PCR amplification, and ssDNA samples as obtained, for example, using Lambda exonuclease digestion. The hybridization isotherms of the two types of samples are different (13). The labeling of the targets can also affect the hybridization behavior (34). Our discussion concerns samples of ssDNA targets assuming ideal labels that do not interfere with the hybridization. It focuses on the role of two quantitative characteristics of the sample: the number of bases in the target, N, and the molar concentration of the target, ct. N is determined by the choice of primers used for the PCR amplification or by the fragmentation step in the sample preparation. Note that the products of the PCR are monodisperse whereas the fragmentation introduces polydispersity in the size of the targets. In this last case it is only possible to control the average size of the targets. Typical reported values for PCR products vary in the range 100 ≤ N ≤ 350. The average N resulting from the fragmentation procedure is not always specified but the range 50 ≤ N ≤ 200 is representative. It is useful to note another distinction between the two procedures. Targets produced by PCR often have the hybridization site situated roughly in the middle of the target. In the case of fragmented targets, the location of the hybridization site is no longer controlled. With regard to ct it is helpful to stress the distinction between bioanalytic experiments, utilizing DNA chips to interrogate biological samples (see, for example, Prix et al. (6)), and physical chemistry experiments aiming to understand the function of DNA chips (see, for example, Peterson et al. (22)). In biology experiments ct is a priori unknown because it is set by the biological sample and its treatment. In marked contrast, in physical chemistry experiments the target concentration is imposed by the experimentalist as is the composition of the sample. In such experiments the target used is often identical in length to the probes, n = N. As noted earlier, our analysis is motivated by bioanalytical experiments where 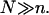
The hybridization conditions include the composition of the hybridization solution, the hybridization temperature, T, and the hybridization time, th. Typical hybridization temperatures vary over the range 30°C ≤ T ≤ 60°C depending on n and the GC fraction. The hybridization times also vary widely with typical values in the range of 2h ≤ th ≤ 16h. In most cases the hybridization solution contains 1 M of NaCl.
Different hybridization regimes are possible, depending on the values of n, N, and Σ0. To distinguish these regimes, it is necessary to first specify the molecular length scales of ssDNA and dsDNA. These are well established for dsDNA (35). In the range of parameters considered, dsDNA is a rod-like molecule with each basepair contributing 3.4 Å to its length. The radius of dsDNA is 9.5 Å and its cross-section area is 284 Å2. We will limit our analysis to Σ0 > 284 Å2 to avoid discussion of steric hindrance to hybridization. The corresponding characteristics of ssDNA are not well established. A typical value of the monomer size is a = 6 Å (36,37). The cited values of the persistence length, lp, vary between lp = 7.5 Å and lp = 35 Å (38). ssDNA is often described as a random coil though long-range interactions are expected to give rise to swollen configurations (39). In the following we will consider ssDNA as a swollen coil characterized by its Flory radius (40). This choice is dictated by our treatment of the brush, where the Flory radius emerges as a natural length scale. Accordingly, an isolated unhybridized probe occupies a hemisphere of radius rF ∼ n3/5a whereas a terminally hybridized target occupies a hemisphere of radius 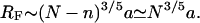 As we shall discuss, the unhybridized probes do not interact when
As we shall discuss, the unhybridized probes do not interact when 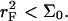 Similarly, when
Similarly, when 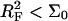 there is no brush regime. It is thus possible to distinguish between three different scenarios. A Langmuir regime is expected when
there is no brush regime. It is thus possible to distinguish between three different scenarios. A Langmuir regime is expected when 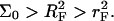 Brush effects, with no interactions between the probes, will occur when
Brush effects, with no interactions between the probes, will occur when 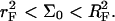 Finally, when
Finally, when 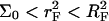 both the brush effect and probe-probe interactions play a role. All three scenarios occur in the reported variety of DNA chips.
both the brush effect and probe-probe interactions play a role. All three scenarios occur in the reported variety of DNA chips.
In the following we consider the role of n, N, and Σ0 in bioanalytical experiments. For brevity we focus on the simplest among the experimentally realistic situations. Thus, we consider monodispersed ssDNA targets and monodispersed oligonucleotide probes. This avoids complication due to unspecified polydispersity and to competitive bulk hybridization. It is convenient to concentrate on the 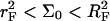 case with
case with 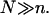 As we shall see, this makes for a simpler discussion of the brush effects. It also allows us to ignore small corrections due to probe-probe interactions. Finally, our analysis assumes DNA chips with a passivated surface and probes anchored to the surface via short, flexible spacer chains.
As we shall see, this makes for a simpler discussion of the brush effects. It also allows us to ignore small corrections due to probe-probe interactions. Finally, our analysis assumes DNA chips with a passivated surface and probes anchored to the surface via short, flexible spacer chains.
Our analysis is concerned with the modifications of the hybridization isotherm and rate equations as Σ0 decreases from the Langmuir range, 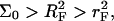 into the brush regime,
into the brush regime,  To implement this program, it is helpful to identify a reference state. In the following we utilize a probe layer that approaches the bulk values for the hybridization rate and equilibrium constants. We argue that this is the case when the following conditions are satisfied. First, the surface is perfectly nonadsorbing to both ss- and dsDNA. Under these conditions adsorbed states are not involved in the hybridization reaction and the two-state approximation for the hybridization reaction is justified. Second, the probes are attached to the surface via long, flexible, and neutral spacers. We argue that the effect of the surface diminishes as the length of the spacers increases. Note that the spacers modify two effects. One is the steric hindrance that occurs when the probes are directly attached to the surface. The other is the reduction in the number of accessible configurations in the vicinity of an impenetrable planar surface. Ideally, the reference state involves spacer chains that do not interact with either the probes and the targets. The third condition is that the distance between the anchored probes ensures zero probe-probe interaction energy, irrespective of their hybridization state. For this reference state, the equilibrium hybridization constant at the surface Kpt approaches
To implement this program, it is helpful to identify a reference state. In the following we utilize a probe layer that approaches the bulk values for the hybridization rate and equilibrium constants. We argue that this is the case when the following conditions are satisfied. First, the surface is perfectly nonadsorbing to both ss- and dsDNA. Under these conditions adsorbed states are not involved in the hybridization reaction and the two-state approximation for the hybridization reaction is justified. Second, the probes are attached to the surface via long, flexible, and neutral spacers. We argue that the effect of the surface diminishes as the length of the spacers increases. Note that the spacers modify two effects. One is the steric hindrance that occurs when the probes are directly attached to the surface. The other is the reduction in the number of accessible configurations in the vicinity of an impenetrable planar surface. Ideally, the reference state involves spacer chains that do not interact with either the probes and the targets. The third condition is that the distance between the anchored probes ensures zero probe-probe interaction energy, irrespective of their hybridization state. For this reference state, the equilibrium hybridization constant at the surface Kpt approaches 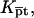 the equilibrium hybridization constant for the bulk reaction between the free chains. Accordingly, the hybridization isotherm in the small spot limit, when the hybridization at the surface has a negligible effect on initial molar concentration of the target ct, is
the equilibrium hybridization constant for the bulk reaction between the free chains. Accordingly, the hybridization isotherm in the small spot limit, when the hybridization at the surface has a negligible effect on initial molar concentration of the target ct, is
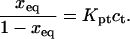 |
(1) |
It is important to distinguish between Kpt and
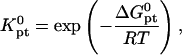 |
(2) |
where 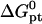 is the molar standard hybridization free energy as obtained from the nearest neighbor model (41), T is the temperature, and R = 1.987 cal/mol.K is the gas constant. First,
is the molar standard hybridization free energy as obtained from the nearest neighbor model (41), T is the temperature, and R = 1.987 cal/mol.K is the gas constant. First, 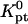 and
and 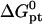 as calculated from the nearest-neighbor model are identical for all N ≥ n + 2. They allow, at most, for the effect of two dangling ends. Second, this model incorporates only nearest-neighbor interactions along the backbone of the chain. It thus assumes that the oligonucleotide adopts the configuration of an ideal random coil. In particular,
as calculated from the nearest-neighbor model are identical for all N ≥ n + 2. They allow, at most, for the effect of two dangling ends. Second, this model incorporates only nearest-neighbor interactions along the backbone of the chain. It thus assumes that the oligonucleotide adopts the configuration of an ideal random coil. In particular,  does not account for excluded volume interactions between the monomers. In addition,
does not account for excluded volume interactions between the monomers. In addition, 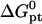 clearly does not allow for the effect of the impenetrable wall or for the interactions between the hybridized targets or between them and the neighboring probes. These additional terms and their effect on the hybridization isotherm will be discussed in the following three sections.
clearly does not allow for the effect of the impenetrable wall or for the interactions between the hybridized targets or between them and the neighboring probes. These additional terms and their effect on the hybridization isotherm will be discussed in the following three sections.
Our choice of the parameters used in the numerical calculations is based on two experimental systems. One, of Guo et al. (5), utilized probes of length n = 15 with PCR produced targets of length N = 157 or 347. Both ssDNA and dsDNA were investigated, the area per probe was varied in the range 300 Å2 ≤ Σ0 ≤ 3000 Å2, and the hybridization was carried out at T = 30°C. The hybridization times varied with N being th = 2–3 h for N = 157 and th = 6–8 h for N = 347. Note that in this study some of the data corresponds to the 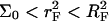 regime where probe-probe interactions are not negligible. The second system is the Affymetrix GeneChip Escherichia coli antisense genome array (31). In this case, probes of length n = 25 hybridize with fragmented, thus polydispersed, ds cDNA targets with average length in the range 50 ≤ N ≤ 200. The hybridization is carried out at T = 45°C for th = 16 h. A rough approximation of Σ0 for Affymetrix chips was obtained from the estimated density of functional groups in the substrate before the synthesis of the probes: 27 pM/cm2, and the stepwise yield of the synthesis, ∼90%. Only 14 pM/cm2 attain n ≥ 6 (15). This estimate yields Σ0 ≥ 1200 Å2. In both systems the hybridization was carried out in a solution containing 1 M of NaCl. The base sequence of the probes considered in the calculations and their thermodynamic parameters for hybridization, as calculated using the nearest-neighbor model with a perfectly matched target (42–44), are specified in Table 1.
regime where probe-probe interactions are not negligible. The second system is the Affymetrix GeneChip Escherichia coli antisense genome array (31). In this case, probes of length n = 25 hybridize with fragmented, thus polydispersed, ds cDNA targets with average length in the range 50 ≤ N ≤ 200. The hybridization is carried out at T = 45°C for th = 16 h. A rough approximation of Σ0 for Affymetrix chips was obtained from the estimated density of functional groups in the substrate before the synthesis of the probes: 27 pM/cm2, and the stepwise yield of the synthesis, ∼90%. Only 14 pM/cm2 attain n ≥ 6 (15). This estimate yields Σ0 ≥ 1200 Å2. In both systems the hybridization was carried out in a solution containing 1 M of NaCl. The base sequence of the probes considered in the calculations and their thermodynamic parameters for hybridization, as calculated using the nearest-neighbor model with a perfectly matched target (42–44), are specified in Table 1.
TABLE 1.
The thermodynamic parameters utilized in the numerical calculations
| Probe | Δ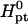 kcal/mol kcal/mol |
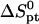 cal/mol.K cal/mol.K |
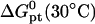 kcal/mol kcal/mol |
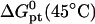 kcal/mol kcal/mol |
|---|---|---|---|---|
| p1 | −121.00 | −334.06 | −19.73 | −14.72 |
| p2 | −203.30 | −546.32 | −37.69 | −29.49 |
Parameters correspond to two probes: i), the n = 15 wild-type probe p1 (5′–CGTCCTCTTCAAGAA–3′) incorporates the codon 406 of exon 4 of the human tyrosinase gene. The N = 157 and 347 targets incorporate the perfect complementary segment 5′–TTCTTGAAGAGGACG–3′ (5). ii), The Affymetrix E. Coli antisense n = 25 probe p2 annotated AFFX-BioB-5_at:242:77, with interrogation point 177, corresponds to the sequence 5′–AGATTGCAAATACTGCCCGCAAACG–3′. The fragmented cDNA targets incorporate the perfect complementary sequence 5′–CGTTTGCGGGCAGTATTTGCAATCT–3′. The parameters are calculated from the nearest-neighbor model (42–44) using the HyTher program with a 1 M NaCl salt concentration. Because the targets are longer than the probes two dangling ends are invoked.
BACKGROUND ON POLYMER BRUSHES
Polymer brushes are formed by chains with one monomer anchored to a planar surface (24,25). In the simplest case, the anchoring moiety is the terminal monomer. When the area per chain, Σ, is large the chains do not crowd each other. In this “mushroom” regime, the chains may be roughly considered as occupying hemispheres whose radius is comparable to the Flory radius of the free chain, RF. When the surface density increases such that 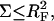 the chains begin to crowd each other, thus forming a “brush”. In the brush regime the chains stretch out along the normal to the surface so as to decrease the monomer concentration, c, and the number of repulsive monomer-monomer contacts. A simple description that captures the leading behavior of brushes is provided by the Alexander model (24,25,45). Within it the concentration profile of the brush is modeled by a step function of height H, i.e., c = N/HΣ at altitudes up to H above the surface and c = 0 for higher altitudes. All the free ends are assumed to straddle the outer boundary of the brush at height H. In the following we will use the Flory version of the model, ignoring scaling corrections. The regime of validity of this mean field approach for semiflexible chains is expanded in comparison to that of flexible polymers (46). This justifies the use of the “Alexander-Flory” model with a free energy per chain in a brush
the chains begin to crowd each other, thus forming a “brush”. In the brush regime the chains stretch out along the normal to the surface so as to decrease the monomer concentration, c, and the number of repulsive monomer-monomer contacts. A simple description that captures the leading behavior of brushes is provided by the Alexander model (24,25,45). Within it the concentration profile of the brush is modeled by a step function of height H, i.e., c = N/HΣ at altitudes up to H above the surface and c = 0 for higher altitudes. All the free ends are assumed to straddle the outer boundary of the brush at height H. In the following we will use the Flory version of the model, ignoring scaling corrections. The regime of validity of this mean field approach for semiflexible chains is expanded in comparison to that of flexible polymers (46). This justifies the use of the “Alexander-Flory” model with a free energy per chain in a brush
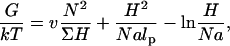 |
(3) |
where k is the Boltzmann constant. The first term allows for the monomer-monomer interactions. It is of the form vc2Vchain where v is the second virial coefficient and Vchain = ΣH is the volume per chain. The second accounts for the entropy loss incurred because of the stretching of a Gaussian chain, comprising of Na/lp persistent sequences of length lp, along the normal to the surface. Here a is the monomer size, lp is the persistence length of the chain, and the span of the Gaussian unswollen coil is 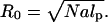 The last term arises because the impenetrable surface carrying the anchoring site reduces the number of accessible configurations of the tethered chain. For a Gaussian chain with a free end at altitude H the number is reduced by a factor of
The last term arises because the impenetrable surface carrying the anchoring site reduces the number of accessible configurations of the tethered chain. For a Gaussian chain with a free end at altitude H the number is reduced by a factor of 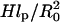 (47). This contribution is often ignored because it has a negligible effect on the equilibrium dimensions of the chains. It leads, however, to a significant modification of the hybridization constant at the surface. The last two terms of Eq. 3 apply, in this form, when
(47). This contribution is often ignored because it has a negligible effect on the equilibrium dimensions of the chains. It leads, however, to a significant modification of the hybridization constant at the surface. The last two terms of Eq. 3 apply, in this form, when 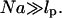 We have omitted a term allowing for the entropy associated with the placement of the free end. This is because the Alexander model assumes that all free ends are constrained to the brush boundary. For simplicity we ignore, here and in the following, numerical factors of order unity. Minimization of G with respect to H yields the equilibrium values of Gbrush and H
We have omitted a term allowing for the entropy associated with the placement of the free end. This is because the Alexander model assumes that all free ends are constrained to the brush boundary. For simplicity we ignore, here and in the following, numerical factors of order unity. Minimization of G with respect to H yields the equilibrium values of Gbrush and H
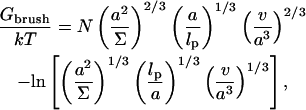 |
(4) |
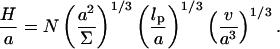 |
(5) |
In the mushroom regime, the chains occupy a hemisphere of radius
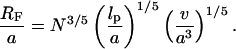 |
(6) |
Accordingly, the free energy per chain in the mushroom regime, Gmush, is set by the requirement Gmush = Gbrush at the mushroom-brush boundary when 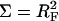 and H = RF thus leading to
and H = RF thus leading to
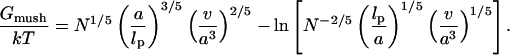 |
(7) |
As noted earlier, the properties of the chains in the mushroom regime are comparable to those of free coils. In turn, the free coil behavior is specified by the free energy (48)
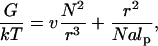 |
(8) |
leading, upon minimization with respect to the radius r, to RF as given by Eq. 6 and to the equilibrium free energy of a coil
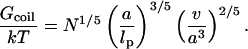 |
(9) |
The difference between Gmush and Gcoil is due to the logarithmic correction −ln(RF/Na) arising from the wall effect.
Within the approach described above, the nature of the grafted chain is specified by three parameters, the monomer size a, the persistence length lp, and the second virial coefficient associated with monomer-monomer interactions v. For the case of a brush formed by polyelectrolyte chains in aqueous solution of high ionic strength, “high salt,” v can be approximated (see Appendix A and Pincus (27)) by
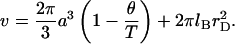 |
(10) |
The first term allows for the hard core repulsion between the monomers and for a weak, long-ranged van der Waals attraction between them. Here θ is the θ-temperature where v of a neutral chain vanishes, thus leading to the behavior of an ideal Gaussian coil. This term by itself is used to describe the behavior of neutral polymers (40). The second term arises from the screened electrostatic interactions between the singly charged monomers. Here lB = e2/εkT is the Bjerrum length (49) where ε is the dielectric constant, k the Boltzmann constant, and T the temperature. In water, with 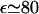 at room temperature,
at room temperature, 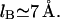 Note that the variation of ε with T contributes to the T dependence of lB. The Debye length rD characterizes the range of the screened electrostatic interactions in a salt solution (49). For a 1:1 salt with number concentration of ions cs,
Note that the variation of ε with T contributes to the T dependence of lB. The Debye length rD characterizes the range of the screened electrostatic interactions in a salt solution (49). For a 1:1 salt with number concentration of ions cs, 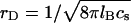 thus, in a 1-M solution rD = 3 Å. In our model, the presence of the
thus, in a 1-M solution rD = 3 Å. In our model, the presence of the 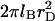 term in v distinguishes polyelectrolyte brushes from neutral ones. It is important to stress the limitations of approximating v by Eq. 10. It corresponds to the interactions between individual charged spherical monomers. For cylindrical noncharged monomers
term in v distinguishes polyelectrolyte brushes from neutral ones. It is important to stress the limitations of approximating v by Eq. 10. It corresponds to the interactions between individual charged spherical monomers. For cylindrical noncharged monomers 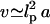 rather than
rather than  (40). Furthermore, this description does not allow for the contribution of hydrogen bonds with water nor for the effect of correlations on the electrostatic interactions. Finally, the appropriate θ temperature remains to be determined. With these caveats in mind, the second term is roughly comparable to 2πa3/3 and should be dominant for
(40). Furthermore, this description does not allow for the contribution of hydrogen bonds with water nor for the effect of correlations on the electrostatic interactions. Finally, the appropriate θ temperature remains to be determined. With these caveats in mind, the second term is roughly comparable to 2πa3/3 and should be dominant for 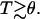 As a result v is comparable to 2πa3/3 and the swelling behavior of the chain is similar to that of a neutral chain in an athermal solvent (48). In other words, even short chains swell to their Flory radius. We should add that by using
As a result v is comparable to 2πa3/3 and the swelling behavior of the chain is similar to that of a neutral chain in an athermal solvent (48). In other words, even short chains swell to their Flory radius. We should add that by using 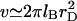 we are able to recover our earlier results (13) for the case of n = N (Appendix A).
we are able to recover our earlier results (13) for the case of n = N (Appendix A).
In the Flory-type approach, described above, the equilibrium state is determined by a global balance of the osmotic pressure of the monomers and the restoring elastic force of the stretched Gaussian chains. A more refined analysis of the brushes, utilizing self-consistent field (SCF) theory, is possible. This avoids the assumptions of uniform stretching and step-like concentration profiles. It yields the same functional forms for the characteristic height, H, and for Gbrush but with somewhat different numerical prefactors. With these reservations in mind we utilize the simplest approach, described earlier, because it typically yields the correct leading behavior in similar systems. A SCF theory is necessary for the description of effects that depend strongly on the details of the concentration profile and the distribution of the free ends.
Our discussion thus far concerned brushes anchored to the surface by the terminal headgroup. In DNA chips the situation is often different in that the hybridization site, the anchoring functionality, is located roughly at the middle of the target. As a result, each hybridized probe carries two unhybridized tails (Fig. 1 d) of length N1 and N2 = N1(1 + α) such that N1 + N2 + n = N. In considering the effect of this feature note that in the brush regime the details of the anchoring functionality are screened with a distance Σ1/2 from the surface. As a result, it is possible to estimate the modification of Gbrush and H in two cases, 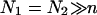 and
and 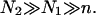 When N1 = N2 the resulting brush is similar to that formed by chains of length N/2 but with an area per chain Σ/2. In this case Gbrush is larger by a factor
When N1 = N2 the resulting brush is similar to that formed by chains of length N/2 but with an area per chain Σ/2. In this case Gbrush is larger by a factor 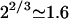 whereas H is smaller by a factor 22/3 in comparison to the values found for a brush of terminally anchored chains of length N and area per chain Σ. In the limit of
whereas H is smaller by a factor 22/3 in comparison to the values found for a brush of terminally anchored chains of length N and area per chain Σ. In the limit of 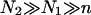 the resulting brush may be considered as bidispersed, comprising an equal number of chains of length N1 and N2. Such a bidispersed brush can be described as a superposition of two brushes (50). A simple two-layer model incorporates an inner brush of chains of length N1 and area per chain of Σ/2 and an outer brush formed by chains of length N2 − N1 = αN1 and area per chain Σ at the distal boundary of the inner brush. Within the Flory approximation this scheme leads to
the resulting brush may be considered as bidispersed, comprising an equal number of chains of length N1 and N2. Such a bidispersed brush can be described as a superposition of two brushes (50). A simple two-layer model incorporates an inner brush of chains of length N1 and area per chain of Σ/2 and an outer brush formed by chains of length N2 − N1 = αN1 and area per chain Σ at the distal boundary of the inner brush. Within the Flory approximation this scheme leads to 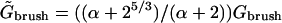 and
and 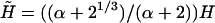 where Gbrush and H correspond to a monodispersed brush of chains of length N with an area per chain Σ. Note that α = 0 corresponds to N1 = N2 whereas
where Gbrush and H correspond to a monodispersed brush of chains of length N with an area per chain Σ. Note that α = 0 corresponds to N1 = N2 whereas 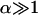 to
to 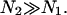 In both cases the effect is to modify Gbrush and H as obtained earlier by a multiplicative factor of order unity. In keeping with our policy we will omit these numerical factors in the interest of simplicity.
In both cases the effect is to modify Gbrush and H as obtained earlier by a multiplicative factor of order unity. In keeping with our policy we will omit these numerical factors in the interest of simplicity.
TARGET-PROBE INTERACTIONS
The preceding discussion of brushes allows for the interactions among the hybridized targets and the effects of the impenetrable wall. However, the brush regime is only attainable when the hybridized targets can interact with neighboring probes, thus giving rise to an additional contribution to the free energy of the system. In discussing the target-probe interactions it is useful to distinguish between three regimes. When 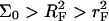 the hybridized targets cannot crowd each other. Roughly speaking, each one may be considered to occupy a hemisphere of radius RF containing a single probe that is hybridized to the target (Fig. 1 a). Because each target interacts with a single probe we will refer to this regime as 1:1. Our principle interest is in the two regimes that occur when
the hybridized targets cannot crowd each other. Roughly speaking, each one may be considered to occupy a hemisphere of radius RF containing a single probe that is hybridized to the target (Fig. 1 a). Because each target interacts with a single probe we will refer to this regime as 1:1. Our principle interest is in the two regimes that occur when 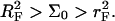 When the hybridization degree x is sufficiently small each target will occupy, as before, a hemisphere of radius RF. However, it will now interact with
When the hybridization degree x is sufficiently small each target will occupy, as before, a hemisphere of radius RF. However, it will now interact with 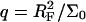 probes (Fig. 1 b). We will thus refer to this regime as 1:q. Note that in the polymer science nomenclature both 1:1 and 1:q regimes fall into the “mushroom” range, when the tethered chains do not overlap. The brush threshold occurs at x = xB when the hemispheres occupied by the different targets come into grazing contact. For a surface of total area AT the area per hybridized target is
probes (Fig. 1 b). We will thus refer to this regime as 1:q. Note that in the polymer science nomenclature both 1:1 and 1:q regimes fall into the “mushroom” range, when the tethered chains do not overlap. The brush threshold occurs at x = xB when the hemispheres occupied by the different targets come into grazing contact. For a surface of total area AT the area per hybridized target is 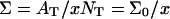 where NT is the total number of probes; xB corresponds to
where NT is the total number of probes; xB corresponds to  or
or 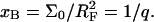 When x exceeds xB the hybridized targets begin to overlap, thus forming a brush (Fig. 1 c). Because the area per chain in this regime decreases as Σ = Σ0/x the target experiences interactions only with x−1 < q probes.
When x exceeds xB the hybridized targets begin to overlap, thus forming a brush (Fig. 1 c). Because the area per chain in this regime decreases as Σ = Σ0/x the target experiences interactions only with x−1 < q probes.
To estimate the free energy of interactions between the target and the probes, in the spirit of the Flory approach, we assume that each probe contributes an interaction free energy Gint/kT = vnc. Here c is the monomer concentration within the monomer cloud formed by the hybridized targets, i.e., we assume the interaction with the probes does not affect c as obtained in our earlier discussion of the mushroom and brush regimes. As we shall elaborate later, this assumption is justified only when  or
or
 |
(11) |
In the 1:1 regime each hybridized target occupies a hemisphere of radius RF incorporating a single probe. Accordingly 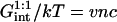 with
with 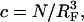 thus leading to
thus leading to
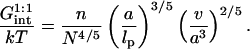 |
(12) |
This estimate is reasonable when 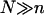 such that the region occupied by the unhybridized target is sufficiently large so as to encompass the hybridized probe. Roughly speaking, this implies (N − n)3/5a > 3.4n Å. Within the 1:q regime each hybridized target interacts with
such that the region occupied by the unhybridized target is sufficiently large so as to encompass the hybridized probe. Roughly speaking, this implies (N − n)3/5a > 3.4n Å. Within the 1:q regime each hybridized target interacts with 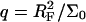 probes. Accordingly
probes. Accordingly 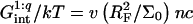 with
with 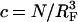 or
or
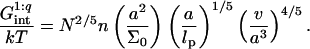 |
(13) |
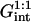 and
and 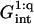 are independent of x. In marked contrast
are independent of x. In marked contrast 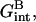 accounting for the target-probe interactions in the brush regime, varies with x. This variation arises because of the x dependence of the monomer concentration within the brush, cbrush = N/ΣH where Σ ∼ 1/x and H ∼ x1/3. Gbrush(x) is obtained from Eq. 4 upon replacing Σ by Σ0/x. Within the Flory approach the total interaction free energy between the targets and the probes is vNTncbrush. The interaction free energy per hybridized target is thus vncbrush/x or
accounting for the target-probe interactions in the brush regime, varies with x. This variation arises because of the x dependence of the monomer concentration within the brush, cbrush = N/ΣH where Σ ∼ 1/x and H ∼ x1/3. Gbrush(x) is obtained from Eq. 4 upon replacing Σ by Σ0/x. Within the Flory approach the total interaction free energy between the targets and the probes is vNTncbrush. The interaction free energy per hybridized target is thus vncbrush/x or
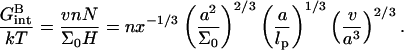 |
(14) |
The condition Eq. 11 ensures that the interaction term  is a weak perturbation to the Flory free energy of the mushroom Gmush(N). When this requirement is not satisfied the chain span exceeds the Flory radius. This is an unphysical result because the interactions driving the extra swelling are confined to the surface. In this case the chain can no longer be assumed to occupy a hemispherical region encompassing the probes. The uniform monomeric distribution inherent to the Flory approach should be refined so as to reflect locally stretched configurations allowing the chain to avoid the probes. For simplicity we will not consider this regime.
is a weak perturbation to the Flory free energy of the mushroom Gmush(N). When this requirement is not satisfied the chain span exceeds the Flory radius. This is an unphysical result because the interactions driving the extra swelling are confined to the surface. In this case the chain can no longer be assumed to occupy a hemispherical region encompassing the probes. The uniform monomeric distribution inherent to the Flory approach should be refined so as to reflect locally stretched configurations allowing the chain to avoid the probes. For simplicity we will not consider this regime.
BRUSH EFFECTS: THERMODYNAMICS OF HYBRIDIZATION
Having obtained the free-energy terms associated with target-target and target-probe interactions at the surface, we are in a position to investigate their effect on the hybridization isotherm. To simplify the equations we set v = a3 and lp = a. The hybridization isotherm is determined by the equilibrium condition of the hybridization reaction 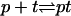 at the probe layer that is, μpt = μp + μt where μi is the chemical potential of species i. Here p and t signify single-stranded probe and target whereas pt is the hybridized probe-target pair. We first consider μt. In practice, the molar concentration of the targets, ct, is only weakly diminished by the hybridization reaction and it is reasonable to assume that ct is constant. The generalization to the opposite case, when this small spot approximation fails, is straightforward (13). Because the target solution is dilute and the ionic strength of the solution is high, electrostatic interactions between the targets are screened. Consequently μt assumes the weak solution form
at the probe layer that is, μpt = μp + μt where μi is the chemical potential of species i. Here p and t signify single-stranded probe and target whereas pt is the hybridized probe-target pair. We first consider μt. In practice, the molar concentration of the targets, ct, is only weakly diminished by the hybridization reaction and it is reasonable to assume that ct is constant. The generalization to the opposite case, when this small spot approximation fails, is straightforward (13). Because the target solution is dilute and the ionic strength of the solution is high, electrostatic interactions between the targets are screened. Consequently μt assumes the weak solution form
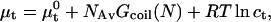 |
(15) |
where 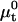 is the chemical potential of the standard state of the hybridization site and NAv is the Avogadro number. We choose a standard state such that
is the chemical potential of the standard state of the hybridization site and NAv is the Avogadro number. We choose a standard state such that 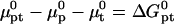 as given by the nearest-neighbor method. As discussed earlier, this implies a standard state having an ideal coil configuration. When the hybridization site is within the target, it also reflects the contribution of two dangling ends. Gcoil(N), as given by Eq. 7, allows for the swelling of the free coil due to excluded volume and electrostatic interactions. Strictly speaking,
as given by the nearest-neighbor method. As discussed earlier, this implies a standard state having an ideal coil configuration. When the hybridization site is within the target, it also reflects the contribution of two dangling ends. Gcoil(N), as given by Eq. 7, allows for the swelling of the free coil due to excluded volume and electrostatic interactions. Strictly speaking,  ln at where at is the activity (51). The dimensionless at is related to the molar concentration of targets ct via at = γct where γ is the activity coefficient. Because
ln at where at is the activity (51). The dimensionless at is related to the molar concentration of targets ct via at = γct where γ is the activity coefficient. Because 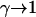 as
as 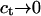 we will, for simplicity, express μt by Eq. 15 noting that the molar ct in this expression is dimensionless.
we will, for simplicity, express μt by Eq. 15 noting that the molar ct in this expression is dimensionless.
It is useful to first specify Kpt of the reference state corresponding, as discussed in “Design of oligonucleotide microarray experiments”, to 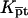 of the bulk reaction
of the bulk reaction 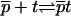 where
where 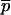 denotes a free probe chain. To this end we need
denotes a free probe chain. To this end we need
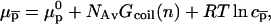 |
(16) |
and
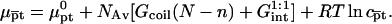 |
(17) |
The equilibrium condition 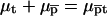 yields
yields 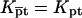 with
with
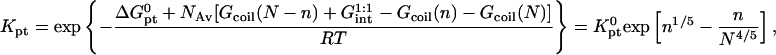 |
(18) |
where 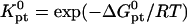 and
and 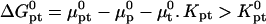 because the hybridization results in the formation of a rodlike ds domain whose monomers experience only short-range interactions with each other and long-range interactions with the monomers of the unhybridized ss tails.
because the hybridization results in the formation of a rodlike ds domain whose monomers experience only short-range interactions with each other and long-range interactions with the monomers of the unhybridized ss tails.
The chemical potentials μpt and μp are specified by the free energy per probe site of the surface, γsite. In the 1:1 regime, when 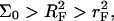 there is no mutual interaction between the probes or between the targets. The molar free energy of probe sites is
there is no mutual interaction between the probes or between the targets. The molar free energy of probe sites is
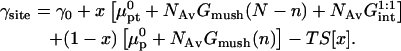 |
(19) |
Here γ0 is the free-energy density of the bare surface whereas 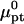 and
and 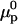 denote the chemical potentials of the hybridized and nonhybridized probes in the standard state. As noted before, the standard state of p is an ideal coil with no excluded volume interactions. The two Gmush terms allow for the excluded volume and screened electrostatic interactions as well as for the effect of the impenetrable wall. Gmush(N − n) accounts for the monomer-monomer interactions of the unhybridized tail of pt whereas Gmush(n) allows for the contribution of the unhybridized probe.
denote the chemical potentials of the hybridized and nonhybridized probes in the standard state. As noted before, the standard state of p is an ideal coil with no excluded volume interactions. The two Gmush terms allow for the excluded volume and screened electrostatic interactions as well as for the effect of the impenetrable wall. Gmush(N − n) accounts for the monomer-monomer interactions of the unhybridized tail of pt whereas Gmush(n) allows for the contribution of the unhybridized probe. 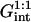 reflects the electrostatic and excluded volume interactions between the hybridized target and its own probe. The mixing entropy per mol of p and pt sites is S[x] = −R[x ln x + (1 − x) ln(1 − x)]. The equilibrium condition μpt = μp + μt can be expressed in terms of the exchange chemical potential of the hybridized probe,
reflects the electrostatic and excluded volume interactions between the hybridized target and its own probe. The mixing entropy per mol of p and pt sites is S[x] = −R[x ln x + (1 − x) ln(1 − x)]. The equilibrium condition μpt = μp + μt can be expressed in terms of the exchange chemical potential of the hybridized probe, 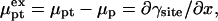 as
as 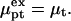 The hybridization isotherm, thus obtained, assumes the familiar Langmuir form
The hybridization isotherm, thus obtained, assumes the familiar Langmuir form
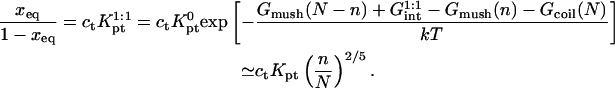 |
(20) |
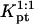 is smaller than Kpt because of the effect of an impenetrable wall giving rise to the (n/N)2/5 factor reflecting the reduction in the number of configurations available to the unhybridized tail of pt.
is smaller than Kpt because of the effect of an impenetrable wall giving rise to the (n/N)2/5 factor reflecting the reduction in the number of configurations available to the unhybridized tail of pt.
In the  range the hybridization behavior is independent of x. As noted earlier, an x dependence is expected when
range the hybridization behavior is independent of x. As noted earlier, an x dependence is expected when  We first discuss the 1:q regime occurring when x < xB; γsite in this range is similar to the one describing the 1:1 regime. The only difference is the replacement of
We first discuss the 1:q regime occurring when x < xB; γsite in this range is similar to the one describing the 1:1 regime. The only difference is the replacement of 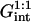 by
by 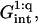 thus allowing for the interactions between a hybridized target and q > 1 probes. The hybridization isotherm as obtained from
thus allowing for the interactions between a hybridized target and q > 1 probes. The hybridization isotherm as obtained from 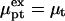 is
is
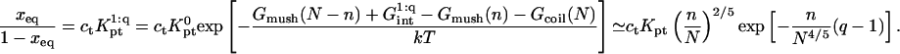 |
(21) |
As in the 1:1 regime, the hybridization isotherm is of the Langmuir form. The equilibrium constant,  is, however, smaller than
is, however, smaller than 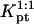 because
because 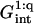 is larger than
is larger than 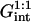 by a factor of
by a factor of 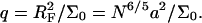
When 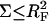 or
or  the hybridized targets begin to crowd each other and form a brush. This crossover occurs at
the hybridized targets begin to crowd each other and form a brush. This crossover occurs at 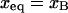 corresponding to
corresponding to
 |
(22) |
The γsite term in the brush regime,
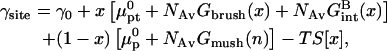 |
(23) |
is distinctive in two respects. First, Gmush(N − n) is replaced by an x dependent free energy of a chain in a brush, Gbrush(x). Second, the term allowing for the target-probe interactions, 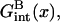 is also a function of x. The hybridization isotherm, obtained as before, is
is also a function of x. The hybridization isotherm, obtained as before, is
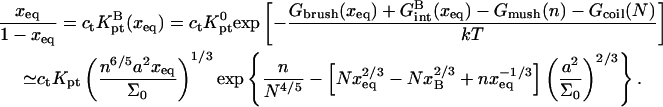 |
(24) |
The N1/5 term, arising from Gcoil(N) is expressed as 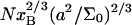 to underline the crossover behavior at xB. By construction, this isotherm is only meaningful when ct > cB so that x > xB. It deviates strongly from the Langmuir form because of the x dependence of Gbrush and
to underline the crossover behavior at xB. By construction, this isotherm is only meaningful when ct > cB so that x > xB. It deviates strongly from the Langmuir form because of the x dependence of Gbrush and 
The complete “long-tail” hybridization isotherm for the 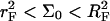 case is obtained from Eqs. 21 and 24. In this isotherm, as in the interaction free Langmuir isotherm (Eq. 1),
case is obtained from Eqs. 21 and 24. In this isotherm, as in the interaction free Langmuir isotherm (Eq. 1), 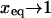 as ct increases. However, the two scenarios differ strongly with respect to the range of ct involved (Fig. 2). The saturation in the long-tail case occurs at a much higher ct. When xeq vs. ct curves of the two scenario are compared over a limited ct range (Fig. 2 A), the long-tail isotherm is superficially similar to a Langmuir isotherm but with apparent saturation at
as ct increases. However, the two scenarios differ strongly with respect to the range of ct involved (Fig. 2). The saturation in the long-tail case occurs at a much higher ct. When xeq vs. ct curves of the two scenario are compared over a limited ct range (Fig. 2 A), the long-tail isotherm is superficially similar to a Langmuir isotherm but with apparent saturation at  A plot of xeq vs. log ct (Fig. 2 B) is necessary to visualize the differences in the saturation behavior.
A plot of xeq vs. log ct (Fig. 2 B) is necessary to visualize the differences in the saturation behavior.
FIGURE 2.

The hybridization isotherms as calculated using Eqs. 21 and 24 for the probe target pairs utilized by Guo et al.(5) with Σ0 = 2500 Å2 and T = 30°C. N = 157 (solid line), N = 347 (dashed line) and the reference state case calculated from Eq. 1 with N = 157 (dotted line). The xeq vs. ct curves are depicted in panel A for the range 0 ≤ ct ≤ 1 pM whereas xeq vs. log ct plots are depicted in panel B (−9 corresponds to 1 nM).
A useful measure of the sensitivity of the DNA chip is the c50 corresponding to the target concentration, ct, needed to obtain at equilibrium xeq = 1/2 (13). The c50 also provides a rough estimate for the onset of saturation, as discussed earlier. In the 1:1 regime, where the hybridization follows a Langmuir isotherm, 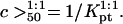 When
When 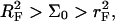 we can distinguish between two scenarios. So long as
we can distinguish between two scenarios. So long as 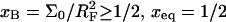 is attained before the onset of the brush and
is attained before the onset of the brush and 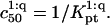 In the opposite case,
In the opposite case, 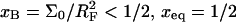 occurs in the brush regime and
occurs in the brush regime and 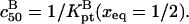 The corresponding experimental guidelines assume a more useful form when considering the logarithm of c50. In particular, these relate the range of expected target concentrations ct, as given by
The corresponding experimental guidelines assume a more useful form when considering the logarithm of c50. In particular, these relate the range of expected target concentrations ct, as given by  or
or 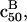 to
to 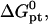 n, N, and Σ0
n, N, and Σ0
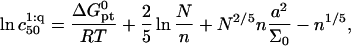 |
(25) |
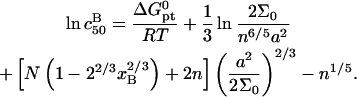 |
(26) |
The 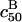 can be significantly higher than
can be significantly higher than 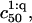
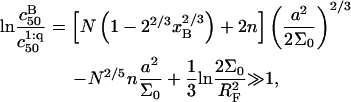 |
(27) |
because it is dominated by the factor 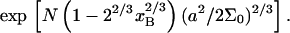 It is helpful to compare Eqs. 25 and 26 with the Langmuir isotherm of the “reference” state, Eq. 1, where
It is helpful to compare Eqs. 25 and 26 with the Langmuir isotherm of the “reference” state, Eq. 1, where 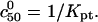 The guideline obtained, following the same procedure, is
The guideline obtained, following the same procedure, is
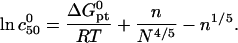 |
(28) |
In this case 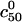 is determined by n, N, and
is determined by n, N, and 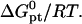 In marked contrast
In marked contrast 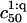 and
and 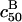 depend explicitly on Σ0. The strong N dependence of
depend explicitly on Σ0. The strong N dependence of 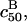 as compared to
as compared to  and
and 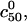 is illustrated in Fig. 3. The increase of
is illustrated in Fig. 3. The increase of 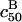 signals a corresponding loss of sensitivity.
signals a corresponding loss of sensitivity.
FIGURE 3.

Plots of log 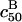 vs. N for the probes utilized by Guo et al. (5) with Σ0 = 2500 Å2 (solid line) and Σ0 = 5000 Å2 (dashed line). T = 30°C and n = 15. The reference state log
vs. N for the probes utilized by Guo et al. (5) with Σ0 = 2500 Å2 (solid line) and Σ0 = 5000 Å2 (dashed line). T = 30°C and n = 15. The reference state log 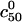 is plotted for comparison (···). The circles correspond to the crossover between 1:1 and 1:q regimes whereas squares correspond to the crossover between 1:q and B regimes.
is plotted for comparison (···). The circles correspond to the crossover between 1:1 and 1:q regimes whereas squares correspond to the crossover between 1:q and B regimes.
To utilize these guidelines one needs 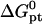 as calculated using the nearest-neighbor model. However, to highlight the role of n as a design parameter, it is helpful to use the Wetmur approximation (52) where average values of the nearest-neighbor contributions are utilized. Accordingly,
as calculated using the nearest-neighbor model. However, to highlight the role of n as a design parameter, it is helpful to use the Wetmur approximation (52) where average values of the nearest-neighbor contributions are utilized. Accordingly, 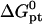 of perfectly matched probe-target pair, when the hybridization site is located within the target, is approximated by
of perfectly matched probe-target pair, when the hybridization site is located within the target, is approximated by
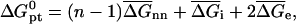 |
(29) |
where 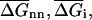 and
and 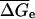 are the average values corresponding to a nearest-neighbor pair, an initiation step and a dangling end. Wetmur estimated the nearest-neighbor contribution by
are the average values corresponding to a nearest-neighbor pair, an initiation step and a dangling end. Wetmur estimated the nearest-neighbor contribution by 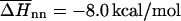 and
and  the initiation term by a temperature independent
the initiation term by a temperature independent 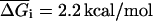 and the dangling end contribution by
and the dangling end contribution by  and
and 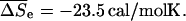 Note that although useful, the Wetmur approximation erroneously predicts identical
Note that although useful, the Wetmur approximation erroneously predicts identical  for all pt pairs with N = n.
for all pt pairs with N = n.
BRUSH EFFECTS: KINETICS OF HYBRIDIZATION
Having obtained the equilibrium constants 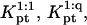 and
and 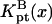 for the hybridization at the surface we are now in a position to consider the corresponding rate constants. To this end we will assume, and later confirm, that the rate is reaction controlled. Again, for simplicity, we set numerical prefactors to unity, v = a3 and lp = a. It is necessary to recall first the relevant features of the kinetics of oligonucleotide hybridization and of the desorption of polymers out of a brush.
for the hybridization at the surface we are now in a position to consider the corresponding rate constants. To this end we will assume, and later confirm, that the rate is reaction controlled. Again, for simplicity, we set numerical prefactors to unity, v = a3 and lp = a. It is necessary to recall first the relevant features of the kinetics of oligonucleotide hybridization and of the desorption of polymers out of a brush.
As discussed in “Design of oligonucleotide microarray experiments”, the reference state of our analysis is a layer of noninteracting probes bound to a passivated surface by long flexible spacers. We assume that the molecular mechanism of hybridization in this case is identical to the bulk one and that the kinetics follow the Langmuir rate law
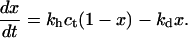 |
(30) |
In this regime the hybridization and denaturation rate constants, kh and kd, are independent of Σ0 or x and approach their bulk values. At equilibrium 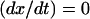 leading to Kpt = kh/kd as required by detailed balance. In turn, the hybridization mechanism of free oligonucleotides in solution is thought to involve the steps outlined below (35,39,53,54). An approach and alignment of the single-stranded oligonucleotides is followed by the hybridization of a single basepair. A stable nucleus, comprising nc + 1 basepairs, is formed by stepwise addition of hybridized pairs. Importantly, a ds sequence of n ≤ nc is unstable. Once nc + 1 is attained, the ds domain is rapidly “zipped up”. For oligonucleotides comprising GC basepairs
leading to Kpt = kh/kd as required by detailed balance. In turn, the hybridization mechanism of free oligonucleotides in solution is thought to involve the steps outlined below (35,39,53,54). An approach and alignment of the single-stranded oligonucleotides is followed by the hybridization of a single basepair. A stable nucleus, comprising nc + 1 basepairs, is formed by stepwise addition of hybridized pairs. Importantly, a ds sequence of n ≤ nc is unstable. Once nc + 1 is attained, the ds domain is rapidly “zipped up”. For oligonucleotides comprising GC basepairs 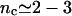 and the hybridization rate constant exhibits the form
and the hybridization rate constant exhibits the form 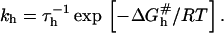 Here τh is a molecular timescale characterizing the formation of the last basepair of the nucleus whereas the activation free energy
Here τh is a molecular timescale characterizing the formation of the last basepair of the nucleus whereas the activation free energy 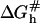 reflects the formation of a ds nucleus of nc basepairs plus the activation free energy for adding the next basepair. Importantly, the reaction is not diffusion controlled but involves a number of activation barriers associated with a corrugated free-energy profile (39). A rough estimate of
reflects the formation of a ds nucleus of nc basepairs plus the activation free energy for adding the next basepair. Importantly, the reaction is not diffusion controlled but involves a number of activation barriers associated with a corrugated free-energy profile (39). A rough estimate of  within the Wetmur approximation (52) yields
within the Wetmur approximation (52) yields 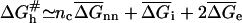 indicating that
indicating that 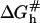 depends on nc rather than n. This last point rationalizes a phenomenological result we will utilize later, namely kh in high ionic strength solutions is
depends on nc rather than n. This last point rationalizes a phenomenological result we will utilize later, namely kh in high ionic strength solutions is
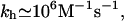 |
(31) |
to within one order of magnitude and with a weak T dependence (39). This, together with the detailed balance requirement Kpt = kh/kd yields
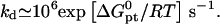 |
(32) |
In terms of the Wetmur approximation kd is expressed as  The activation barrier for denaturation involves, thus, the break up of n − nc basepairs so as to form an unstable ds domain. Importantly, for 15 ≤ n ≤ 25, the denaturation life time at 37°C is measured in years.
The activation barrier for denaturation involves, thus, the break up of n − nc basepairs so as to form an unstable ds domain. Importantly, for 15 ≤ n ≤ 25, the denaturation life time at 37°C is measured in years.
At this point it is of interest to comment on a result, obtained from computer simulations, concerning the kinetics of desorption out of a brush (55). It concerns a planar brush formed from flexible and neutral chains with one terminal monomer experiencing a short-range attraction to the wall. The attraction was modeled as a well of width a, a monomer size, and depth Gwell. In this system the expulsion rate constant is
 |
(33) |
where τ(Σ) is the time required by the headgroup to diffuse across a distance Σ1/2, corresponding to the innermost blob of the brush. Importantly, kout although Σ dependent was found to be independent of N. Once the surface bond is broken, the expulsion of the chain out of the brush is driven by repulsive monomer-monomer interactions with neighboring chains. This last stage is a fast process and thus not rate controlling. The system studied in Wittmer et al. (55) differs from ours in two respects. First, in this study the attractive potential is laterally invariant, i.e., the surface is uniformly attractive. As a result, the reaction coordinate is the distance between the terminal end group and the surface z. In our case the attractive potential is localized at the immediate vicinity of the probe and the early steps of denaturation involve lateral separation of the two strands. Consequently the reaction coordinate at the vicinity of the surface is no longer z. Second, in Wittmer et al. (55), the barrier to adsorption is due to the brush. There is no barrier in the mushroom regime where the reaction is diffusion controlled. This is also the case in the brush regime when the terminal group resides within a distance Σ1/2 from the surface. However, as noted earlier, the hybridization reaction in the bulk is not diffusion controlled. Accordingly, one should consider the possibility that the rate of hybridization at the surface is similarly not controlled by diffusion. In such a case the denaturation rate constant, corresponding to kout, will be independent of both N and Σ.
In the following we will assume, and later confirm, that the rate of hybridization at the surface is reaction controlled rather than diffusion controlled. In quantitative terms, the implementation of the reaction control hypothesis involves three ingredients. First, we assume that the rate equation may be written as
 |
(34) |
where ct* is the local concentration of target hybridization sites in the vicinity of the probes; 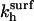 and
and 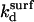 denote the rate constants at the surface. In writing this equation we make a number of straightforward microscopic hypotheses. First, that the hybridization and denaturation reactions at the surface are, respectively, monomolecular and bimolecular. Second, the encounter probability between a probe and a target is proportional to ct*. Note that ct* is laterally invariant in the 1:q and brush regimes, thus implying that the diffusion is fast enough to ensure lateral homogeneity. The second ingredient requires a lengthier discussion. We will argue that
denote the rate constants at the surface. In writing this equation we make a number of straightforward microscopic hypotheses. First, that the hybridization and denaturation reactions at the surface are, respectively, monomolecular and bimolecular. Second, the encounter probability between a probe and a target is proportional to ct*. Note that ct* is laterally invariant in the 1:q and brush regimes, thus implying that the diffusion is fast enough to ensure lateral homogeneity. The second ingredient requires a lengthier discussion. We will argue that 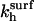 differs from the corresponding bulk value whereas
differs from the corresponding bulk value whereas 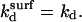 The physical justification for this conjecture is as follows. The denaturation process is mostly local, reflecting reorganization of hydrogen bonds and stacking interactions (39). Accordingly, the breaking of the basepairs should not be influenced by the presence of the impenetrable wall. In contrast, we expect the wall to affect the hybridization process. In particular, the free-energy price for the approach and alignment of the single-stranded oligonucleotides should be lower because the entropy of the reacting chains at the impenetrable surface is lower. The last ingredient is the assumption that ct* for any x is equal to the equilibrium concentration of unhybridized terminal groups at the surface. In other words, the diffusion of chains is sufficiently fast in comparison to the hybridization reaction to ensure that a Boltzmann distribution is maintained. This condition is especially stringent in the brush regime, where inbound diffusion must overcome a potential barrier due to interactions with the previously tethered chains. The equilibrium condition requires that ct*/ct = exp(−Δμ/RT) where Δμ(x) is the difference between the chemical potential of a fully inserted chain and a free one. Altogether, for each of the three regimes
The physical justification for this conjecture is as follows. The denaturation process is mostly local, reflecting reorganization of hydrogen bonds and stacking interactions (39). Accordingly, the breaking of the basepairs should not be influenced by the presence of the impenetrable wall. In contrast, we expect the wall to affect the hybridization process. In particular, the free-energy price for the approach and alignment of the single-stranded oligonucleotides should be lower because the entropy of the reacting chains at the impenetrable surface is lower. The last ingredient is the assumption that ct* for any x is equal to the equilibrium concentration of unhybridized terminal groups at the surface. In other words, the diffusion of chains is sufficiently fast in comparison to the hybridization reaction to ensure that a Boltzmann distribution is maintained. This condition is especially stringent in the brush regime, where inbound diffusion must overcome a potential barrier due to interactions with the previously tethered chains. The equilibrium condition requires that ct*/ct = exp(−Δμ/RT) where Δμ(x) is the difference between the chemical potential of a fully inserted chain and a free one. Altogether, for each of the three regimes
 |
(35) |
where 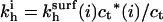 is the observable hybridization rate constant;
is the observable hybridization rate constant; 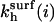 and
and  specify, respectively, the values of
specify, respectively, the values of 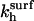 and ct* in the i regime. At equilibrium the condition
and ct* in the i regime. At equilibrium the condition  yields
yields
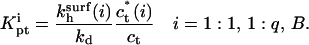 |
(36) |
Because Kpt = kh/kd the rate constants for the three regimes, i = 1:1, 1:q and B, are
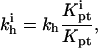 |
(37) |
leading, up to numerical prefactors, to
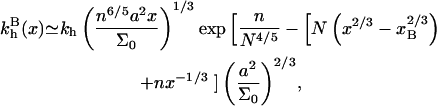 |
(38) |
 |
(39) |
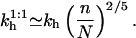 |
(40) |
Note that 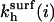 are independent of the regime i and larger than kh. In particular,
are independent of the regime i and larger than kh. In particular, 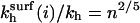 exp(n/N4/5). The n2/5 factor is due to the higher free energy of the probe as compared to that of the corresponding free oligonucleotide and the exponential term arises from probe-target interactions.
exp(n/N4/5). The n2/5 factor is due to the higher free energy of the probe as compared to that of the corresponding free oligonucleotide and the exponential term arises from probe-target interactions.
The results above were obtained assuming that the hybridization rate is controlled by the reaction rather than by the diffusion toward the surface. To check the consistency of this approach we consider the corresponding Damköhler number Da = Jreac/Jdif (56). Here Jreac and Jdif are the maximal fluxes associated with the reaction and the inbound diffusion, assuming reaction control. Reaction control implies 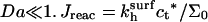 is an upper bound on the reaction flux. The inbound flux of chain through the brush is Jdif = ct*vbarrier where vbarrier is the diffusion velocity of a single chain at the vicinity of the surface where the brush potential is essentially flat. Recent experimental results and a unified picture of theoretical models of Jdif are presented by Titmuss et al. (57). Altogether
is an upper bound on the reaction flux. The inbound flux of chain through the brush is Jdif = ct*vbarrier where vbarrier is the diffusion velocity of a single chain at the vicinity of the surface where the brush potential is essentially flat. Recent experimental results and a unified picture of theoretical models of Jdif are presented by Titmuss et al. (57). Altogether
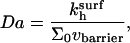 |
(41) |
where vbarrier = αkT/ηNa2. Here η is the solvent viscosity and α is a polymer specific numerical constant; α of ssDNA has not yet been determined but for flexible synthetic polymers  For water at 25°C η = 0.89 × 10−3 Nm−2s. The Damköhler number at 25°C, when both fluxes are expressed in units of chains m−2s−1, is
For water at 25°C η = 0.89 × 10−3 Nm−2s. The Damköhler number at 25°C, when both fluxes are expressed in units of chains m−2s−1, is
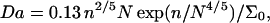 |
(42) |
where we assumed α = 0.1, kh = 106 M−1s−1, a = 6 Å, and expressed Σ0 in Å2. For 100 ≤ N ≤ 600, n = 15 and T = 25°C, the Damköhler number varies in the range 4 × 10−2 ≤ Da ≤ 0.2 when Σ0 = 1500 Å2 and 0.1 ≤ Da ≤ 0.5 when Σ0 = 500 Å2. The variation of water viscosity in the range 0°C ≤ T ≤ 70°C affects the Da values at most by a factor 2. Accordingly, the assumption of reaction control of the hybridization rate is justified for typical values of N and Σ0. It will, however, fail eventually for high N values. One should note that the issue of reaction versus diffusion also arise for the diffusion from the bulk toward the surface. When the hybridization chamber is agitated this is not an issue and we will not discuss it further.
As required, the rate constants Eqs. 38–40 obey detailed balance and exhibit the proper crossover behavior. In particular, 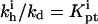 as well as
as well as 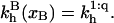 The x dependence of
The x dependence of 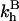 slows down the adsorption rate (Fig. 4);
slows down the adsorption rate (Fig. 4); 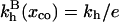 is a possible measure for the onset of significant slow down due to the brush formation. In the limit of
is a possible measure for the onset of significant slow down due to the brush formation. In the limit of 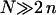 the x−1/3 term is negligible and the onset occurs roughly at
the x−1/3 term is negligible and the onset occurs roughly at
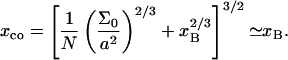 |
(43) |
It thus affects the whole brush regime. The slower kinetics in the brush regime can affect the attained hybridization even after long hybridization periods (Fig. 5). This is of practical importance because samples of identical ct but different N will vary in their signal intensity.
FIGURE 4.

The hybridization kinetics, as described by a plot of x versus the time t in hours, for the probe target pairs of Guo et al. (5): n = 15, T = 30°C, ct = 1 nM, and Σ0 = 5000 Å2. The N = 157 (solid line) and N = 347 (dashed line) curves are compared to the reference state case with N = 157 (dotted line).
FIGURE 5.

The hybridization fraction attained after th = 16 h as a function of N for the Affimetrix probe p2, n = 25, T = 45°C, ct = 0.1 nM with Σ0 = 2500 Å2 (solid line) and Σ0 = 5000 Å2 (dashed line).
DISCUSSION
The relative size of the targets and probes is an important characteristic of oligonucleotide microarrays. When the two are of equal size, N = n, the onset of interaction between the probes is roughly set by the span of the probes as determined by n. In biology experiments the targets are much larger, 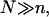 and the onset of interactions is controlled by N. The progress of hybridization can give rise to crowding of the nonhybridized tails when
and the onset of interactions is controlled by N. The progress of hybridization can give rise to crowding of the nonhybridized tails when 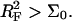 The polyelectrolyte brush thus formed affects the hybridization isotherm and the rate equations. In particular, it lowers both the hybridization rate and the attainable hybridization for a given concentration of targets. It is important to allow for this effect in the design of DNA microarrays, in the formulating of the protocols of sample preparation and hybridization as well as in the analysis of the results. With regard to design of DNA chips the brush effect is important in choosing the desired density of oligonucleotide probes, or equivalently Σ0. The brush effect will lower the fraction of probes that actually hybridize. As a result, the benefits of increasing the surface density of oligonucleotide probes diminish when the intended targets are long. When Σ0 is set, these considerations suggest a criteria for tuning the length of the targets, N, as controlled by the choice of the PCR primers or of the fragmentation procedure. In particular, it is beneficial to shorten N so as to avoid crowding. When brush effects do occur the analysis of the results should allow for the ensuing deviations from the Langmuir behavior. This is an important point for the implementation of model-based algorithms.
The polyelectrolyte brush thus formed affects the hybridization isotherm and the rate equations. In particular, it lowers both the hybridization rate and the attainable hybridization for a given concentration of targets. It is important to allow for this effect in the design of DNA microarrays, in the formulating of the protocols of sample preparation and hybridization as well as in the analysis of the results. With regard to design of DNA chips the brush effect is important in choosing the desired density of oligonucleotide probes, or equivalently Σ0. The brush effect will lower the fraction of probes that actually hybridize. As a result, the benefits of increasing the surface density of oligonucleotide probes diminish when the intended targets are long. When Σ0 is set, these considerations suggest a criteria for tuning the length of the targets, N, as controlled by the choice of the PCR primers or of the fragmentation procedure. In particular, it is beneficial to shorten N so as to avoid crowding. When brush effects do occur the analysis of the results should allow for the ensuing deviations from the Langmuir behavior. This is an important point for the implementation of model-based algorithms.
Physical-chemistry-type experiments that aim to investigate the function of DNA microarrays tend to focus on the symmetric case of N = n. Our discussion highlights the merit of studying the kinetics and the equilibrium behavior in the asymmetric case, 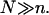 In this case it is of interest to correlate the hybridization behavior with measurements of the brush thickness.
In this case it is of interest to correlate the hybridization behavior with measurements of the brush thickness.
Our analysis focused on the case of ssDNA targets so as to avoid complications due to the secondary structure of RNA molecules. The importance of the secondary structure of RNA targets, as used in gene expression experiments, is yet to be established because the effect of labeling by biotin is not well understood. The effect of the fragmentation on the kinetics of hybridization suggests, however, that a crowding effect of some sort is indeed involved.
Acknowledgments
The authors benefited from instructive discussions with E. Southern, E. Katz, and T. Livache.
E.B.Z. was funded by the Commissariat à l'Energie Atomique with additional support from the Russian Fund for Fundamental Research (RFBR 02-0333127).
APPENDIX A: THE VIRIAL COEFFICIENT AND THE CASE OF A BRUSH OF RODS
Consider first the second virial coefficient
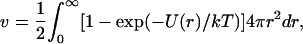 |
(44) |
for spherical monomers of radius a when their interactions are purely repulsive. In particular, the interaction potential, U(r), comprises a hard-core repulsion together with a screened electrostatic repulsion, that is
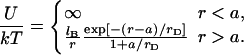 |
(45) |
Here rD is the Debye screening length and lB is the Bjerrum length (58). The hard-core contribution to v is 2πa3/3. The electrostatic contribution, assuming that 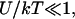 is
is 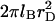 and altogether
and altogether
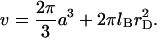 |
(46) |
If one supplements the electrostatic repulsion U by a weak van der Waals attraction the first term assumes the form 2πa3(1 − θ/T)/3 where θ is the θ-temperature (40) thus leading to Eq. 10. For 0.1 M of NaCl salt, rD = 10 Å and assuming a = 6 Å we find that the electrostatic term dominates. When the salt concentration is 1 M the screening length diminishes to rD = 3 Å and the two terms are comparable.
In the case of probes and targets of equal length, n = N, the probe layer consists of a mixture of single-stranded probes and hybridized, double-stranded ones. The associated interaction free energy for this case can be obtained (13) upon assuming, following Korolev et al. (59), that both adopt rod-like configurations of equal length L = nb where 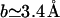 is the contribution of a base or basepair to the length of the rod. The hybridized probes are rodlike because a dsDNA is rigid on the length scales of a typical probe (10 ⩽ n ≤ 30). Viewing the unhybridized probes as rigid rods is an approximation justified, for the short probes, by two related observations. One is the tendency of ssDNA to form rigid domains of single-stranded helices due to stacking interactions (35,39,60). The second is that the persistence length attributed to ssDNA is comparable to the length of the probes. It is important, however, to stress that the configurations of ssDNA are not yet fully characterized. As noted in "Design of oligonucleotide microarray experiments", the reported values of the persistence length of ssDNA vary over a wide range 7.5 Å ≤ lp ≤ 35 Å. In any case, the approximation of the brush height by L is sensible for short oligonucleotides with n such that
is the contribution of a base or basepair to the length of the rod. The hybridized probes are rodlike because a dsDNA is rigid on the length scales of a typical probe (10 ⩽ n ≤ 30). Viewing the unhybridized probes as rigid rods is an approximation justified, for the short probes, by two related observations. One is the tendency of ssDNA to form rigid domains of single-stranded helices due to stacking interactions (35,39,60). The second is that the persistence length attributed to ssDNA is comparable to the length of the probes. It is important, however, to stress that the configurations of ssDNA are not yet fully characterized. As noted in "Design of oligonucleotide microarray experiments", the reported values of the persistence length of ssDNA vary over a wide range 7.5 Å ≤ lp ≤ 35 Å. In any case, the approximation of the brush height by L is sensible for short oligonucleotides with n such that  For reasonable values of lp/a this condition is fulfilled for
For reasonable values of lp/a this condition is fulfilled for  Similarly, the thermodynamic parameters of the stacking interactions are not fully established. With these reservations in mind, this picture provides a convenient approximation because it allows us to assign to the probe layer a unique thickness, independent of x. In particular, the thickness of the probe layer is comparable to L. The interaction free-energy density within the probe layer is accordingly, Fint = vc2 where c = n(1 + x)/Σ0L is the number concentration of monomers within the layer and the interaction free-energy density per unit area is
Similarly, the thermodynamic parameters of the stacking interactions are not fully established. With these reservations in mind, this picture provides a convenient approximation because it allows us to assign to the probe layer a unique thickness, independent of x. In particular, the thickness of the probe layer is comparable to L. The interaction free-energy density within the probe layer is accordingly, Fint = vc2 where c = n(1 + x)/Σ0L is the number concentration of monomers within the layer and the interaction free-energy density per unit area is
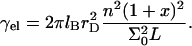 |
(47) |
Accordingly, the overall free energy per probe site
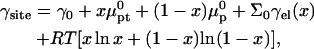 |
(48) |
and the equilibrium condition 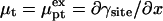 yields
yields
 |
(49) |
with  This recovers, up to a factor of 2, results we obtained earlier using the box approximation for the solution of the Poisson-Boltzmann equation (13). The isotherm obtained above differs from the “brush isotherm” because the chain elasticity does not play a role and the layer thickness does not exhibit an x dependence.
This recovers, up to a factor of 2, results we obtained earlier using the box approximation for the solution of the Poisson-Boltzmann equation (13). The isotherm obtained above differs from the “brush isotherm” because the chain elasticity does not play a role and the layer thickness does not exhibit an x dependence.
APPENDIX B: EFFECT OF CHAIN SELF-AVOIDANCE ON THE HYBRIDIZATION CONSTANTS
The Flory approximation as used in the text overestimates both the elastic and interaction free energies. Another delicate point concerns the entropy of the free ends. At the same time, the Flory approximation is known to be robust and its performance for the brush has been studied showing relatively mild deviation from the exact results obtained by SCF theory (24). With these points in mind it is of interest to confirm the results obtained utilizing the Alexander-Flory approximation by a more rigorous approach. In the following we present exact results concerning  In particular, the alternative derivation allows for the chain self-avoidance while ignoring the small correction due to interactions between the hybridized ds domain and unhybridized tail of the target. To this end we utilize the partition function of a self-avoiding chain (61,62). The partition function Zcoil(N) of a free self-avoiding chain of N monomers is
In particular, the alternative derivation allows for the chain self-avoidance while ignoring the small correction due to interactions between the hybridized ds domain and unhybridized tail of the target. To this end we utilize the partition function of a self-avoiding chain (61,62). The partition function Zcoil(N) of a free self-avoiding chain of N monomers is
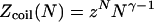 |
(50) |
where z is the model-dependent effective partition function of a monomer, and γ is a universal configurational exponent. For a self-avoiding chain with a terminal monomer anchored to an impenetrable planar surface, a “mushroom”, the partition function is
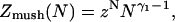 |
(51) |
where γ1 is a different universal configurational exponent.
When a probe and a target hybridize, the ds domain can be envisioned as a rigid rod with a partition function
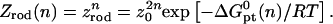 |
(52) |
Here, zrod is the partition function of a pair of hybridized monomers, z0 is partition function of a single monomer in an ideal Gaussian coil, n is number of basepairs in the ds domain, and 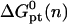 is the free-energy difference between the rigid ds and ideal coil ss domains. The free-energy G is related to the partition function Z by G = −RT ln Z.
is the free-energy difference between the rigid ds and ideal coil ss domains. The free-energy G is related to the partition function Z by G = −RT ln Z.
The hybridization constant Kpt in a solution of targets and probes whose respective lengths are N and  is
is
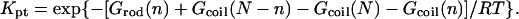 |
(53) |
Using Eqs. 50, 52, and 53, we obtain
 |
(54) |
where 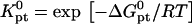 as introduced earlier.
as introduced earlier.
For the hybridization at a surface in the 1:1 regime
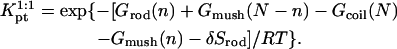 |
(55) |
Here, δ Srod ≡ ln(β) is the reduction in the rod entropy due to its attachment to the surface. The specific value of β, of order unity, depends on the length and flexibility of the spacer. In the simplest case, of a short flexible spacer, the surface eliminates half of the space available to a free rod in the solution, thus yielding β = 1/2. Equations 50, 51, and 55 lead to
 |
(56) |
The ratio of hybridization constants at the surface and in solution, as determined from Eqs. 54 and 56 is
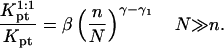 |
(57) |
The values of γ ≈ 1.167 and γ1 ≈ 0.695 were obtained using field theoretical methods and numerical calculations (61,62). Therefore, γ − γ1 ≈ 0.47 is in close agreement with 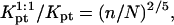 Eq. 20.
Eq. 20.
References
- 1.Graves, D. J. 1999. Powerful tools for genetic analysis come of age. Trends Biotechnol. 17:127–134. [DOI] [PubMed] [Google Scholar]
- 2.Wang, J. 2000. From DNA biosensors to gene chips. Nucleic Acids Res. 28:3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lockhart, D. J., and E. A. Winzeler. 2000. Genomics, gene expression and DNA arrays. Nature. 405:827–836. [DOI] [PubMed] [Google Scholar]
- 4.Heller, M. J. 2002. DNA microarray technology: devices, systems and applications. Annu. Rev. Biomed. Eng. 4:129–153. [DOI] [PubMed] [Google Scholar]
- 5.Guo, Z., R. A. Guilfoyle, A. J. Thiel, R. Wang, and L. M. Smith. 1994. Direct fluorescence analysis of genetic polymorphism by hybridization with oligonucleotide arrays on glass supports. Nucleic Acids Res. 22:5456–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prix, L., P. Uciechowski, B. Böckmann, M. Giesing, and A. J. Schuetz. 2002. Diagnostic biochip array for fast and sensitive detection of K-ras mutations in stool. Clin. Chem. 48:428–435. [PubMed] [Google Scholar]
- 7.Chan, V., D. J. Graves, and S. McKenzie. 1995. The biophysics of DNA hybridization with immobilized oligonucleotide probes. Biophys. J. 69:2243–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Livshits, M. A., and A. D. Mirzabekov. 1996. Theoretical analysis of the kinetics of DNA hybridization with gel-immobilized oligonucleotides. Biophys. J. 71:2795–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vainrub, A., and M. B. Pettitt. 2002. Coulomb blockage of hybridization in two-dimensional DNA arrays. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 66:041905. [DOI] [PubMed] [Google Scholar]
- 10.Bhanot, G., Y. Louzoun, J. Zhu, and C. DeLisi. 2003. The importance of thermodynamic equilibrium for high throughput gene expression arrays. Biophys. J. 84:124–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Held, G. A., G. Grinstein, and Y. Tu. 2003. Modeling of DNA microarray data by using physical properties of hybridization. Proc. Natl. Acad. Sci. USA. 100:7575–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang, L., M. F. Miles, and K. D. Aldape. 2003. A model of molecular interactions on short oligonucleotide microarrays. Nat. Biotechnol. 21:818–821. [DOI] [PubMed] [Google Scholar]
- 13.Halperin, A., A. Buhot, and E. B. Zhulina. 2004. Sensitivity, specificity, and the hybridization isotherms of DNA chips. Biophys. J. 86:718–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halperin, A., A. Buhot, and E. B. Zhulina. 2004. b. Hybridization isotherms of DNA chips and the quantification of mutation studies. Clin. Chem. 50:2254–2262. [DOI] [PubMed] [Google Scholar]
- 15.Forman, J. E., I. D. Walton, D. Stern, R. P. Rava, and M. O. Trulson. 1998. Thermodynamics of duplex formation and mismatch discrimination on photolithographically synthesized oligonucleotide arrays. ACS Symp. Ser. 682:206–228. [Google Scholar]
- 16.Okahata, Y., M. Kawase, K. Niikura, F. Ohtake, H. Furusawa, and Y. Ebara. 1998. Kinetic measurements of DNA hybridization on an oligonucleotide-immobilized 27-MHz quartz crystal microbalance. Anal. Chem. 70:1288–1296. [DOI] [PubMed] [Google Scholar]
- 17.Steel, A. B., T. M. Herne, and M. J. Tarlov. 1998. Electrochemical quantitation of DNA immobilized on gold. Anal. Chem. 70:4670–4677. [DOI] [PubMed] [Google Scholar]
- 18.Georgiadis, R., K. P. Peterlinz, and A. W. Peterson. 2000. Quantitative measurements and modeling of kinetics in nucleic acid monolayer films using SPR spectroscopy. J. Am. Chem. Soc. 122:3166–3173. [Google Scholar]
- 19.Nelson, B. P., T. E. Grimsrud, M. R. Liles, R. M. Goodman, and R. M. Corn. 2001. Surface plasmon resonance imaging measurements of DNA and RNA hybridization adsorption onto DNA microarrays. Anal. Chem. 73:1–7. [DOI] [PubMed] [Google Scholar]
- 20.Dai, H., M. Meyer, S. Stepaniants, M. Ziman, and R. Soughton. 2002. Use of hybridization kinetics for differentiating specific from non-specific binding to oligonucleotide microarrays. Nucleic Acids Res. 30:e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kepler, T. B., L. Crosby, and K. T. Morgan. 2002. Normalization and analysis of DNA microarray data by self consistency and local regression. Genome Biol. 3:0037.1–0037.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peterson, A. W., L. K. Wolf, and R. M. Georgiadis. 2002. Hybridization of mismatched or partially matched DNA at surfaces. J. Am. Chem. Soc. 124:14601–14607. [DOI] [PubMed] [Google Scholar]
- 23.Hekstra, D., A. R. Taussig, M. Magnasco, and F. Naef. 2003. Absolute mRNA concentrations from sequence-specific calibration of oligonucleotide arrays. Nucleic Acids Res. 31:1962–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milner, S. T. 1991. Polymer brushes. Science. 251:905–914. [DOI] [PubMed] [Google Scholar]
- 25.Halperin, A., M. Tirrell, and T. P. Lodge. 1992. Tethered chains in polymer microstructures. Adv. Polym. Sci. 100:31–71. [Google Scholar]
- 26.Rühe, J., M. Ballauff, M. Biesalski, P. Dziezok, F. Grohn, D. Johannsmann, N. Houbenov, N. Hugenberg, R. Konradi, S. Minko, M. Motornov, R. R. Netz, et al. 2004. Polyelectrolyte brushes. Adv. Polym. Sci. 165:77–150. [Google Scholar]
- 27.Pincus, P. 1991. Colloid stabilization with grafted polyelectrolytes. Macromolecules. 24:2912–2919. [Google Scholar]
- 28.Borisov, O. V., T. M. Birshtein, and E. B. Zhulina. 1991. Collapse of grafted polyelectrolyte layer. J. Phys. II. 1:521–526. [Google Scholar]
- 29.Su, H.-J., S. Surrey, S. E. McKenzie, P. Fortina, and D. J. Graves. 2002. Kinetics of heterogeneous hybridization on indium tin oxide surfaces with and without an applied potential. Electrophoresis. 23:1551–1557. [DOI] [PubMed] [Google Scholar]
- 30.Rosenow, C., R. M. Saxena, M. Durst, and T. R. Gingeras. 2001. Prokaryotic RNA preparation methods useful for high density array analysis: comparison of two approaches. Nucleic Acids Res. 29:E112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Affymetrix. 2004. GeneChip Expression Analysis. Technical Manual. Available at http://www.affymetrix.com/support/technical/manuals.affx [Online].
- 32.Pirrung, M. C. 2002. How to make a DNA chip? Angew. Chem. Int. Ed. Engl. 41:1277–1289. [DOI] [PubMed] [Google Scholar]
- 33.Southern, E., K. Mir, and M. Shchepinov. 1999. Molecular interactions on microarrays. Nat. Genet. 21:5–9. [DOI] [PubMed] [Google Scholar]
- 34.Naef, F., and M. Magnasco. 2003. Solving the riddle of the bright mismatches: labeling and effective binding in oligonucleotide arrays. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 68:011906. [DOI] [PubMed] [Google Scholar]
- 35.Cantor, C. R., and P. R. Schimmel. 1980. Biophysical Chemistry. W. H. Freeman, New York, NY.
- 36.Smith, S. B., Y. J. Cui, and C. Bustamante. 1996. Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science. 271:795–799. [DOI] [PubMed] [Google Scholar]
- 37.Strick, T. R., M.-N. Dessinges, G. Charvin, N. H. Dekker, J.-F. Allemand, D. Bensimon, and V. Croquette. 2003. Stretching of macromolecules and proteins. Rep. Prog. Phys. 66:1–45. [Google Scholar]
- 38.Mills, J. B., E. Vacano, and P. J. Hagerman. 1999. Flexibility of single-stranded DNA: use of gapped duplex helices to determine the persistence lengths of poly(dT) and poly(dA). J. Mol. Biol. 285:245–257. [DOI] [PubMed] [Google Scholar]
- 39.Turner, D. H. 2000. Chap.8 Conformational Changes. In Nucleic Acids: Structures, Properties and Functions. V. A. Bloomfield, D. Crothers, and I. Tinoco, Jr. University Science Books, Sausalito, CA.
- 40.Rubinstein, M., and R. H. Colby. 2003. Polymer Physics. Oxford University Press, Oxford, UK.
- 41.SantaLucia, J., and D. Hicks. 2004. The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct. 33:415–450. [DOI] [PubMed] [Google Scholar]
- 42.SantaLucia, J. 1998. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA. 95:1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peyret, N., P. A. Seneviratne, H. T. Allawi, and J. SantaLucia. 1999. Nearest-neighbor thermodynamics and NMR of DNA sequences with internal A.A, C.C, G.G, and T.T mismatches. Biochemistry. 38:3468–3477. [DOI] [PubMed] [Google Scholar]
- 44.Peyret, N., and J. SantaLucia, Jr. HyTer, Version 1.0. Wayne State University, Detroit, MI. Available at http://ozone2.chem.wayne.edu/hyther/hytherm1main.html [Online].
- 45.Alexander, S. 1977. Adsorption of chain molecules with a polar head—a scaling description. J. Phys. (Paris). 38:983–987. [Google Scholar]
- 46.Birshtein, T. M., and E. B. Zhulina. 1984. Conformations of star-branched macromolecules. Polym. 25:1453–1461. [Google Scholar]
- 47.DiMarzio, E. A. 1965. Proper accounting of conformations of a polymer near a surface. J. Chem. Phys. 42:2101–2106. [Google Scholar]
- 48.De Gennes, P. G. 1979. Scaling Concepts in Polymer Physics. Cornell University Press, Ithaca, NY.
- 49.Evans, D. F., and H. Wennerström. 1994. The Colloid Domain. VHC, New York, NY.
- 50.Birshtein, T. M., Yu. V. Liatskaya, and E. B. Zhulina. 1990. Theory of supermolecular structure of polydisperse block copolymers. 1. Planar layers of grafted chains. Polym. 31:2185–2196. [Google Scholar]
- 51.Moore, W. J. 1972. Physical Chemistry. Longman, London, UK.
- 52.Wetmur, J. G. 1991. DNA probes: applications of the principles of nucleic acid hybridization. Crit. Rev. Biochem. Mol. Biol. 26:227–259. [DOI] [PubMed] [Google Scholar]
- 53.Craig, M. E., D. M. Crothers, and P. Doty. 1971. Relaxation kinetics of dimer formation by self complementary oligonucleotides. J. Mol. Biol. 62:383–392. [DOI] [PubMed] [Google Scholar]
- 54.Pörschke, D., and M. Eigen. 1971. Co-operative non-enzymatic base recognition. III. Kinetics of the helix-coil transition of the oligoribouridylic·oligoriboadenylic acid system and of oligoriboadenylic acid alone at acidic pH. J. Mol. Biol. 62:361–364. [DOI] [PubMed] [Google Scholar]
- 55.Wittmer, J., A. Johner, J. F. Joanny, and K. Binder. 1994. Chain desorption from semidilute polymer brush: a Monte Carlo simulation. J. Chem. Phys. 101:4379–4390. [Google Scholar]
- 56.Blanch, H. W., and D. S. Clark. 1996. Biochemical Engineering. Marcel Dekker, New York, NY.
- 57.Titmuss, S., W. H. Briscoe, I. E. Dunlop, G. Sakellariou, N. Hadjichristidis, and J. Klein. 2004. Effect of end-group sticking energy on the properties of polymer brushes: comparing experiment and theory. J. Chem. Phys. 121:11408–11419. [DOI] [PubMed] [Google Scholar]
- 58.Fowler, R., and E. A. Gugenheim. 1960. Statistical Thermodynamics. Cambridge, UK.
- 59.Korolev, N., A. P. Lyubartsev, and L. Nordenskiöld. 1998. Application of polyelectrolyte theories for analysis of DNA melting in the presence of Na+ and Mg2+ Ions. Biophys. J. 75:3041–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buhot, A., and A. Halperin. 2004. Effects of stacking on the configurations and elasticity of single-stranded nucleic acids. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 70:020902. [DOI] [PubMed] [Google Scholar]
- 61.Duplantier, B. 1989. Statistical mechanics of polymer networks of any topology. J. Stat. Phys. 54:581–680. [Google Scholar]
- 62.Eisenriegler, E., K. Kremer, and K. Binder. 1982. adsorption of polymer chains at surfaces: scaling and Monte Carlo analyses. J. Chem. Phys. 77:6296–6320. [Google Scholar]