

Abstract
Osmolytes are small organic solutes accumulated at high concentrations by cells/tissues in response to osmotic stress. Osmolytes increase thermodynamic stability of folded proteins and provide protection against denaturing stresses. The mechanism of osmolyte compatibility and osmolyte-induced stability has, therefore, attracted considerable attention in recent years. However, to our knowledge, no quantitative study of osmolyte effects on the strength of hydrophobic interactions has been reported. Here, we present a detailed molecular dynamics simulation study of the effect of the osmolyte trimethylamine-N-oxide (TMAO) on hydrophobic phenomena at molecular and nanoscopic length scales. Specifically, we investigate the effects of TMAO on the thermodynamics of hydrophobic hydration and interactions of small solutes as well as on the folding-unfolding conformational equilibrium of a hydrophobic polymer in water. The major conclusion of our study is that TMAO has almost no effect either on the thermodynamics of hydration of small nonpolar solutes or on the hydrophobic interactions at the pair and many-body level. We propose that this neutrality of TMAO toward hydrophobic interactions—one of the primary driving forces in protein folding—is at least partially responsible for making TMAO a “compatible” osmolyte. That is, TMAO can be tolerated at high concentrations in organisms without affecting nonspecific hydrophobic effects. Our study implies that protein stabilization by TMAO occurs through other mechanisms, such as unfavorable water-mediated interaction of TMAO with the protein backbone, as suggested by recent experimental studies. We complement the above calculations with analysis of TMAO hydration and changes in water structure in the presence of TMAO molecules. TMAO is an amphiphilic molecule containing both hydrophobic and hydrophilic parts. The precise balance of the effects of hydrophobic and hydrophilic segments of the molecule appears to explain the virtual noneffect of TMAO on the strength of hydrophobic interactions.
INTRODUCTION
Folded structures of most proteins are sensitive to changes in environmental conditions such as temperature, pressure, moisture content, and the presence of salts and other solutes. Significant perturbations in thermodynamic conditions can cause changes in secondary and tertiary structure, leading to a partial or complete loss of their activity. Organisms are known to adapt to such perturbations in different ways, including evolutionary adaptations that endow stability/activity under extreme conditions (e.g., as in extremophiles) or through accumulation of small organic solutes called osmolytes. Specifically, the accumulation of osmolytes, such as trimethylamine-N-oxide (TMAO), is observed when cells or tissues are subjected to osmotic or water stress resulting from an exposure to high salinity, high hydrostatic pressures, and dessication or dehydration (1–7).
Osmolytes are typically accumulated in the intracellular environment at relatively high concentrations. At these concentrations, osmolytes increase thermodynamic stability of folded proteins without perturbing other cellular processes or biomolecular interactions. Simultaneously achieving both of these objectives restricts the physicochemical nature of the osmolyte molecules. Indeed, previous studies have shown that nature has converged on a few osmolytes (e.g., TMAO) that are common to a variety of organisms, including microorganisms (e.g., bacteria and fungi), plants, and animals (8).
With regards to osmolyte induced stability, perhaps the most clear argument has emerged from studies of Bolen et al. (1,9–11) which focus on the role of backbone solvation in protein stability. In particular, Bolen et al. have shown that the protein backbone is effectively osmophobic; and hiding the backbone into the core of folded proteins can provide significant stability in the presence of osmolytes. The osmophobic nature of the protein backbone results from differences in the hydration of backbone and osmolyte molecules, which depend on local water structure and interactions. A few studies in this direction have been reported (12,13). In addition to the ability of osmolytes to stabilize folded proteins, their otherwise nonperturbing nature, i.e., the property of being “compatible solutes”, is equally important, especially at high concentrations. However, the origin of osmolyte compatibility yet remains to be understood.
If unfavorable interaction of osmolytes with the protein backbone accounts for the increased protein stability, then, to be simultaneously compatible requires that osmolytes have negligible effects on other factors important in biomolecular stability and interactions. These other factors include hydrophobic interactions which are believed to play a major role in protein stability and interactions, molecular recognition, and micelle and membrane formation (14–17). Indeed, increasing or decreasing the strength of hydrophobic interactions could lead to nonspecific effects, such as aggregation, and will be detrimental to protein structure and function in vivo. To our knowledge, no prior study has reported a detailed quantitative analysis of TMAO effects, specifically on hydrophobic phenomena. Here we report results from detailed molecular dynamics simulations of hydrophobic hydration/interactions in aqueous TMAO solutions at the molecular as well as nanoscopic length scales. We focus on the effect of TMAO on vacuum-to-water transfer free energies and water-mediated hydrophobic interactions between molecular solutes. We also study the effect of TMAO on folding/unfolding free-energy landscape of a relatively large hydrophobic polymer. Major conclusion of our study is that TMAO has virtually no effect on the strength of hydrophobic interactions. We complement these thermodynamic studies by focusing on the structure of water in the hydration shell of TMAO molecules. Further, by systematically changing the partial charges on the TMAO molecules, we are able to pinpoint the origin of TMAO neutrality toward hydrophobic interactions, which likely explains the intracellular compatibility of TMAO at high concentrations.
METHODS
Molecular dynamics simulations of aqueous solutions of TMAO
We performed molecular dynamics (MD) simulations of aqueous solutions of varying TMAO concentrations (1.0, 2.0, and 3.0 mol/L) using GROMACS (18,19). Extended simple point charge (SPC/E) (20) model of water and the recently published force-field parameters for TMAO (21) were used in all the simulations. The number of each species used in the simulations are given in Table 1. Table 2 lists the Lennard Jones (LJ) parameters and the partial charges used in this study. Lorentz-Berthelot mixing rules were used to model LJ interactions between species of different types (22). Periodic boundary conditions were applied and the particle mesh Ewald method (23) was used to calculate the electrostatic interactions with a grid spacing of 0.1 nm. A time step of 2 fs was used and Berendsen's coupling algorithm was employed to maintain constant temperature (24) with the time constant for heat bath coupling set at 0.5 ps. The SETTLE algorithm (25) was used to constrain OH and HH distances in water with a geometric tolerance of 0.001 nm. All the systems were equilibrated at a pressure of 1 atm using Berendsen's pressure coupling algorithm (24) with a pressure relaxation time of 0.5 ps. Equilibration runs of 200 ps were followed by production runs of 3 ns in the NPT ensemble at 1 atm and 300 K. Configurations of molecules were saved every 0.3 ps for further analysis. At 1 atm, 1, 2, and 3 mol/L TMAO-water solutions have equilibrium densities of 1.021, 1.042, and 1.061 gm/cm3, respectively, which are in good agreement with experimental values (see also 21).
TABLE 1.
Number of species of each type used in MD simulations
| Hydrophobic solutes | Water | TMAO | [TMAO], mol/L |
|---|---|---|---|
| - | 550 | 0 | 0 |
| - | 550 | 18 | 1 |
| - | 550 | 38 | 2 |
| - | 550 | 60 | 3 |
| 10 (Me) | 550 | 0 | 0 |
| 10 (Me) | 550 | 18 | 1 |
| 10 (Me) | 550 | 38 | 2 |
| 10 (Me) | 550 | 60 | 3 |
| 1 (25-mer) | 2000 | 135 | 2 |
Me indicates methanes and 25-mer is the hydrophobic polymer. The last column gives an approximate concentration of TMAO in the solution.
TABLE 2.
Partial charges and Lennard-Jones interaction parameters (ε and σ) of the various atom types used in our MD simulations
| Atom type | q/e | σ, nm | ε, kJ/mol |
|---|---|---|---|
| O (SPC/E) | −0.8476 | 0.3165 | 0.6502 |
| H (SPC/E) | 0.4238 | 0.00 | 0.00 |
| N (TMAO) | 0.44 | 0.2926 | 0.8368 |
| O (TMAO) | −0.65 | 0.3266 | 0.6385 |
| C (TMAO) | −0.26 | 0.3041 | 0.2828 |
| H (TMAO) | 0.11 | 0.1775 | 0.0774 |
| Me | 0.00 | 0.371 | 1.234 |
| Monomer | 0.00 | 0.373 | 0.5856 |
“Monomer” in the table indicates the united atom methylene unit that makes up the hydrophobic polymer.
To monitor structural changes in water, we calculated various water-water (e.g., OO, OH, HH) radial distribution functions (rdf) as well as the tetrahedral orientational order parameter of water molecules (26–29) as a function of TMAO concentration. We also studied the hydration of TMAO molecules through calculations of various TMAO-water site-site rdfs.
Thermodynamics of hydrophobic hydration
We calculated the vacuum-to-solvent transfer free energies of LJ solutes in pure water and in TMAO solutions of varying concentrations using the test particle insertion method (30,31). A total of 27,000 test particle insertions were performed by placing a cubic grid in each configuration. The corresponding packing fractions or the point solute limit of Widom insertion probability are 0.66, 0.62, and 0.58 in 1, 2, and 3 mol/L TMAO solutions, respectively. The excess chemical potential for hydration of LJ solutes is given by
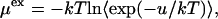 |
(1) |
where u is the solute-solvent energy of interaction (30,31), k is the Boltzmann constant, T is the temperature, and 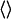 denotes ensemble average over the TMAO-water aqueous solution configurations. We used solute-solute ε = 1.234 kJ/mol, with the solute σ values ranging from 0.2 to 0.5 nm (in steps of 0.03 nm) to calculate the corresponding values of μex. Such LJ solutes have been used in the literature to represent spherical nonpolar/hydrophobic solutes (32,33).
denotes ensemble average over the TMAO-water aqueous solution configurations. We used solute-solute ε = 1.234 kJ/mol, with the solute σ values ranging from 0.2 to 0.5 nm (in steps of 0.03 nm) to calculate the corresponding values of μex. Such LJ solutes have been used in the literature to represent spherical nonpolar/hydrophobic solutes (32,33).
Thermodynamics of pair and many-body hydrophobic interactions
To characterize the effect of TMAO on hydrophobic interactions at the molecular and larger length scales, we studied two different systems as described below. Methodological details including water and TMAO interaction parameters, periodic boundary conditions, treatment of electrostatic interactions, temperature and pressure controls, and bond constraints in water are identical to those described above.
Small solute simulations
MD simulations of 10 methane molecules (Me) in aqueous solutions of varying TMAO concentrations (1.0, 2.0, and 3.0 mol/L) were performed. Methane molecules were represented as spherically symmetric united atom LJ solutes (32). Table 2 lists the corresponding LJ parameters used in these simulations. For all systems, equilibration runs of one nanosecond were followed by production runs of ∼20 ns in the NPT ensemble at 300 K and 1 atm. Configurations of methane molecules were saved every 0.2 picoseconds for further analysis. Methane-methane pair correlation functions, g(r), and the potentials of mean force (PMFs), W(r) = −kTlng(r), were calculated in pure water and in aqueous solutions of TMAO. The PMF, W(r), is the reversible work done in bringing a methane molecule from infinity to a given distance r from a methane at origin. Me-Me PMFs provide a quantitative measure of the strength of hydrophobic interactions at the pair level.
Simulations of a hydrophobic polymer
A recent study on the collapse of a hydrophobic polymer in a coarse-grained model of water (34) highlighted the relevance of large-scale hydrophobicity to realistic self-assembly processes. Motivated by that study our group recently studied thermodynamics of folding-unfolding transitions of a hydrophobic polymer and salt effects on polymer conformational equilibria in solution (35). The details of parameter development and thermodynamics of folding of this polymer are given elsewhere (35). The polymer is a chain of 25 united atom hydrophobic monomers (Table 1) connected by harmonic bond length and angle potentials. The monomers of this polymer interact with each other and with water and TMAO sites through LJ interactions. The interactions with the first and the second nearest neighbors along the chain-making bonds [i, (i + 1)] and bond angles [i, (i + 2)] with a given monomer were excluded. Harmonic potentials were used for monomer-monomer bond stretching [Vb = 0.5k(r − r0)2, where k = 334720.0 kJ mol−1 nm−2 and r0 = 0.153 nm] and bond angle stretching [Vθ = 0.5kθ(θ − θ0)2, where kθ = 462.0 kJ mol−1 deg−2 and θ0 = 111.0 deg]. Intrapolymer torsion potential was turned off in simulations of this polymer.
MD simulations of the polymer were performed in pure water and in 2 mol/L aqueous solution of TMAO. The simulations were carried out in the NPT ensemble at a pressure of 1 atm and a temperature of 300 K. To obtain efficient sampling of the phase space, umbrella sampling technique (36–38) was employed with a restraining potential Wumbrella = ku(Rg − Rg0)2 applied to the radius of gyration of the polymer. Here ku is the umbrella constant in kJ mol−1 nm−2, Rg is the radius of gyration in nm, and Rg0 is the reference radius of gyration in nm. ku was chosen independently in each Rg window to obtain efficient sampling (35).
Polymer conformational equilibria were monitored by calculating probability distribution of the radius of gyration, p(Rg), of the polymer. The low Rg conformations of the polymer are compact or folded whereas the large Rg conformers are extended. We calculated the PMF along the Rg coordinate, W(Rg) = −kTlnp(Rg), in pure water and in 2 mol/L TMAO solution.
RESULTS AND DISCUSSION
Effect of TMAO on thermodynamics of hydration and interaction of hydrophobic solutes
Fig. 1 shows excess chemical potentials (or free energies of hydration) of different nonpolar solutes in water and in aqueous solutions of TMAO. In all solutions, the chemical potential increases with increasing solute size. The free energy of hydration of nonpolar solutes in aqueous solution contains two contributions: i), reversible work required to form a cavity of the size and shape of the solute; and ii), free energy arising from solute-solvent attractive interactions. It is the first contribution that typically dominates nonpolar solute hydration thermodynamics (33,39,40). With increasing solute size, the work required to form a cavity in solvent increases leading to the monotonic increase of chemical potential with solute size shown in Fig. 1.
FIGURE 1.

Excess chemical potential, μex, of LJ solutes in kT units as a function of solute size σss/2 with increasing TMAO concentration of the solution (0, 1, 2, and 3M). Calculations of μex for hard-sphere solutes in these solutions are shown in the inset (rHS is the hard sphere radius).
The addition of an additive to water can change the excess chemical potential of hydration of a nonpolar solute. For example, salts typically increase the chemical potential leading to salting out (i.e., decrease in solubility) of nonpolar solutes from water. Interestingly, Fig. 1 shows that addition of TMAO has negligible effect on the chemical potential of hydrophobic solutes in water. Similar calculations for purely repulsive (hard-sphere) solutes (inset of Fig. 1) indicate that addition of TMAO (at 3 mol/L concentration) increases the chemical potential of those solutes by 10–15%. This suggests that van der Waals interactions between TMAO and nonpolar hydrophobic (LJ) solutes compensate for that increase leading to the almost negligible effect on the thermodynamics of hydrophobic hydration observed in Fig. 1.
Fig. 2 shows the free energy of interaction of methanes (i.e., the pair potentials of mean force) obtained from MD simulations of methanes in aqueous solutions of varying TMAO concentration. The characteristic features of these profiles, their physical origin, and significance has been discussed in detail previously (41–43). We observe a primary minimum at 0.39 nm corresponding to the direct contact of methanes in water (contact minimum) and a secondary minimum at 0.73 nm corresponding to configurations of methane molecules separated by a water molecule (solvent-separated minimum). The two minima are separated by a desolvation barrier located at 0.57 nm. Any thermodynamic perturbation such as temperature or pressure changes (41,42) or addition of additives (35) are expected to change the relative stability of the contact and solvent-separated minima as well as the height of the desolvation barrier. For example, addition of salts (such as NaCl) leads to a significant relative stabilization of contact configurations, characterizing the strengthening of hydrophobic interactions at the pair level (35,44,45). Interestingly, PMF profiles in Fig. 2 indicate that TMAO has a negligible effect on the relative thermodynamic stability of contact and solvent-separated conformations compared to that in pure water. Any change (if present) is within the error bars of our calculations. This observation is consistent with the negligible effect of TMAO on the thermodynamics of vacuum-to-solvent transfer of LJ solutes reported above.
FIGURE 2.

Methane-methane potentials of mean force, W(r)/kT = − lng(r), in pure water (0M TMAO) and in aqueous TMAO solutions of concentrations 1, 2, and 3M. PMFs show that TMAO has negligible effect on the free energy of interaction of methanes in water.
Effect of TMAO on the conformational equilibria of a hydrophobic polymer
Many recent studies have stressed the importance of hydrophobic interactions at the nanoscopic (1 nm or larger) level in realistic self-assembly processes (34,46). To make connections with many-body hydrophobic interactions present in macromolecular collapse and folding, we recently studied thermodynamics of conformational equilibria of a model hydrophobic polymer in water and in salt solutions (35). This model polymer comprises a chain of 25 monomers, is 36 Å long in its extended state, and buries over 200 Å2 area in its compact states compared to extended states. The monomers interact with water and TMAO sites with LJ interactions (see Methods) and intrapolymer potentials include harmonic bond length and angle potentials, but no torsion potentials. In water, this model polymer displays a two-state folding-unfolding behavior with the free-energy minimum corresponding to the compact folded states separated by a barrier from the minimum corresponding to the unfolded states (see Fig. 3).
FIGURE 3.

Conformational free-energy profile of a 25-mer hydrophobic polymer in pure water and 2 M TMAO solution along the Rg coordinate. We reference the PMF to zero for Rg = 0.805, W(Rg = 0.805) = 0.0.
Our calculations of the thermodynamics of conformational equilibria in water and in salt solutions show that addition of salt significantly increases the thermodynamic stability of folded states relative to extended states. In contrast, addition of TMAO to water has a negligible effect on the thermodynamics of conformational equilibria of the hydrophobic polymer. The free energy along the Rg coordinate remains essentially unchanged up on addition of TMAO. This observation is again consistent with the negligible effect of TMAO on the thermodynamics of hydrophobic hydration and interactions at the pair PMF level as well as with its negligible effects on the vapor-liquid surface tension of aqueous TMAO solutions (47).
Analysis of TMAO effects using preferential interactions
Preferential interactions analysis used extensively by Timasheff and co-workers (7,48,49,50) provides an alternative approach to probe molecular origins of the observed neutrality of TMAO toward hydrophobic effects. We recently performed such an analysis using configurations obtained from MD simulations to understand the salt-induced strengthening of hydrophobic interactions (35). In general, if an additive is depleted from the vicinity of a macromolecule, it increases the chemical potential of that macromolecule. For example, depletion of salt ions in the vicinity of hydrophobic polymer increases its chemical potential and stabilizes the compact states relative to the extended ones. The extent of additive depletion or enhancement in the vicinity of a macromolecule is quantified by the preferential interaction coefficient Γ defined as (35,51,52,53,54),
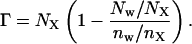 |
(2) |
The region surrounding the macromolecule is divided into vicinal and bulk domains by placing a boundary between the two (35,51,52). Nw, NX represent the number of water and additive molecules in the vicinal region and nW and nX, represent water and additive molecules in the bulk region, respectively. A negative numerical value of Γ indicates depletion of additive from the vicinal region, whereas a positive value represents preferential enhancement or binding of the additive to the macromolecule. Γ values in different conformational states a and b can be used to calculate the change in free-energy difference between those two states upon addition of the solute (35,52) as
 |
(3) |
We calculated the number of water and TMAO molecules within a cutoff distance, rcut, of the polymer atoms, which were used to obtain the Γ values. Fig. 4 shows Γ profiles for the hydrophobic polymer with two different Rg values which represent compact and extended conformations of the polymer. The Γ values are plotted as a function of cutoff distance that separates the vicinal region of the polymer from the bulk. Γ values are small at low values of rcut and attain their asymptotic values at a distance of ∼1 nm. The maximum cutoff distance is restricted to nanometer length scales due to the limited system size and the use of periodic boundary conditions, leading to a somewhat poor convergence. Fig. 4 shows that Γ values for both compact and extended states are positive but small in magnitude compared to those observed for salt solutions (35), indicating a small enhancement in the concentration of TMAO molecules in the vicinity of both folded and unfolded states of the polymer. More importantly, differences between Γ values beyond a reasonable value of rcut are even smaller, comparable to the error bars of our calculations. This relatively small difference in the Γ profiles of TMAO near folded and unfolded conformations of the polymer leads to the negligible relative free-energy difference between them (according to Eq. 3), again consistent with our observations of the neutrality of TMAO toward hydrophobic interactions.
FIGURE 4.

Preferential interaction coefficient, Γ, of TMAO near a hydrophobic polymer in water. The Γ profile is plotted as a function of the cutoff distance from polymer sites. Γ profiles are shown for two conformational ensembles of the polymer: compact (Rg = 0.42 ± 0.04 nm) and extended (Rg = 0.9 ± 0.04 nm).
The lack of significant strengthening or weakening of the pair and many-body hydrophobic interactions in high-concentration TMAO solutions is, indeed, remarkable. TMAO is accumulated in cells of certain organisms at rather high concentrations (2,8). Significant strengthening of hydrophobic interactions could lead to deleterious effects such as unwanted nonspecific aggregation of partially unfolded or misfolded proteins. Instead, at high concentrations, TMAO can be compatible with cellular machinery through its neutrality toward hydrophobic interactions and impart high stability through the so-called osmophobic interactions with the protein backbone (1,9–11). What makes TMAO neutral toward hydrophobic interactions? Molecular mechanisms based on water structural changes have been invoked historically to address this and similar questions. Below we investigate systematically the effect of TMAO on water structure with specific focus on the packing and orientations of water molecules in the vicinity of TMAO molecules. A successful molecular theory that relates water structural changes to thermodynamic changes is not currently available (55). In addition, our results below indicate that different measures of “water structure” can display opposite trends in presence of additives, making water structure based perspective less useful.
Effect of TMAO on water structure
Fig. 5 shows water oxygen-oxygen (OW-OW) and oxygen-hydrogen (OW-HW) site-site radial distribution functions (rdfs) in pure water and in solutions of increasing TMAO concentration. Both profiles show behavior typical of water-water correlation functions (20). Namely, the locations of the first and second peaks in OW-OW rdf at 0.28 and 0.45 nm characterize the hydrogen bonded first neighbor and the tetrahedrally located second neighbor distances, respectively. With increasing TMAO concentration, the height of the first peak in both rdfs increases monotonically, consistent with the observation of Zou et al. (12). Increase in the peak heights of water-water rdfs could be interpreted as “an enhancement of water structure” induced by TMAO. However, we note that the rdfs are normalized by the bulk water number density in a given system, which decreases with increasing TMAO concentration, and can lead to an enhancement in the rdf peak heights which is partly artificial. Further, it is difficult to quantify the orientational order of water from site-site rdfs alone. To this end, we calculated probability distribution of the orientational order parameter q (26–29) for water molecules in solutions of increasing TMAO concentration. Four nearest neighbor water molecules of a water molecule k are identified and six angles, θikj, subtended by neighbors i and j about k are calculated to obtain q using
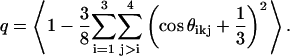 |
(4) |
By definition, 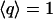 for ideal tetrahedral arrangement of water molecules (such as in ice Ih) and
for ideal tetrahedral arrangement of water molecules (such as in ice Ih) and 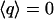 for orientationally uncorrelated ideal gas like configurations (27,29).
for orientationally uncorrelated ideal gas like configurations (27,29).
FIGURE 5.

Water oxygen-oxygen (a) and oxygen-hydrogen (b) rdfs obtained from MD simulations of 0, 1, 2, and 3M aqueous TMAO solutions. The inset in a focuses on the first peak of the oxygen-oxygen rdf.
Fig. 6 a shows the probability distribution, p(q), of the orientational order parameter q as a function of TMAO concentration. p(q) is bimodal with peaks at q ≈ 0.8 and q ≈ 0.5, indicating somewhat distinct populations of water molecules with high as well as low tetrahedral order (29). With increasing TMAO concentration, population of orientationally less ordered water molecules (q <0.5) increases monotonically at the expense of orientationally more ordered water molecules. The probability distribution of angle θikj, p(θikj), also shows a similar trend (see Fig. 6 b). The height of the peak in θikj near the ideal tetrahedral angle of 109.5° decreases with the addition of TMAO, whereas the population of θikj values lower than ∼105° increases monotonically. Interestingly, probability of orientations corresponding to θikj ≈ 105 or q ≈ 0.55 appears to be unaffected by the addition of TMAO. Thus, quantification of structure using site-site rdfs indicates enhanced structuring, whereas calculations of orientational order parameter leads to the opposite conclusion. Together, these calculations highlight the limitations of arguments that relate “enhanced” or “decreased” water structure to macromolecular thermodynamics.
FIGURE 6.

Probability distributions of the orientational order parameter q (a) and angle θikj (b) for water molecules in pure water and in 0, 1, 2 and 3M aqueous TMAO solutions.
With respect to the observed neutrality of TMAO toward hydrophobic interactions, details of the hydration of a TMAO molecule are more instructive (see Fig. 7). A TMAO molecule comprises three methyl groups, each with a total partial charge of +0.07e, an oxygen atom with a partial charge of −0.65e, and a nitrogen atom with a partial charge of +0.44e making the molecule electrically neutral. The central nitrogen atom is buried away from the vicinal water molecules and has little direct effect on the surrounding water structure, which is determined primarily by the methyl groups and the oxygen atom of TMAO.
FIGURE 7.

Radial distribution functions of water oxygen and hydrogen atoms with TMAO oxygen (a) and carbon (b), obtained from MD simulations of 1M aqueous TMAO solution. A schematic based on the snapshot from MD simulation shows a typical surface parallel orientation of water near methyl groups and a negative ion like hydration of the TMAO oxygen atom (c).
Fig. 7 shows rdfs that characterize the hydration of methyl groups and of oxygen atom of a TMAO molecule. Although, the spherical averaging inherent in the rdf calculation leads to the loss of some local structural information, several interesting features can be noted. The hydration of methyl groups displays the well-known characteristics of hydrophobic hydration; specifically, the peaks of oxygen and hydrogen densities are at similar locations, indicating orientations of vicinal water molecules parallel to the TMAO surface. A tail of hydrogen densities is also seen for r < 0.3 nm, which has been observed previously in simulations of methane, neopentane, and tetramethyl ammonium ions in water (56–58). Inspection of snapshots from MD simulations also shows surface-parallel orientations of water molecules near methyl group of TMAO (see Fig. 7 c) consistent with the rdfs.
In contrast, near the oxygen atom, there is a significant orientational polarization of vicinal water molecules. The distance between the first peaks of hydrogen and oxygen rdfs is exactly 0.1 nm, equal to the OH bond length, and indicates orientation of the vicinal water molecules similar to that near a negative ion or near a water oxygen in bulk water. On average, three water molecules are observed within a distance of 0.35 nm of the TMAO oxygen that donate hydrogen bonds (see Fig. 7 a). Thus from water structure perspective, a TMAO molecule appears like a short (almost spherical) amphiphile comprising spatially distinct hydrophobic and hydrophilic regions. Such local hydration patterns are critical in determining if water-mediated interaction between TMAO and other solutes (hydrophobic, polar, or ionic) will be favorable or unfavorable (as in TMAO interactions with protein backbone).
Understanding the neutrality of TMAO toward hydrophobic interactions
The above results show that TMAO has negligible effect on a variety of hydrophobic phenomena—vacuum-to-water transfer, methane-methane pair PMFs, and folding-unfolding of a hydrophobic polymer. In contrast, most other additive solutes or cosolvents such as salts or alcohols have a stabilizing or destabilizing effect on hydrophobic phenomena. The neutrality of TMAO likely arises from its specific chemistry and the consequent hydration patterns. These factors can be systematically varied in molecular simulations by changing the parameters of force field describing the interactions of TMAO with other molecules in the system. To this end, we performed simulations of folding-unfolding of the hydrophobic polymer in aqueous solutions of TMAO analogs. We generated the TMAO analogs by systematically scaling the partial charge on each TMAO atom by a factor λ, such that q(λ) = λ × q(λ = 1) for λ = 0, 0.5, 1.0, and 1.5. The λ = 1 state corresponds exactly to the TMAO molecule studied above. TMAO is electrically neutral for all values of λ. Varying partial charge in this manner thus helps sampling the range of chemistries from purely hydrophobic to hydrophilic/amphiphilic.
Fig. 8 shows the folding-unfolding free-energy profile for the hydrophobic polymer in 2 mol/L aqueous solutions of TMAO analogs. For λ = 0, the TMAO-analog is completely hydrophobic, binds the unfolded states of the polymer strongly, and unfolds the polymer. Consequently, the free-energy profile shows a single broad minimum in extended states near Rg ≈ 0.73 nm. Increasing the numerical value of partial charges reduces that preferential binding and stabilizes the compact states slightly for λ = 0.5. For λ = 1.0, that is, for TMAO solutes, interestingly, the free-energy profile is identical to that in pure water, indicating the precise balance of hydrophobic/hydrophilic parts of the molecule in terms of their effect on hydrophobic interactions. Increasing the value of λ further does not significantly change the free-energy profile except for inducing somewhat increased stabilization of very compact states with low Rg values. Preliminary calculations indicate that the neutrality of TMAO toward hydrophobic interactions is observed over a relatively broad range of λ values near λ = 1. That is, precise choice of λ = 1 may not be necessary for the neutrality toward hydrophobic interactions to be observed, thus making our observations less sensitive of the precise choice of partial charges on TMAO atoms in our force field. These calculations highlight the role of additive molecule chemistry and specifically its hydrophobic/hydrophilic nature in influencing the water-mediated interactions between hydrophobic solutes.
FIGURE 8.

Free-energy profile of a hydrophobic polymer,  in 2 M aqueous solutions of different TMAO analogs. The partial charge on each TMAO atom is scaled by λ. For all curves W(Rg = 0.805) = 0.0.
in 2 M aqueous solutions of different TMAO analogs. The partial charge on each TMAO atom is scaled by λ. For all curves W(Rg = 0.805) = 0.0.
CONCLUSIONS
Osmolytes are small organic molecules that are accumulated at high concentration in cells/tissues of certain organisms when subjected to osmotic or water stress, high salinity, hydrostatic pressures, or dessication or dehydration stresses (8). Two properties of osmolyte molecules are particularly important—their ability to impart increased thermodynamic stability to folded proteins and their compatibility in the intracellular environment at high concentrations.
To understand the compatibility, we focused on the effects of a well-known osmolyte, TMAO, on a variety of hydrophobic phenomena in aqueous solutions using molecular dynamics simulations. Our calculations clearly show that TMAO has a negligible effect on the thermodynamics of hydrophobic effects, including vacuum-to-water transfer free energies, methane-methane pair potentials of mean force, and on the folding-unfolding free energy of a hydrophobic polymer. At the molecular level, the neutrality of TMAO toward hydrophobic interactions is manifested in the lack of strong preferential binding or depletion of TMAO in the vicinity of hydrophobic solutes. The neutrality of TMAO is also consistent with the negligible dependence of water liquid-vapor surface tension on TMAO concentration in the aqueous solution (47). Significant strengthening of hydrophobic interactions could lead to deleterious effects such as unwanted nonspecific aggregation of partially unfolded or misfolded proteins. Instead, at high concentrations TMAO can be compatible with cellular machinery through its neutrality toward hydrophobic interactions.
Fundamentally understanding what makes TMAO neutral toward hydrophobic interactions is a complex problem. The hydration patterns of TMAO indicate an amphiphilic character of the TMAO molecular surface comprising hydrophilic oxygen atom and hydrophobic methyl groups. To test whether an approximate balance of the effect of these two opposite chemistries on the strength of hydrophobic interactions makes TMAO neutral toward hydrophobic interactions, we performed simulations of TMAO analogs. We generated the TMAO analogs by systematically scaling the partial charges of TMAO molecule. These calculations indicate that the amphiphilic character of TMAO is likely responsible for its neutrality toward hydrophobic interactions.
The neutrality of TMAO toward hydrophobic effects, however, does not explain the increased stability of proteins in TMAO solutions. Important insights in this direction have come from the work of Bolen et al. (1,9–11) which has emphasized the role of protein backbone, especially its “osmophobic” nature, in determining thermodynamic stability of proteins in osmolyte solutions. Specifically, Bolen et al. (1,9) have shown that the water-mediated interaction between protein backbone and TMAO molecules is unfavorable making the unfolded states of the protein less stable relative to folded states in TMAO solutions. Designing small molecules that have unfavorable interactions with the protein backbone appears to be an excellent strategy toward protein stabilization. Each protein has a backbone, no matter what the composition of hydrophobic, polar, and ionic side chains; therefore, stabilization will likely be universal. In addition, the neutrality toward hydrophobic interactions confers “compatibility” to osmolytes, which can be accumulated at high concentrations. Whether these characteristics of osmolyte molecules are universal remains to be seen. Molecular simulations of osmolyte systems that include protein backbone like chemistries will help provide insights into the osmophobic effect and the concomitant effects on the stability of proteins (59).
Acknowledgments
We thank Drs. Amrit Kalra, Tuhin Ghosh, and Hank Ashbaugh for a critical reading of and comments on the manuscript.
S.G. acknowledges financial support of the National Science Foundation-CAREER (CTS-0134023) grant. J.S.D. and S.G. also acknowledge partial financial support of the grant National Institutes of Health-GM66712.
References
- 1.Wang, A., and D. W. Bolen. 1997. A naturally occurring protective system in urea-rich cells: mechanism of osmolyte protection of proteins against urea denaturation. Biochemistry. 36:9101–9108. [DOI] [PubMed] [Google Scholar]
- 2.Yancey, P. H., M. E. Clark, S. C. Hand, R. D. Bowlus, and G. N. Somero. 1982. Living with water stress: evolution of osmolyte systems. Science. 217:1214–1222. [DOI] [PubMed] [Google Scholar]
- 3.Lin, T.-Y., and S. N. Timasheff. 1994. Why do some organisms use a urea-methylamine mixture as osmolyte? Thermodynamic compensation of urea and trimethylamine n-oxide interactions with protein. Biochemistry. 33:12695–12701. [DOI] [PubMed] [Google Scholar]
- 4.Gillett, M. B., J. R. Suko, F. O. Santoso, and P. H. Yancey. 1997. Elevated levels of trimethylamine oxide in muscles of deep-sea gadiform teleosts: A high-pressure adaptation. J. Exp. Zool. 279:386–391. [Google Scholar]
- 5.Yancey, P. H. 2001. Water stress, osmolytes and proteins. Am. Zool. 41:669–709. [Google Scholar]
- 6.Yancey, P. H., W. R. Blake, and J. Conley. 2002. Unusual organic osmolytes in deep-sea animals: adaptations to hydrostatic pressure and other perturbants. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 133:667–676. [DOI] [PubMed] [Google Scholar]
- 7.Timasheff, S. N. 2003. Preferential interactions of urea with lysozyme and their linkage to protein denaturation. Biophys. Chem. 105:421–448. [DOI] [PubMed] [Google Scholar]
- 8.Somero, G. N., C. B. Osmond, and C. L. Bolis. 2001. Water and Life. Springer-Verlag, New York.
- 9.Liu, Y., and D. W. Bolen. 1995. The peptide backbone plays a dominant role in protein stabilization by naturally occurring osmolytes. Biochemistry. 34:12884–12891. [DOI] [PubMed] [Google Scholar]
- 10.Baskakov, I., and D. W. Bolen. 1998. Forcing thermodynamically unfolded proteins to fold. J. Biol. Chem. 273:4831–4834. [DOI] [PubMed] [Google Scholar]
- 11.Bolen, D. W., and I. V. Baskakov. 2001. The osmophobic effect: natural selection of a thermodynamics force in protein folding. J. Mol. Biol. 310:955–963. [DOI] [PubMed] [Google Scholar]
- 12.Zou, Q., B. J. Bennion, V. Daggett, and K. P. Murphy. 2002. The molecular mechanism of stabilization of proteins by TMAO and its ability to counteract the effects of urea. J. Am. Chem. Soc. 124:1192–1202. [DOI] [PubMed] [Google Scholar]
- 13.Bennion, B. J., and V. Daggett. 2004. Counteraction of urea-induced protein denaturation by trimethylamine n-oxide: a chemical chaperone at atomic resolution. Proc. Natl. Acad. Sci. USA. 101:6433–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dill, K. A. 1990. Dominant forces in protein folding. Biochemistry. 29:7133–7135. [DOI] [PubMed] [Google Scholar]
- 15.Tanford, C. 1973. The Hydrophobic Effect: Formation of Micelles and Biological Membranes. John Wiley, New York.
- 16.Kauzmann, W. 1959. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 14:1–63. [DOI] [PubMed] [Google Scholar]
- 17.Pratt, L. R., and A. Pohorille. 2002. Hydrophobic effects and modeling of biophysical aqueous solution interfaces. Chem. Rev. 102:2671–2692. [DOI] [PubMed] [Google Scholar]
- 18.Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. Gromacs: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91:43–56. [Google Scholar]
- 19.Lindahl, E., B. Hess, and D. van der Spoel. 2001. Gromacs 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7:306–317. [Google Scholar]
- 20.Berendsen, H. J. C., J. R. Grigera, and T. P. Straatsma. 1987. The missing term in effective pair potentials. J. Phys. Chem. 91:6269–6271. [Google Scholar]
- 21.Kast, K. M., J. Brickmann, S. M. Kast, and R. S. Berry. 2003. Binary phases of aliphatic n-oxides and water: Force field development and molecular dynamics simulation. J. Phys. Chem. A. 107:5342–5351. [Google Scholar]
- 22.Allen, M. P., and D. J. Tildesley. 1987. Computer Simulation of Liquids. Clarendon Press, Oxford.
- 23.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: An n log(n) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 24.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 25.Miyamoto, S., and P. A. Kollman. 1992. Settle: an analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 13:952–962. [Google Scholar]
- 26.Chau, P. L., and A. J. Hardwick. 1998. A new order parameter for tetrahedral configurations. Mol. Phys. 92:511–518. [Google Scholar]
- 27.Truskett, T. M., S. Torquato, and P. G. Debenedetti. 2000. Towards a quantification of disorder in materials: distinguishing equilibrium and glassy sphere packings. Phys. Rev. E. 62:993–1001. [DOI] [PubMed] [Google Scholar]
- 28.Torquato, S., T. M. Truskett, and P. G. Debenedetti. 2000. Is random close packing of spheres well defined? Phys. Rev. Lett. 84:2064–2067. [DOI] [PubMed] [Google Scholar]
- 29.Errington, J. R., and P. G. Debenedetti. 2001. Relationship between structural order and the anomalies of liquid water. Nature. 409:318–321. [DOI] [PubMed] [Google Scholar]
- 30.Widom, B. 1963. Some topics in the theory of fluids. J. Chem. Phys. 39:2808–2812. [Google Scholar]
- 31.Widom, B. 1982. Potential distribution theory and statistical mechanics of fluids. J. Phys. Chem. 86:869–872. [Google Scholar]
- 32.Verlet, L., and J. J. Weis. 1972. Perturbation-theory for thermodynamic properties of simple liquids. Mol. Phys. 24:1013–1024. [Google Scholar]
- 33.Garde, S., A. E. Garcia, L. R. Pratt, and G. Hummer. 1999. Temperature dependence of the solubility of nonpolar gases in water. Biophys. Chem. 78:21–32. [DOI] [PubMed] [Google Scholar]
- 34.ten Wolde, P. R., and D. Chandler. 2002. Drying-induced hydrophobic polymer collapse. Proc. Natl. Acad. Sci. USA. 99:6539–6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghosh, T., A. Kalra, and S. Garde. 2005. On the salt-induced stabilization of pair and many-body hydrophobic interactions. J. Phys. Chem. B. 109:642–651. [DOI] [PubMed] [Google Scholar]
- 36.Beutler, T. C., and W. F. V. Gunsteren. 1994. The computation of a potential of mean force: choice of the biasing potential in the umbrella sampling technique. J. Chem. Phys. 100:1492–1497. [Google Scholar]
- 37.Ferrenberg, A. M., and R. H. Swendsen. 1989. Optimized Monte Carlo data analysis. Phys. Rev. Lett. 63:1195–1198. [DOI] [PubMed] [Google Scholar]
- 38.Frenkel, D., and B. Smit. 2001. Understanding Molecular Simulation. Academic Press, New York.
- 39.Pratt, L. R., and D. Chandler. 1977. Theory of the hydrophobic effect. J. Chem. Phys. 67:3683–3704. [Google Scholar]
- 40.Pratt, L. R., and D. Chandler. 1980. Effects of solute-solvent attractive forces on hydrophobic correlations. J. Chem. Phys. 73:3434–3441. [Google Scholar]
- 41.Ghosh, T., A. E. Garcia, and S. Garde. 2002. Enthalpy and entropy contributions to the pressure dependence of hydrophobic interactions. J. Chem. Phys. 116:2480–2486. [Google Scholar]
- 42.Ghosh, T., A. E. Garcia, and S. Garde. 2001. Molecular dynamics simulations of pressure effects on hydrophobic interactions. J. Am. Chem. Soc. 123:10997–11003. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh, T., A. E. Garcia, and S. Garde. 2003. Water-mediated three-particle hydrophobic interactions between hydrophobic solutes: size, pressure, and salt dependences. J. Phys. Chem. B. 107:612–617. [Google Scholar]
- 44.Kalra, A., N. Tugcu, S. M. Cramer, and S. Garde. 2001. Salting-in and salting-out of hydrophobic solutes in aqueous salt solutions. J. Phys. Chem. B. 105:6380–6386. [Google Scholar]
- 45.Smith, P. E. 1999. Computer simulation of cosolvent effects on hydrophobic hydration. J. Phys. Chem. B. 103:525–534. [Google Scholar]
- 46.Hummer, G., and S. Garde. 1998. Cavity expulsion and weak dewetting of hydrophobic solutes in water. Phys. Rev. Lett. 80:4193–4196. [Google Scholar]
- 47.Kita, Y., T. Arakawa, T.-Y. Lin, and S. N. Timasheff. 1994. Contribution of the surface free energy perturbation to protein-solvent interactions. Biochemistry. 33:15178–15189. [DOI] [PubMed] [Google Scholar]
- 48.Arakawa, T., and S. N. Timasheff. 1985. The stabilization of proteins by osmolytes. Biophys. J. 47:411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arakawa, T., and S. N. Timasheff. 1982. Stabilization of protein structure by sugars. Biochemistry. 21:6536–6544. [DOI] [PubMed] [Google Scholar]
- 50.Arakawa, T., and S. N. Timasheff. 1984. Mechanism of protein salting in and salting out by divalent cation salts: balance between hydration and salt binding. Biochemistry. 25:5912–5923. [DOI] [PubMed] [Google Scholar]
- 51.Parsegian, V. A., R. P. Rand, and D. C. Rau. 2000. Osmotic stress, crowding, preferential hydration, and binding: a comparison of perspectives. Proc. Natl. Acad. Sci. USA. 97:3987–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baynes, B. M., and B. L. Trout. 2003. Proteins in mixed solvents: a molecular-level perspective. J. Phys. Chem. B. 107:14058–14067. [Google Scholar]
- 53.Shimizu, S. 2004. Estimating hydration changes upon biomolecular reactions from osmotic stress, high pressure, and preferential hydration experiments. Proc. Natl. Acad. Sci. USA. 101:1195–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schellman, J. A. 1978. Solvent denaturation. Biopolymers. 17:1305–1322. [DOI] [PubMed] [Google Scholar]
- 55.Ashbaugh, H. S., D. Asthagiri, L. R. Pratt, and S. B. Rempe. 2003. Hydration of krypton and consideration of clathrate models of hydrophobic effects from the perspective of quasi-chemical theory. Biophys. Chem. 105:323–338. [DOI] [PubMed] [Google Scholar]
- 56.Garde, S., G. Hummer, and M. E. Paulaitis. 1996. Hydrophobic interactions: conformational equilibria and the association of nonpolar molecules in water. Faraday Discuss. 103:125–139. [Google Scholar]
- 57.Garde, S., G. Hummer, and M. E. Paulaitis. 1998. Free energy of hydration of a molecular ionic solute: tetramethylammonium ion. J. Chem. Phys. 108:1552–1561. [Google Scholar]
- 58.Rajamani, S., T. Ghosh, and S. Garde. 2004. Size dependent ion hydration, its asymmetry, and convergence to macroscopic behavior. J. Chem. Phys. 120:4457–4466. [DOI] [PubMed] [Google Scholar]
- 59.Bennion, B. J., M. L. DeMarco, and V. Daggett. 2004. Preventing misfolding of the prion protein by trimethylamine n-oxide. Biochemistry. 43:12955–12963. [DOI] [PubMed] [Google Scholar]