

Abstract
The increasing prevalence of tuberculosis in many areas of the world, associated with the rise in drug-resistant Mycobacterium tuberculosis (MTB) strains, presents a major threat to global health. InhA, the enoyl-ACP reductase from MTB, catalyzes the nicotinamide adenine dinucleotide (NADH)-dependent reduction of long-chain trans-2-enoyl-ACP fatty acids, an intermediate in mycolic acid biosynthesis. Mutations in the structural gene for InhA are associated with isoniazid resistance in vivo due to a reduced affinity for NADH, suggesting that the mechanism of drug resistance may be related to specific interactions between enzyme and cofactor within the NADH binding site. To compare the molecular events underlying ligand affinity in the wild-type, I21V, and I16T mutant enzymes and to identify the molecular aspects related to resistance, molecular dynamics simulations of fully solvated NADH-InhA (wild-type and mutants) were performed. Although very flexible, in the wild-type InhA-NADH complex, the NADH molecule keeps its extended conformation firmly bound to the enzyme's binding site. In the mutant complexes, the NADH pyrophosphate moiety undergoes considerable conformational changes, reducing its interactions with its binding site and probably indicating the initial phase of ligand expulsion from the cavity. This study should contribute to our understanding of specific molecular mechanisms of drug resistance, which is central to the design of more potent antimycobacterial agents for controlling tuberculosis.
INTRODUCTION
Despite more than five decades of effective chemotherapy, tuberculosis (TB) caused by Mycobacterium tuberculosis (MTB) still remains an important problem of public health. The World Health Organization estimates that there were 8.8 million new cases of TB in 2002. The global incidence rate of TB was growing at ∼1.1% per year and the number of cases at 2.4% per year (1).
Infection with drug-sensitive strains of MTB can be effectively treated with a combination of drugs including isoniazid (INH), rifampicin, pyrazinamide, and ethambutol or streptomycin (2–5). However, the emergence of multidrug-resistant strains of MTB, defined as resistant to at least INH and rifampicin (the two most powerful anti-TB drugs), makes it urgent to identify the mechanisms of drug action and resistance whose understanding should contribute to the design of more potent antimycobacterial agents for controlling drug-sensitive and drug-resistant TB.
INH has been the cornerstone in TB chemotherapy since it was first found to exhibit a powerful bactericidal activity against MTB in 1952 (6,7). This drug causes the loss of acid-fastness of the tubercle bacilli as a result of the inhibition of mycolic acid synthesis, α-alkyl β-hydroxy long-chain fatty acids (60–90 carbons in length), important components of the mycobacterial cell wall (8,9).
Using a genetic approach, Jacobs and co-workers identified the protein product of the mycobacterial inhA gene as the putative target for INH inhibition (10). The InhA enzyme, or trans-2-enoyl-ACP-reductase (EC 1.3.1.9), carries out the stereospecific reduction of α,β-unsaturated fatty acids bound to the acyl carrier protein in an NADH-dependent reaction (11) and is a member of the type II dissociated fatty acid biosynthesis pathway (FAS II) in MTB.
INH itself does not inhibit the enzyme directly but requires conversion to an activated form of the drug by a mycobacterial catalase-peroxidase (KatG) (12–15) and becomes covalently attached to the nicotinamide ring of the NADH bound within the active site of the enzyme (16).
Studies of clinical isolates from MTB resistant to INH were found to have mutations in the inhA structural gene, leading to an increase in dissociation constant for NADH cofactor binding to the purified enzyme (17). Consistent with these results, x-ray crystallographic studies (16,18,19) have revealed that all mutations in the enoyl reductase conferring INH resistance were located in the nucleotide binding site. This observation suggested that the mechanism of drug resistance may be related to specific interactions between enzyme and cofactor within the NADH binding site.
Although INH resistance has also been attributed to another FAS II enzyme (KasA) (20,21), thermal inactivation of InhA in Mycobacterium smegmatis resulted in identical phenotypic response to that treated with INH (22), therefore validating InhA as the primary target of INH and as an important target for drug discovery.
InhA is a member of the short chain dehydrogenase/reductase (SDR) family of enzymes (23,24), with a polypeptide backbone Rossmann fold topology, in which a parallel β-sheet is flanked by α-helices forming the central core of the NADH binding site.
Although dinucleotide-binding shows very low overall sequence homology, their protein backbones superimpose fairly well (25,26) and have some common sequence features, one of which is a glycine-rich phosphate binding loop that connects the C-terminus of β1 with the N-terminus of α1 (23,26,27).
In a computational comparison of 102 high-resolution Rossmann fold enzymes, Tanner and co-workers (28) identified that even though the majority of the enzymes display the pyrophosphate binding loop sequence having three Gly, the two enoyl-ACP-reductases analyzed (from Brassica napus and Escherichia coli) have a G-X7-G pattern. In InhA, the last Gly is substituted by an Ala, but it has the same NADH mode of binding described elsewhere (28–30).
As mutations in the glycine-rich loop have been correlated with attenuation or elimination of enzyme activity (31–33) and two mutations in this loop (I21V and I16T) are related to INH resistance and decrease of cofactor affinity (wild-type InhA: Km NADH = 56 ± 4 μM, Kd NADH = 0,57 ± 0,04 μM; I21V InhA: Km NADH = 104 ± 7 μM, Kd NADH = 13,9 ± 1,7 μM; I16T InhA: Km NADH = 149 ± 10 μM, Kd NADH = 5,95 ± 0,33 μM) (17), in this work we report the molecular dynamics (MD) simulations of wild-type InhA and isoniazid-resistant clinical isolates I21V and I16T mutants in complex with the NADH molecule to identify the molecular aspects of NADH affinity differences and predict the events related to INH resistance.
MATERIALS AND METHODS
MD simulations
MD simulations were performed with the SANDER module of AMBER 6.0 (34) using the Cornell et al. all-atom model force field (35), except for the atomic charges in the NADH molecule which were assigned by ab initio calculations in the HF 6-31G* level (36) and fitted with the RESP procedure (37).
The initial structure for the wild-type InhA-NADH complex was taken from the 2,2 Å crystal structure 1ENY (18) without the crystallographic water molecules. The NADH pyrophosphate moiety was considered deprotonated at physiological pH. The all-atom model of the protein in complex with the cofactor contains 4079 atoms and a net molecular charge of −6. Hence, six sodium counterions were added near surface Asp residues (5 Å apart) to neutralize the negative charge density of the complex, which was then immersed in a rectangular box containing a total of 9132 TIP3P (38) water molecules. The initial simulation cell dimensions were 77.5 Å × 64.3 Å × 70.0 Å and had the complex solvated by a layer of water molecules of at least 10 Å in all directions, as previously described (39). The simulation cell contained a total of 31,481 atoms.
In the MD protocol, all hydrogen atoms, ions, and water molecules were first subjected to 100 steps of energy minimization by steepest descent to remove close van der Waals contacts. The system was then relaxed with sequential steps of energy minimization (100 steps of steepest descent plus 400 steps of conjugate gradient) where the solute atoms were gradually allowed to move until no restraints were applied. The temperature of the system was then increased from 10 K to 298,16 K in 6 steps (10 K to 50 K, 50 K to 100 K, 100 K to 150 K, 150 K to 200 K, 200 K to 250 K, and 250 K to 298,16 K), and the velocities at each step were reassigned according to the Maxwell-Boltzmann distribution at that temperature and equilibrated for 2 ps.
Energy minimization and MD were carried out under periodic boundary conditions. The simulation was computed in the NPT ensemble at 298.16 K with the Berendsen temperature coupling and constant pressure of 1 atm with isotropic molecule-based scaling (40). The SHAKE algorithm (41), with a 10−5 Å tolerance, was applied to fix all bonds containing a hydrogen atom, allowing the use of a time step of 2.0 fs in the integration of the equations of motion. No extra restraints were applied after the equilibration phase. The electrostatic interactions between nonligand atoms were evaluated by the particle-mesh Ewald method with a charge grid spacing of ∼1.0 Å, and the charge grid was interpolated on a cubic grid with the direct sum tolerance set to 4.0 × 10−6. The Lennard-Jones interactions were evaluated using a 9.0 Å atom-based cutoff (42). Data were collected for 3.1 ns, and structures for analysis were saved every 0.5 ps. The initial system preparation and results analysis were performed on a 2.4 GHz Pentium IV PC, and the MD simulations were performed on a Pentium III PC cluster with 16 single nodes of 1GHz and 256 Mb memory each.
The I21V and I16T mutants were constructed by in silico mutations of the 1.9 ns instantaneous wild-type InhA-NADH conformation, in a way that simulates a spontaneous mutation. As the velocities were taken from the wild-type InhA-NADH simulation, the mutants' MD proceeded with no new thermalization, thus simulating what we called on-the-fly mutation.
The convergence of the different simulations were analyzed in terms of the energy components, secondary structure, root mean-square deviation (RMSD) from the initial x-ray structure (1ENY), and root mean-square-fluctuation (RMSF) to estimate the B-factor. For the B-factor calculation, the RMSFs were calculated relative to the last 2 ns averaged backbone structures, and all coordinate frames from the trajectories were first superimposed on the initial conformation to remove any effect of overall translation and rotation. Atomic isotropic B-factors were calculated from the trajectories using the equation
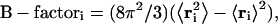 |
where  is the mean-square positional fluctuation of atom i (43,44).
is the mean-square positional fluctuation of atom i (43,44).
Hydrogen bonds analysis
Because the MD simulations include hydrogen atoms, it was possible to make an explicit analysis of hydrogen bond properties, such as donor-acceptor assignments and hydrogen bond occupancies.
For the occupancies calculations, the NADH hydrogen bonding interactions were assumed to be present if the participating heavy atoms were <3.9 Å apart and the angle formed between the heavy atoms and donated hydrogen was larger than 120° and was performed with the carnal module of AMBER 6.0. The strongest hydrogen bonds have the highest occupancies, the smallest standard deviations, and the shortest heavy atoms separations. For this reason, we assumed an occupancy cutoff of 50% for the most important H-bonds.
The total number of direct H-bonds between the cofactor and the protein, or those mediated by water molecules, were calculated with the HBPLUS program and default parameters (distance D-A = 3.9 Å and minimum angle = 90°) (45).
Dictionary of secondary structure of proteins (46) and PROCHECK-NMR (47) were used to evaluate the secondary structure pattern during the simulations.
Figures were generated with the SwissPDB Viewer v3.7 b2 program (48). Graphics, averages, and standard deviations were calculated with Origin 6.1 Scientific Graphing and Analysis Software (Microcal Software, OriginLab, Northampton, MA).
RESULTS AND DISCUSSION
Protein structural features
To obtain an estimate of the MD trajectory quality and convergence, the backbone RMSD from the starting crystal structure (1ENY) were calculated (Fig. 1). After a rapid increase during the first 250 ps, the protein backbone RMSD average and standard deviation over the last 2 ns of the wild-type InhA-NADH trajectory was 1.6 ± 0.1 Å. In the I21V InhA-NADH and I16T InhA-NADH complexes, mutations of the 1.9 ns instantaneous wild-type structure caused a perturbation in these systems, and the simulation had to be prolonged for another 6.5 and 5.5 ns for I21V and I16T, respectively. The I21V InhA-NADH system was most affected by mutation, and only 4.0 ns after the I21V mutation, the backbone RMSD reached a plateau around 2.1 ± 0.1 Å during the last 2 ns. The instability caused by mutation was not so evident in the I16T mutant, but its backbone RMSD average over the last 2 ns was 2.0 ± 0.1 Å.
FIGURE 1.

Backbone RMSD of wild-type InhA, I21V, and I16T mutants from the starting crystal structure (1ENY). The on-the-fly mutation moment is identified by a vertical line at 1.9 ns.
The total number of protein hydrogen bonds (TNHB) was also monitored along the trajectories and compared with the crystal structure as a way to access the overall stabilization of the structures. Dictionary of Secondary Structure of Proteins (45) analysis of the last 2 ns snapshots of each enzyme showed that there were no significant differences among the systems (TNHBwild-type = 178 ± 5, TNHBI21V = 174 ± 5, TNHBI16T = 175 ± 5). This lower number of hydrogen bonds related to the crystal structure (TNHB1ENY = 203) was expected as, in solution, some residue-residue interactions can be substituted by residue-water interactions.
Because the overall Rossmann fold is important for the cofactor binding and InhA activity, the last eight averaged structures over a 250 ps window of each trajectory were also examined in terms of their secondary structure with PROCHECK-NMR (47) to characterize conformational changes during the MD simulations (data not shown). In all systems, the secondary structures were well maintained, especially the β-sheet core (Fig. 2). In the I21V and I16T mutants, the helical organization of the N-terminal positions of both α7 were lost, being more noticeable for the I21V mutant whose α6 was also less stable.
FIGURE 2.
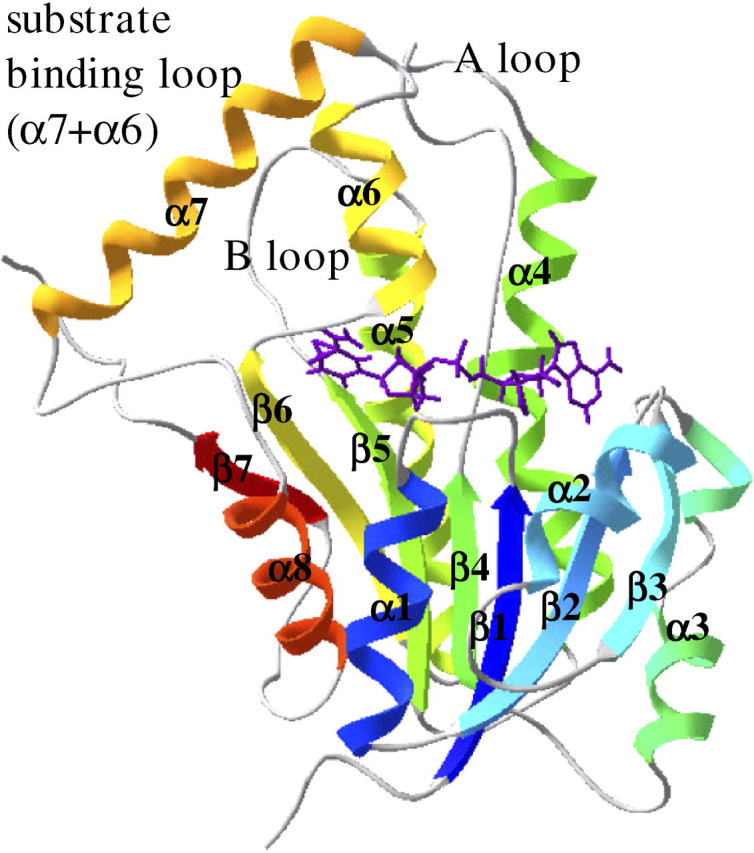
Secondary structure identification of the wild-type InhA initial structure colored according to secondary structure succession, from blue (N-terminal) to red (C-terminal). The NADH molecule is in an extended conformation over the Rossmann fold β-sheet core.
As indicated by the B-factors calculated over the last 2 ns time window (Fig. 3), the most flexible regions correspond to the substrate binding loop (α6 and α7 helices, residues 196–219), the A loop (residues 100–114), and the B loop (residues 150–161). The large mobility of these loops is consistent with the necessity of this region to undergo conformational changes upon fatty acyl substrate binding (19). Even though the substrate binding loop also has higher B-factors in the crystal structure, they are much lower than observed for the dynamic structures, in which crystal packing is not present. The A and B loops, which were involved in subunit contacts in the crystallographic tetrameric structure, were also shown to have higher B-factors. The same is true for residues 80–90 from the loop between α3 and β4. Yet, the mobility of the core residues remain fairly close to those derived from the crystallographic data.
FIGURE 3.

Backbone residue-based B-factors calculated over the last 2 ns time window for wild-type (blue), I21V (green), and I16T (red) InhA enzymes. Crystallographic B-factors (black).
As the substrate binding loop and A and B loops movements are involved in cofactor and substrate entrance and exit, the distance between α6 helix and loop A and α7 helix and loop B were monitored during the trajectories. From this analysis it was observed that although all α6-A loop distances fluctuate near or below the crystal distance (except for a 250 ps period for the wild-type InhA), the α7-B loop distance on the I21V (13.5 ± 0.4 Å) and I16T (13.2 ± 0.3 Å) mutants were above that observed for the crystal structure (12.6 Å), probably induced by the instability introduced by these mutations.
NADH association
Even though the overall Rossmann fold structure was well maintained during the MD trajectories, the NADH association within each binding site was different.
The NADH binding mode within the InhA enzyme is similar to that of the other SDR enzymes with a Rossmann fold structure (23,26,49–52). The NADH molecule binds to the enzyme in an extended conformation along the top of the C-termini of the core β-sheet, with the nicotinamide ring pointing deep into a hydrophobic cavity formed by the strands β4, β5, and β6 and by the helixes α5, α6, and α7 (Fig. 2) (18).
As mutations in the InhA enzyme are associated to an increase in the NADH dissociation constant (17), of particular interest are the hydrogen bonds between the NADH molecule (Fig. 4) and the enzyme since they monitor position and movements of the ligand within the binding site. These H-bonds were monitored during all MD simulations, and Table 1 summarizes their occupancies during the last 2 ns.
FIGURE 4.

Atom names and dihedral angles for the NADH molecule.
TABLE 1.
Hydrogen bond contacts between the NADH ligand and InhA enzymes with >50% occupancies during the production phase
| Occupancy* (%)
|
|||||
|---|---|---|---|---|---|
| Residue | Amino acid group | NADH atom† | Wild-type InhA | I21V | I16T |
| Ile15 | C=O | O′A3 | 70 | — | 54 |
| N | O′A3 | 72 | — | — | |
| Ser20 | OH | OPA2 | 100 | — | — |
| Ile21 | N | OPN2 | 95 | — | — |
| Asp64 | COO− | N6A | 99 | — | 100 |
| Val65 | N | N1A | 100 | — | 92 |
| Gln66 | OE1 | N6A | — | — | 64 |
| Ile95 | C=O | O′N3 | 100 | 100 | 100 |
| Gly96 | N | O′A4 | 100 | 59 | 100 |
| Tyr158 | OH | O7N | — | — | 98 |
| Lys165 | NZ | O′N2 | 83 | 86 | 65 |
| NZ | O′N3 | 99 | 86 | 63 | |
| Ile194 | C=O | N7N | 95 | 84 | 86 |
| N | O7N | 100 | 100 | 100 | |
| Thr196 | OG1 | N7N | 67 | — | — |
| OG1 | OPN1 | 61 | — | — | |
Occupancies were calculated with the CARNAL module from AMBER 6.0.
Atom names from the NADH molecule in Fig. 4.
For the wild-type enzyme, 10 residues make conserved hydrogen bonds with the cofactor molecule (occupancies ≥50%). In the nicotinamide cavity, residues Ile194 and Thr196 interact with the 3-carboxamide group of the nicotinamide ring (N ring) fixing the B-side of the ring (Fig. 5) and thus the H4S hydrogen, facing the substrate cavity, consistent with the proposed InhA mechanism (11,49).
FIGURE 5.

Stereo view of the NADH association in its binding pocket, including the glycine-rich loop, in the production phase average structure of the wild-type InhA simulation. NADH is blue, and hydrogen bonds are represented as dashed green lines. NADH atoms and amino acid groups involved in the interactions are listed in Table 1.
Residues Lys165 and Ile95 interact with the nicotimamide ribose (NR). Initially, the role of the conserved Lys165 residue was attributed to stabilization of the intermediate enolate formed during substrate reduction (11). During the wild-type InhA-NADH dynamics, the Lys165 side-chain hydrogen bonds to the RN hydroxy groups lasted almost the whole trajectory, reinforcing the site-directed mutagenesis studies that indicated that this residue has a primary role in cofactor binding (53).
Next to the Ile95 residue, the Gly96 H-bond to the oxygen atom of the adenine ribose (AR) was maintained for the whole trajectory. The adenine moiety of the NADH cofactor molecule also makes important hydrogen bonds with Asp64 and Val65 located in the adenine ring (A ring) recognition site (54).
In the wild-type enzyme, the glycine-rich loop residues Ser20 and Ile21 are involved in the pyrophosphate moiety interactions and are responsible for keeping this flexible cofactor region bound to the enzyme, whereas the Ile15 residue hydrogen bonds to the AR hydroxy group next to the pyrophosphate moiety (Fig. 5).
Conformational changes caused by the substitution of an apolar residue for a less bulky one (I21V mutation) or for a slightly polar but also less bulky one (I16T mutant) result in a different pattern for NADH interaction.
In the I21V mutant simulation, only four (Ile95, Gly96, Lys165, and Ile194) of the 10 residues that make important bonds with the cofactor molecule are maintained with occurrence ≥50%. Of these residues, only Gly96 is involved in H-bonding with the adenine portion. The other three interact with the nicotinamide portion, and only Ile95 and Ile194 H-bond to O'N3 and O7N, respectively, keeping 100% occurrence.
In the I21V mutant, all direct interactions between the protein and the pyrophosphate moiety became less important during the trajectory. The same is true for those residues that form the adenine recognition region. The occurrence of H-bonds with residues Asp64 and Val65 changed from 99% and 100% to 21% and 46%, respectively.
In the I16T mutant simulation, there were no such changes in the H-bonds pattern between the NADH molecule and the enzyme binding site. From the 10 residues that originally make H-bonds with occurrence ≥50%, 7 are present in this mutant. In the adenine recognition moiety, Asp64, Val65, and now Gln66 are H-bonded to the A ring nitrogens.
Interactions with residues Ile95, Gly96, and Ile194 remained within the same level as in the wild-type enzyme, but both NR hydroxy groups make weaker H-bonds with Lys165 residues.
Because of the NADH conformational changes, the Thr196 H-bonds to the 3-carboxy group from the N ring were substituted by the Tyr158 residue. Originally, the Tyr158 OH group makes an indirect H-bond to this cofactor region through a conserved water molecule that is lost with the I16T mutation. In this mutant, all original interactions between the glycine-rich loop and the pyrophosphate moiety also became less important during the trajectory, and there are no strong H-bonds between the protein and this cofactor region. From the glycine-rich loop, only Ile15 makes a 54% occupancy H-bond with the other hydroxy group from AR (O'A3).
From the MD simulations, it became clear that mutation in the glycine-rich loop residues I21V and I16T change the pattern of direct H-bonds contacts with the pyrophosphate moiety from the NADH molecule. Being less attached to its binding site, this flexible cofactor region undergoes conformational changes, becoming more distant from the glycine-rich loop (Fig. 6).
FIGURE 6.
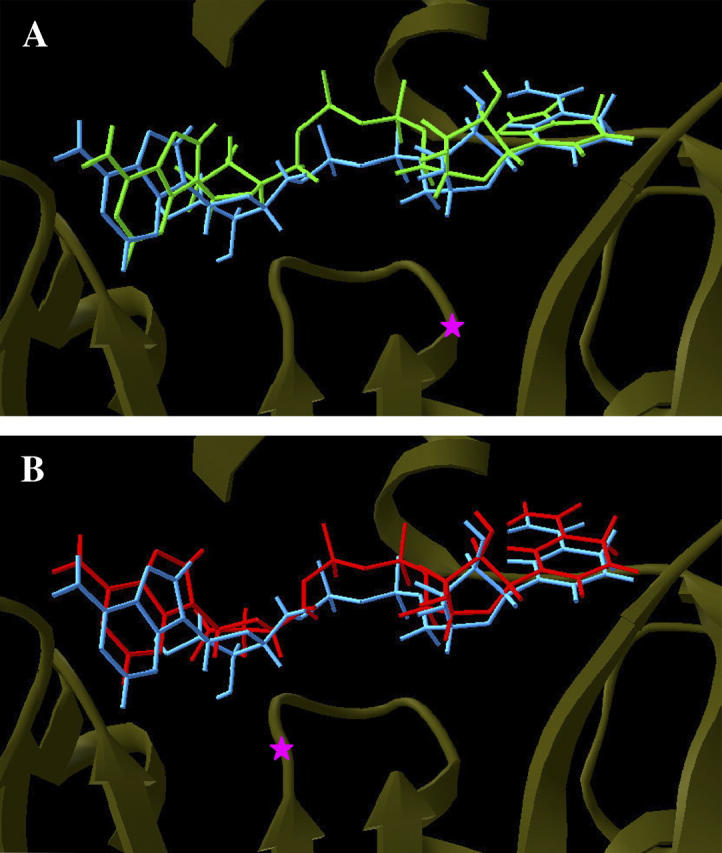
Superposition of the NADH production phase average structures from wild-type (blue) and (A) I21V InhA-NADH (green) complex and (B) I16T InhA-NADH complex (red). Magenta stars indicate the locations of the mutated residues.
The NADH dihedral angles were monitored during the trajectories and showed that while attached to the wild-type enzyme, the cofactor molecule was flexible but its dihedrals fluctuated around values observed in the crystal structures. In the mutant enzymes, NADH undergoes considerable conformational changes in the pyrophosphate moiety, being more prominent for αA and γA dihedrals for both mutants (Fig. 7). The transitions in αA and γA dihedrals, separated by βA, are highly correlated and characteristic of a “crankshaft motion”. This movement is similar to that observed for the phosphodiester torsions in MD trajectories of DNA (55).
FIGURE 7.
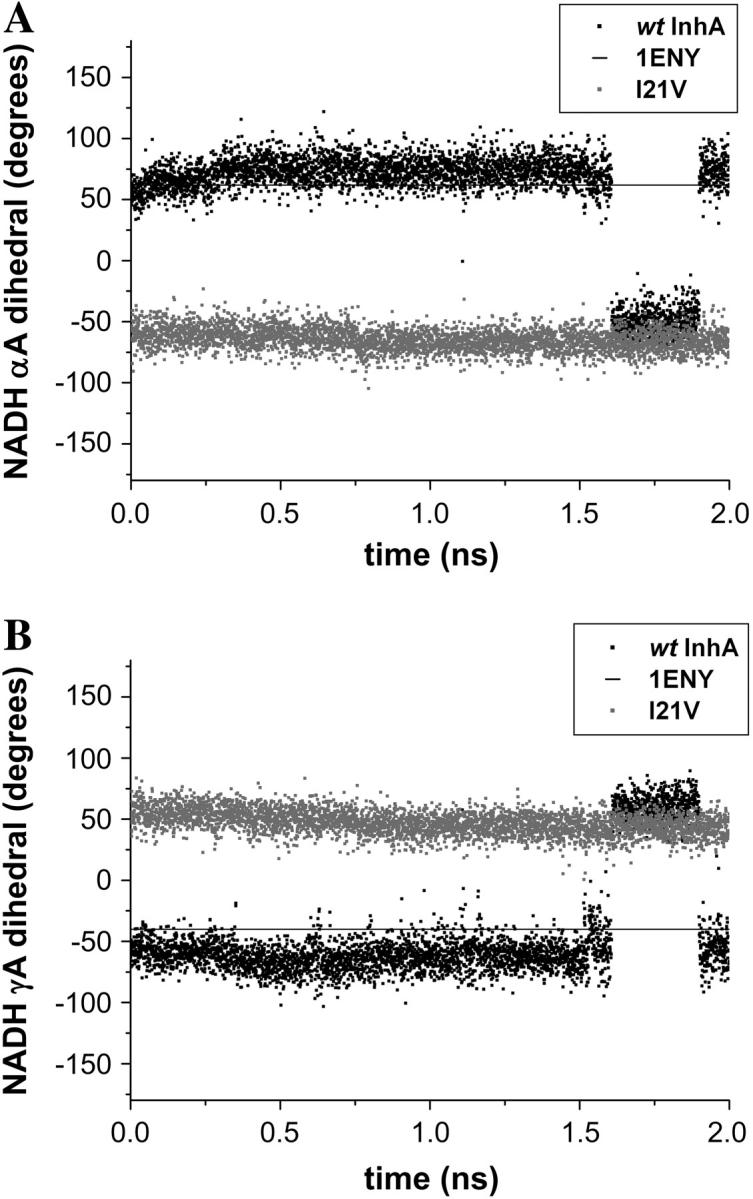
Comparison of the NADH dihedral angles (A) αA and (B) γA for the last 2 ns of the wild-type (black) and I21V InhA-NADH (shaded) trajectories. The horizontal line represents the crystallographic values from 1ENY.
The conformational changes experienced by the cofactor molecule and the opening movement of the B loop region allowed a greater interaction with water molecules. As a consequence, in the mutant enzymes the number of water molecules in the NADH first solvation shell is greater than in the wild-type system (Table 2).
TABLE 2.
Number of water molecules in the first solvation shell (3.5 Å) calculated for the last 2 ns of the wild-type, I21V, and I16T InhA-NADH trajectories using the program ptraj-6.4 from AMBER6.0
| NADH moiety | Wild-type InhA-NADH | I21V-NADH | I16T-NADH |
|---|---|---|---|
| N ring | 1.9 ± 0.3 | 3.3 ± 1.1 | 2.9 ± 1.0 |
| N ribose | 1.7 ± 0.6 | 1.5 ± 0.6 | 2.0 ± 0.7 |
| Pyrophosphate | 8.1 ± 0.8 | 11.2 ± 1.0 | 11.9 ± 1.0 |
| A ribose | 3.6 ± 0.9 | 4.0 ± 1.0 | 4.0 ± 1.2 |
| A ring | 4.4 ± 1.0 | 4.8 ± 1.1 | 4.1 ± 1.1 |
The larger opening between the substrate-binding loop and the B loop in the mutant enzyme as compared to the wild-type enzyme allows more water molecules to enter the nicotinamide binding site. This site is a hydrophobic cavity and in the wild-type enzyme, only 1.9 ± 0.3 water molecules are within a 3.5 Å distance from this cofactor region. In the mutant enzymes, the average number of water molecules became 3.3 ± 1.1 and 2.0 ± 1.0 for I21V and I16T mutant, respectively.
Another significant difference among wild-type and mutant enzymes was in the pyrophosphate solvation shell. This moiety of cofactor moves apart from its binding site, becoming more hydrated, and the average number of water molecules surrounding it changes from 8.1 ± 0.8 to 11.2 ± 1.0 and 11.9 ± 1.0 for the I21V and I16T mutants, respectively.
Because the AR and ring are close to the cofactor binding crevice entrance and accessible to bulk water even in the wild-type enzyme, no differences in this region's solvation shells were observed among the enzymes.
With a larger number of water molecules surrounding the NADH within the mutant binding sites, some direct interactions between cofactor and protein amino acids are substituted by these water molecules. This fact can be observed in the H-bond analysis between the cofactor and the enzymes' amino acids performed with the HBPLUS program (56) for the last 2 ns of each trajectory (Fig. 8).
FIGURE 8.

H-bond analysis of wild-type, I21V, and I16T InhA-NADH, in the production phase, using the HBPLUS program: (A) Total number of H-bonds between the NADH molecule and amino acids from the enzyme binding sites (crystallographic reference in black). (B) Total number of H-bonds with water molecules that mediate cofactor interactions with the enzyme binding sites (average number of water mediated H-bonds in wild-type-InhA system in black).
In the wild-type system, the number of direct H-bonds between the NADH molecule and the enzyme binding site are almost constant, and fluctuations around the average of 11.6 ± 1.3 are slightly higher than in 1ENY crystal structure (8 NADH-enzyme H-bonds). In the mutated enzymes, the large conformational changes make the total number of direct H-bonds decrease. Only in this analysis, the average number was calculated over the last 500 ps of each trajectory where the I21V and I16T mutants make only 5.6 ± 1.1 and 10.5 ± 1.1 direct H-bonds, respectively.
It is known that water molecules are important for cofactor binding within Rossmann fold proteins (28). In the InhA crystal structures (16,18,19), there are at least seven conserved water molecules within the binding site. H-bond analysis between cofactor and bridging water molecules revealed that there is a good number of high occupancy H-bond interactions with these molecules (data not shown), but analyses of the number of H-bonds between the NADH molecule and water molecules that mediate the interaction with the enzymes are different among the systems. Whereas in the wild-type complex the cofactor molecules make an average of 12.0 ± 1.6 H-bonds with water molecules that mediate interactions with the enzyme amino acids, in the I21V and I16T mutants, only 5.5 ± 0.7 and 5.3 ± 1.1 H-bonds are formed, respectively, between these molecules.
Based on this analysis of wild-type InhA-NADH, I21V InhA-NADH, and I16T InhA-NADH complex trajectories, the increase in the NADH dissociation constant in the I21V and I16T mutant enzymes observed in experimental studies (17) can be attributed to the decrease in the number of direct H-bond interactions between the cofactor molecule and amino acids in the binding pocket and between the cofactor molecule and water molecules that mediate interactions with the enzyme. These structural variations, which lead to different affinities for NADH by wild-type and mutant enzymes, were also quantitatively attested by molecular docking experiments with the AutoDock 3.0 program (57). The NADH cofactor was flexibly docked to the average structures of each system using a genetic algorithm protocol (57). As a result we obtained estimates of the NADH free energy of binding (ΔGbind). The docking experiments showed that wild-type InhA has a higher affinity for NADH than the mutant enzymes (wild-type InhA ΔGbind = −15,8 ± 1,2 kcal/mol, I21V InhA ΔGbind = −14,7 ± 1,0 kcal/mol, I16T InhA ΔGbind = −10,3 ± 1,4 kcal/mol), indeed corroborating the structural observations.
CONCLUSIONS
In this work, we reported fully solvated, all-atom MD simulations of the wild-type InhA enzyme and two INH-resistant clinical mutants (I21V and I16T) associated with the cofactor NADH molecule to investigate the molecular events related to drug cofactor affinities that could lead to drug resistance. It was noticed from the MD simulations that these mutations cause macromolecular instabilities that were reflected in the long time necessary for trajectory convergence.
The mutations caused a different pattern of cofactor interaction within the enzyme binding site. Whereas in the wild-type enzyme the NADH remains tightly bounded to its pocket, in an extended conformation in the I21V and I16T it undergoes considerable conformational changes. Mutations in the glycine-rich loop affect cofactor pyrophosphate moiety binding and allow its movement apart from its binding site.
Since in aqueous solution the NADH molecule preferentially adopts a more folded conformation with an interring distance between 5 and 10 Å (58,59; E. Schroeder and O. Norberto de Souza, unpublished data) and has to unfold to bind to the enzyme in an extended conformation, it is likely that the conformational changes experienced by the pyrophosphate moiety inside the mutant enzymes could be indicative of an initial phase of NADH folding and expulsion from its binding cavity as a result of the reduced affinity for the mutants' binding pocket.
As InhA inhibition by INH depends on the covalent INH-NAD adduct and mutations in the glycine-rich loop affects the NADH mode of binding within these enzymes, we have predicted that the lower affinity for the pyrophosphate moiety should contribute to INH resistance.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We are grateful to Dr. Ulf Ryde for providing the NADH parameters, and Dr. César A. F. De Rose and the CPAD/PUCRS team for the Ombrófila cluster administration.
This project was supported by grants from FINEP and Millennium Institute, CNPq/MCT to D.S.S. and FAPERGS to O.N.S. D.S.S. and L.A.B. are CNPq research fellow awardees. E.K.S. was supported by a PhD scholarship from CAPES.
References
- 1.World Health Organization Ninth Annual Report on Tuberculosis. 2004. Global tuberculosis control: surveillance, planning, financing. http://www.who.int/tb/publications/2004/en/index1.html.
- 2.Mitchison, D. A. 1985. The action of antitubercular drugs in short-course chemotherapy. Tubercle. 66:219–225. [DOI] [PubMed] [Google Scholar]
- 3.Stratton, M. A., and M. T. Reed. 1986. Short-course drug therapy for tuberculosis. Clin. Pharm. 5:977–987. [PubMed] [Google Scholar]
- 4.Combs, D. L., R. J. O'Brien, and L. J. Geiter. 1990. USPHS tuberculosis short-course chemotherapy trial-21— effectiveness, toxicity, and acceptability—the report of final results. Ann. Intern. Med. 122:397–406. [DOI] [PubMed] [Google Scholar]
- 5.Schroeder, E. K., O. Norberto de Souza, D. S. Santos, J. S. Blanchard, and L. A. Basso. 2002. Drugs that inhibit mycolic acid biosynthesis in Mycobacterium tuberculosis. Curr. Pharm. Biotechnol. 3:197–225. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein, J., W. A. Lott, B. A. Steinberg, and H. L. Yale. 1952. Chemotherapy of experimental tuberculosis—V. Isonicotinic acid hydrazide (nydrazid) and related compounds. Am. Rev. Tuberc. 65:357–364. [DOI] [PubMed] [Google Scholar]
- 7.Fox, H. H. 1952. The chemical approach to the control of tuberculosis. Science. 116:129–134. [DOI] [PubMed] [Google Scholar]
- 8.Takayama, K., L. Wang, and H. L. David. 1972. Effect of isoniazid on the in vivo mycolic acid biosynthesis, cell growth and viability of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winder, F. G., and P. B. Collins. 1970. Inhibition by isoniazid of synthesis of mycolic acids in Mycobacterium tuberculosis. J. Gen. Microbiol. 63:41–48. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee, A., E. Dubnau, A. Quémard, V. Balasubramanian, K. S. Um, T. Wilson, D. Collins, G. de Lisle, and W. R. Jacobs Jr. 1994. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 263:227–230. [DOI] [PubMed] [Google Scholar]
- 11.Quémard, A., J. C. Sacchettini, A. Dessen, C. Vilchèze, R. Bittman, W. R. Jacobs Jr., and J. S. Blanchard. 1995. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry. 34:8235–8241. [DOI] [PubMed] [Google Scholar]
- 12.Johnsson, K., and P. G. Schultz. 1994. Mechanistic studies of the oxidation of isoniazid by the catalase peroxidase from Mycobacterium tuberculosis. J. Am. Chem. Soc. 116:7425–7426. [Google Scholar]
- 13.Johnsson, K., D. S. King, and P. G. Schultz. 1995. Studies on the mechanism of action of isoniazid and ethionamide in the chemotherapy of tuberculosis. J. Am. Chem. Soc. 117:5009–5010. [Google Scholar]
- 14.Basso, L. A., R. Zheng, and J. S. Blanchard. 1996. Kinetics of inactivation of WILD-TYPE and C243S mutant of Mycobacterium tuberculosis: enoyl reductase by activated isoniazid. J. Am. Chem. Soc. 118:11301–11302. [Google Scholar]
- 15.Quémard, A., A. Dessen, M. Sugantino, W. R. Jacobs Jr., J. C. Sacchettini, and J. S. Blanchard. 1996. Binding of catalase-peroxidase-activated isoniazid to wild-type and mutant Mycobacterium tuberculosis enoyl-ACP reductases. J. Am. Chem. Soc. 118:1561–1562. [Google Scholar]
- 16.Rozwarski, D. A., G. A. Grant, D. H. R. Barton, W. R. Jacobs Jr., and J. C. Sacchettini. 1998. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science. 279:98–102. [DOI] [PubMed] [Google Scholar]
- 17.Basso, L. A., R. Zheng, J. M. Musser, W. R. Jacobs Jr., and J. S. Blanchard. 1998. Mechanisms of isoniazid resistance in Mycobacterium tuberculosis: enzymatic characterization of enoyl reductase mutants identified in isoniazid-resistant clinical isolates. J. Infect. Dis. 178:769–775. [DOI] [PubMed] [Google Scholar]
- 18.Dessen, A., A. Quémard, J. S. Blanchard, W. R. Jacobs Jr., and J. C. Sacchettini. 1995. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science. 267:1638–1641. [DOI] [PubMed] [Google Scholar]
- 19.Rozwarski, D. A., C. Vilchèze, M. Sugantino, R. Bittman, and J. C. Sacchettini. 1999. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J. Biol. Chem. 274:15582–15589. [DOI] [PubMed] [Google Scholar]
- 20.Mdluli, K., D. R. Sherman, M. J. Hickey, B. N. Kreiswirth, S. Morris, C. K. Stover, and C. E. Barry III. 1996. Biochemical and genetic data suggest that InhA is not the primary target for activated isoniazid in Mycobacterium tuberculosis. J. Infect. Dis. 174:1085–1090. [DOI] [PubMed] [Google Scholar]
- 21.Mdluli, K., R. A. Slayden, Y. Zhu, S. Ramaswamy, X. Pan, D. Mead, D. D. Crane, J. M. Musser, and C. E. Barry III. 1998. Inhibition of a Mycobacterium tuberculosis β-ketoacyl ACP synthase by isoniazid. Science. 280:1607–1610. [DOI] [PubMed] [Google Scholar]
- 22.Vilchèze, C., H. R. Morbidoni, T. R. Weisbrod, H. Iwamoto, M. Kuo, J. C. Sacchettini, and W. R. Jacobs Jr. 2000. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J. Bacteriol. 182:4059–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jornvall, H., B. Persson, M. Krook, S. Atrian, R. Gonzalez-Duarte, J. Jeffery, and D. Ghosh. 1995. Short-chain dehydrogenases/reductases (SDR). Biochemistry. 18:6003–6013. [DOI] [PubMed] [Google Scholar]
- 24.Duax, W. L., J. F. Griffin, and D. Ghosh. 1996. The fascinating complexities of steroid-binding enzymes. Curr. Opin. Struct. Biol. 6:813–823. [DOI] [PubMed] [Google Scholar]
- 25.Branden, C. I., and J. Toose. 1999. Introduction to Protein Structure, 2nd ed. Garland Publishing, New York.
- 26.Oppermann, U., C. Filling, M. Hult, N. Shafqat, X. Wu, M. Lindh, J. Shafqat, E. Nordling, Y. Kallberg, B. Persson, and H. Jörnvall. 2003. Short-chain dehydrogenase/reductases (SDR): the 2002 update. Chem. Biol. Interact. 143:247–253. [DOI] [PubMed] [Google Scholar]
- 27.Wierenga, R. K., M. C. H. De Maeyer, and W. G. J. Hol. 1985. Interaction of pyrophosphate moieties with α-helixes in dinucleotide binding proteins. Biochemistry. 24:1346–1357. [Google Scholar]
- 28.Bottoms, C. A., P. E. Smith, and J. J. Tanner. 2002. A structurally conserved water molecule in Rossmann dinucleotide-binding domains. Protein Sci. 11:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossmann, M. G., A. Liljas, C.-I. Branden, and L. J. Banaszak. 1975. Evolutionary and structural relationships among dehydrogenases. Enzymes. 11A:61–102. [Google Scholar]
- 30.Lesk, A. M. 1995. NAD-binding domains of dehydrogenases. Curr. Opin. Struct. Biol. 5:775–783. [DOI] [PubMed] [Google Scholar]
- 31.Rescigno, M., and R. N. Perham. 1994. Structure of the NADPH-binding motif of glutathione reductase: efficiency determined by evolution. Biochemistry. 33:5721–5727. [DOI] [PubMed] [Google Scholar]
- 32.Nishiya, Y., and T. Imanaka. 1996. Analysis of interaction between the Arthrobacter sarcosine oxidase and the coenzyme flavin adenine dinucleotide by site-directed mutagenesis. Appl. Environ. Microbiol. 62:2405–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eschenbrenner, M., L. J. Chlumsky, P. Khanna, F. Strasser, and M. S. Jorns. 2001. Organization of the multiple coenzyme and subunits and role of the covalent flavin link in the complex heterotetrameric sarcosine oxidase. Biochemistry. 40:5352–5367. [DOI] [PubMed] [Google Scholar]
- 34.Pearlman, D. A., D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham III, S. DeBolt, D. Ferguson, G. L. Seibel, and P. A. Kollman. 1995. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91:1–41. [Google Scholar]
- 35.Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz Jr., D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- 36.Ryde, U. 1995. Molecular dynamic simulations of alcohol dehydrogenase with a four- or five-coordinate catalytic zinc ion. Proteins. 21:40–56. [DOI] [PubMed] [Google Scholar]
- 37.Bayly, C. I., P. Cieplak, W. D. Cornell, and P. A. Kollman. 1993. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—the RESP model. J. Phys. Chem. 97:10269–10280. [Google Scholar]
- 38.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 39.Norberto de Souza, O., and R. L. Ornstein. 1997. Effect of periodic box size on aqueous molecular dynamics simulations of a DNA dodecamer with particle-mash Ewald method. Biophys. J. 72:2395–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 41.Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- 42.Norberto de Souza, O., and R. L. Ornstein. 1999. Molecular dynamics simulations of a protein-protein dimmer: particle-mesh Ewald electrostatic model yields far superior results to standard cutoff model. J. Biomol. Struct. Dyn. 16:1205–1218. [DOI] [PubMed] [Google Scholar]
- 43.Hünenberger, P. H., A. E. Mark, and W. F. van Gunsteren. 1995. Fluctuation and cross-correlation analysis of protein motions observed in nanosecond molecular dynamics simulations. J. Mol. Biol. 252:492–503. [DOI] [PubMed] [Google Scholar]
- 44.van Gunsteren, W. F., and A. E. Mark. 1998. Validation of molecular dynamics simulation. J. Chem. Phys. 108:6109–6116. [Google Scholar]
- 45.Baker, E. N., and R. E. Hubbard. 1984. Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 44:97–179. [DOI] [PubMed] [Google Scholar]
- 46.Kabsch, W., and C. Sander. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 47.Laskowski, R. A., M. W. McArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291. [Google Scholar]
- 48.Guex, N., and M. C. Peitsch. 1997. SWISS_MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 18:2714–2723. [DOI] [PubMed] [Google Scholar]
- 49.Fillgrove, K. L., and V. E. Anderson. 2000. Orientation of coenzyme A substrates, nicotinamide and active site functional groups in (di)enoyl-coenzyme A reductases. Biochemistry. 39:7001–7011. [DOI] [PubMed] [Google Scholar]
- 50.Grimm, C., E. Maser, E. Mobs, G. Klebe, K. Reuter, and R. Ficner. 2000. The crystal structure of 3α-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosterone shows a novel oligomerization pattern within the short chain dehydrogenase/reductase family. J. Biol. Chem. 275:41333–41339. [DOI] [PubMed] [Google Scholar]
- 51.Gerratana, B., W. W. Cleland, and P. A. Frey. 2001. Mechanistic roles of thr134, tyr160 and Lees 164 in the reaction catalyzed by did-glucose 4,6-dehydratase. Biochemistry. 40:9187–9195. [DOI] [PubMed] [Google Scholar]
- 52.Kavanagh, K. L., M. Klimacek, B. Nidetzky, and D. K. Wilson. 2002. Crystal structure of Pseudomonas fluorescens mannitol 2-dehydrogenase binary and ternary complexes. J. Biol. Chem. 277:43433–43442. [DOI] [PubMed] [Google Scholar]
- 53.Parikh, S., D. P. Moynihan, G. Xiao, and P. J. Tonge. 1999. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochemistry. 38:13623–13634. [DOI] [PubMed] [Google Scholar]
- 54.Denessiouk, K. A., V.-V. Rantanen, and M. S. Johnson. 2001. Adenine recognition: a motif present in ATP-, CoA-, NAD-, NADP-, and FAD-dependent proteins. Proteins. 44:282–291. [DOI] [PubMed] [Google Scholar]
- 55.Swaminathan, S., G. Ravishanker, and D. L. Beveridge. 1991. Molecular dynamics of B-DNA including water and counterions: a 140-ps trajectory for d(CGCGAATTCGCG) based on the GROMOS force field. J. Am. Chem. Soc. 113:5027–5040. [Google Scholar]
- 56.McDonald, I. K., and J. M. Thornton. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238:777–793. [DOI] [PubMed] [Google Scholar]
- 57.Morris, G. M., D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew, and A. J. Olson. 1998. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19:1639–1662. [Google Scholar]
- 58.Smith, P. E., and J. J. Tanner. 1999. Molecular dynamics simulations of NAD+ in solution. J. Am. Chem. Soc. 121:8637–8644. [Google Scholar]
- 59.Smith, P. E., and J. J. Tanner. 2000. Conformations of nicotinamide adenine dinucleotide (NAD+) in various environments. J. Mol. Recognit. 13:27–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.