

Abstract
At low to moderate ambient salt concentrations, DNA-binding proteins bind relatively tightly to DNA, and only very rarely detach. Intersegmental transfer due to DNA-looping can be excluded by applying an external pulling force to the DNA molecule. Under such conditions, we explore the targeting dynamics of N proteins sliding diffusively along DNA in search of their specific target sequence. At lower densities of binding proteins, we find a reduction of the characteristic search time proportional to N−2, with corrections at higher concentrations. Rates for detachment and attachment of binding proteins are incorporated in the model. Our findings are in agreement with recent single molecule studies in the presence of bacteriophage T4 gene 32 protein for which the unbinding rate is much lower than the specific binding rate.
INTRODUCTION
Many cellular processes involving DNA require a search for binding sites at particular locations on DNA molecules. For example, gene expression, i.e., the reading out of the genetic code and its conversion into either messenger-RNA and subsequent translation into proteins, or into transfer- and ribosomal-RNA that is not translated into proteins, is controlled by a large number of different, rather specific regulatory proteins—so-called transcription factors (TFs). On binding to a specific site (operator) on the genome, they either activate (recruitment) or repress the transcription of the associated gene by RNA polymerase (1,2). Apart from binding to the specific target site, with a lower binding affinity TFs can also attach to nonspecific regions on the genome, the nonspecific binding (3). Both specific and nonspecific binding make sure that a non-negligible portion of TFs is bound on the genome. In fact, it has been found that the majority of molecules of a certain TF can be bound nonspecifically, such as more than 90% of the λ-repressor CI under unperturbed lysogenic conditions in vivo (4,5).
The high accuracy of gene expression control by TFs such as in the famed genetic switch of the bacteriophage λ-Escherichia coli system (2,6–8) requires a fast search and recognition of the target sequence by the TFs. A simple three-dimensional search of the target sequence by the TFs is not sufficient to explain experimentally measured target search rates. It has been suggested relatively early (9,10) that additional search mechanisms such as one-dimensional sliding along the genome are needed to account for the actual efficiency of the search process. In their pioneering work, Berg, von Hippel, and co-workers (11,12) established a statistical model for target search comprising the four fundamental steps, as shown in Fig. 1:
Three-dimensional macrohops during which the TF fully detaches from the genome, until after a volume excursion it rebinds to the DNA (as a good approximation, the landing site on the DNA after a macrohop can be assumed to be equidistributed).
Microhops during which the TF detaches from the DNA but always stays very close to it (i.e., the microhop takes place within a cylinder whose radius corresponds to the escape distance of the TF from the DNA; see Ref. 11).
One-dimensional sliding along the genome (while preserving a certain bonding to the genome).
Intersegmental jumps.
FIGURE 1.

Classical Berg/von Hippel model of target search.
The latter are mediated by DNA-loops bringing two chemically remote segments of the DNA close in Euclidean space (see Ref. 13 and references therein). A TF like Lac repressor, which can establish bonds to two different stretches of dsDNA simultaneously, can then jump from one to the neighboring segment. This process might lead to a paradoxical diffusion behavior (14). However, if the conformational changes in the DNA are not too slow, both the bulk-mediated macrohops and the intersegmental transfer lead to fast mixing of the enzymes' positions along the chain, as it was shown for the related problem in Ref. (15), and on the mean-field level can be described by a desorption followed by the absorption at a random place.
Recently, there has been renewed interest in the targeting problem, both theoretically (16–19) and experimentally (e.g., 20,21), including single molecule studies (22–24). Despite the extensive knowledge of specific binding rates and both specific and nonspecific binding free energies, the precise relative contributions of the different search mechanisms (and, to some extent, the stringent criteria to define these four elementary interactions) are not fully resolved. Moreover, it has been suggested that under tight(er) binding conditions, the sliding of the protein becomes subdiffusive due to the local structure landscape of a heteropolymer DNA (25). This complication, however, is expected to be relaxed in a more loosely bound search mode of the TF (19). We here adopt the latter view of normal diffusion, which we then compare with the experiments reported by Pant et al. (22,23).
In previous studies, the one-dimensional sliding problem has always been considered as a problem of three-dimensional diffusion which is enhanced by one-dimensional diffusion. Thus, workers such as Berg, Winter, and von Hippel (11) assumed that proteins nonspecifically bound would, on average, unbind before finding their specific binding sites. This results in an enhancement of specific binding rates that is proportional to the one-dimensional sliding rate, but the overall specific binding rate depends linearly on protein concentration. These studies neglect the possibility that the protein finds its specific site before unbinding. Given the experimental conditions under which TF binding has been previously studied, this approximation is appropriate. However, as we will show below, this mechanism, in which the unbinding rate is much lower than the specific binding rate, occurs for the one-dimensional search of DNA by the single-stranded DNA binding protein T4 gene 32 protein (gp32). This fast one-dimensional search rate is essential for gp32 to be able to quickly find specific locations on DNA molecules that are undergoing replication, and which have large sections of single-stranded DNA exposed for gp32 binding. The resulting nonlinear concentration dependence of gp32 binding will likely have significant effects on gp32's ability to find its replication sites as well as its ability to recruit other proteins during replication. If these nonlinear effects also occur for TFs, this characteristic will strongly affect regulatory processes governed by protein binding.
Because this case has not been previously systematically investigated, in what follows, we concentrate on the sliding mechanism, which can experimentally be singled out by lowering the salt concentration in solution, leading to higher binding affinity to the DNA due to lack of counterions (22,23). Contributions from looping can be suppressed by use of rather short DNA segments, or by holding the DNA slightly stretched as, for instance, is done in optical tweezers experiments. Under such conditions, the typical time it takes for a TF of a certain species to locate its target sequence will decrease with the number N of nonspecifically bound molecules. We show by scaling arguments and analytic derivation that at relatively low concentrations of TFs, the characteristic targeting time decreases like N−2 in agreement with recent single molecule experiments, and obvious corrections occur at higher concentrations. We stop to mention that, surprisingly, such a detailed theoretical study on the influence of the number N on the search time to our knowledge has not been carried out, particularly for the case of pure one-dimensional sliding.
EXPERIMENTAL EVIDENCE
The concentration of nonspecifically bound proteins on DNA, n0, is determined by the concentration of protein in solution. At low concentrations, n0 = KnsμC, where C is the concentration in solution, μ is the size of the protein in units of nucleotides, and Kns is the nonspecific binding constant of the protein to the DNA lattice. In pure three-dimensional diffusion, the rate of finding a specific site (specific binding rate) is directly proportional to C, whereas in pure one-dimensional diffusion, the rate of specific binding is proportional to C2. Therefore, the concentration-dependence of specific binding can be used to determine the mechanism (three-dimensional or one-dimensional) by which a protein finds its specific binding sites. Recently, Pant et al. (22,23) showed that the rate at which T4 gene 32 protein finds its specific single-stranded binding sites is proportional to C2. In that experiment, optical tweezers were used to stretch single DNA molecules in the presence of protein, as sketched in Fig. 2. Pant et al. showed that the dependence of the DNA melting force on pulling rate could be used to directly determine the rate at which individual gp32 molecules bind at the ends of the DNA molecule. A truncated form of gp32, denoted *I, was found to exhibit binding rates that exceeded the three-dimensional diffusion limit. This result suggests that the protein is initially bound nonspecifically to the double-stranded DNA lattice. After the DNA is stretched, the ends of the molecule fluctuate open, creating new binding sites on single-stranded DNA, to which the protein binds specifically and cooperatively. The rate of specific binding appeared to depend on the square of the protein concentration.
FIGURE 2.

Optical tweezers setup (30). Single λ-DNA molecules are attached at the 3′ ends of each strand to two polystyrene beads. One bead is held by a glass micropipette by suction, whereas the other bead is held in an optical trap, formed by two counter-propagating laser beams focused to a common point. Force-extension curves were obtained by moving the micropipette and measuring the resulting force on the bead via the displacement relative to the focus of the optical trap. From the force-extension data, the binding rates displayed in Fig. 3 could be determined as described in Pant et al. (22,23). (Inset) Melting of the double-strand by gp32 or *I occurs from the pre-existing boundaries at the ends of the molecule.
To test this mechanism further, we have here obtained measurements of the *I specific binding rate, ka, as a function of protein concentration under a variety of solution conditions. According to the calculations below, if the one-dimensional diffusion mechanism dominates the specific binding process, ka should have a C2 dependence. Fig. 3 shows log(ka) (decadic logarithm, log = log10) as a function of log(C) for salt concentrations ranging from 75 mM NaCl to 200 mM NaCl. Within experimental error, the slope is equal to two for this entire range of salt concentrations. These experimental results were obtained on DNA molecules that were approximately 50,000 basepairs (bp) in length. The binding site size for *I is known to be about 7 bp. In addition, under these conditions, the fractional binding of the proteins to the DNA lattice is less than 0.1, so this corresponds to the dilute case discussed below, and size exclusion is not expected to be important. Below, we discuss the general case in which we can have both one-dimensional and three-dimensional diffusion, depending on the on- and off-rates of the protein under specific conditions. However, the results of Fig. 3 show that it is possible to experimentally obtain conditions under which the off-rate is very low, leading to a C2-dependence of the specific binding rates. In the case of gp32, those conditions are obtained under physiological salt concentrations.
FIGURE 3.

Measured rate of binding of the T4 gene 32 protein truncate *I as a function of protein concentrations in 75 mM salt (solid square), 100 mM salt (solid triangle), 150 mM salt (open square), and 200 mM salt (open triangle). The fitted lines have slopes of 1.74 ± 0.35, 1.85 ± 0.24, 2.08 ± 0.39, and 1.95 ± 0.17, respectively. The data obtained at 100 mM salt are fitted by the theoretical model in Fig. 5.
SCALING
The mechanism revealed in the previous section is based on the creation of new specific binding sites at the boundary between single-stranded and double-stranded DNA, due to mechanical unzipping of the DNA at some time t = 0. Due to random search of the SSBs already bound on the DNA, the specific binding site is subsequently filled after the characteristic time T, which depends on the number N of bound proteins. In turn, N is proportional to the volume concentration C of proteins under the experimental conditions studied in the previous section.
Let us at first consider the simplest case when the N identical proteins are constantly attached to the DNA of length L = Mb, and we choose the basepair-to-basepair distance b ≈ 3.5 Å as unit of length. A single TF occupies some 10–20 basepairs (single-stranded DNA binding proteins are typically somewhat smaller, such as 7 bp for gp32), and we denote the corresponding length by λ = μb. This picture corresponds to the situation observed for gp32 searching a single DNA molecule, but it likely also applies to double-stranded DNA binding TFs under certain conditions.
To cross a distance L by bias-free diffusion, a particle on average consumes a time T ≃ L2/D1d where D1d is the corresponding diffusion coefficient for one-dimensional sliding motion on the DNA, and the symbol ≃ indicates that we neglect constant prefactors. If we deal with N identical particles, on average each of them has a free diffusion length of L/N, so that the characteristic search time of any one TF to find the target sequence scales like
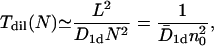 |
(1) |
where n0 = N/M is the number concentration of the TFs on the DNA, and 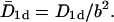 The index is meant to indicate that this result can only hold for the dilute case, in which the length occupied by the TFs is much smaller than the length of the DNA, Nλ ≪ L. In what follows we show that the prefactor in Eq. 1 becomes π/2 for the one-sided situation, and π/8 for the two-sided situation in the case of a ring DNA. In Fig. 4 we display results for T(n0) of a simulation of particles diffusing on a discrete lattice for various n0 under excluded volume conditions, i.e., a given lattice site can be occupied by, at most, one particle. The inverse square-dependence of T(n0) as predicted by Eq. 1 is nicely fulfilled. Our simulation corresponds to a random walk picture in which a particle makes, on the average, one attempted step to the right or to the left per unit time. If the corresponding site is occupied, the step is not performed. We note that taking the step length to be a unit length of the problem leads to the value of the diffusion coefficient of a single particle
The index is meant to indicate that this result can only hold for the dilute case, in which the length occupied by the TFs is much smaller than the length of the DNA, Nλ ≪ L. In what follows we show that the prefactor in Eq. 1 becomes π/2 for the one-sided situation, and π/8 for the two-sided situation in the case of a ring DNA. In Fig. 4 we display results for T(n0) of a simulation of particles diffusing on a discrete lattice for various n0 under excluded volume conditions, i.e., a given lattice site can be occupied by, at most, one particle. The inverse square-dependence of T(n0) as predicted by Eq. 1 is nicely fulfilled. Our simulation corresponds to a random walk picture in which a particle makes, on the average, one attempted step to the right or to the left per unit time. If the corresponding site is occupied, the step is not performed. We note that taking the step length to be a unit length of the problem leads to the value of the diffusion coefficient of a single particle 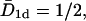 so that if the continuous approximation works correctly, the product
so that if the continuous approximation works correctly, the product 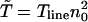 would be constant and equal to π (compare Eq. 16). The results show that the theoretical approximation leading to the 1/N2 behavior remains reasonable even at rather high concentrations, at which the interparticle distance is of the order of the step lengths. In the following section, we employ a continuum approximation to analytically derive the N−2 scaling.
would be constant and equal to π (compare Eq. 16). The results show that the theoretical approximation leading to the 1/N2 behavior remains reasonable even at rather high concentrations, at which the interparticle distance is of the order of the step lengths. In the following section, we employ a continuum approximation to analytically derive the N−2 scaling.
FIGURE 4.

Mean first passage time T(n0) of target search on a large, one-sided system (one target located at x = 0), as a function of the density n0 of excluding walkers that cannot occupy the same lattice site. The maximum density is n0 = 30%. The dashed line corresponds to the exact result 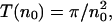 for dimensionless diffusion coefficient
for dimensionless diffusion coefficient 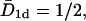 from Eq. 16 obtained in the continuum approximation. We see a slight deviation for larger densities. Each data point corresponds to 105 runs, except for 103 realizations for the lowest density. Note the comparatively small error bars.
from Eq. 16 obtained in the continuum approximation. We see a slight deviation for larger densities. Each data point corresponds to 105 runs, except for 103 realizations for the lowest density. Note the comparatively small error bars.
For direct comparison with the experimental data, Fig. 5 shows an alternative way to present the numerical data from Fig. 4, in dimensional form of the rate ka in units of 1/s versus the volume protein concentration C in units of M. For the conversion, we use the relation n0 = KnsμC, with the nonspecific binding constant Kns = 2.5 × 105 M−1, and the SSB binding size μ = 7 in units of nucleotides (22,23). By logarithmic least-squares fit to the shown data measured at 100 mM salt, we obtain for the diffusion constant D1d of one-dimensional sliding along the dsDNA the value D1d = 3.3 × 10−9cm2/s, which is nicely within the experimental value 10−8…10−9cm2/s for this salt concentration reported in Pant et al. (22,23). This corroborates the validity of our rather simple analytical model for the target search of the *I truncate. Note that the experimental situation with two target sites at either end of the DNA molecule corresponds to Eq. 22 derived below.
FIGURE 5.

Dimensional binding rate ka in 1/s as function of protein concentration C in M, converted from Fig. 4 for parameters corresponding to 100 mM salt. The fitted one-dimensional diffusion constant for sliding along the dsDNA is D1d = 3.3 · 10−9cm2/s, located nicely within the experimental value 10−8…10−9 cm2/s (see Refs. 22,23).
Expressed in terms of the dimensionless occupation ratio f = Nλ/L = Nμ/M = μn0, the diluteness condition becomes f ≪ 1. Although the dilute case may correspond to realistic situations (such as the case of the *I mutant at the salt concentrations we measured) as prepared in the in vitro experiments, nonspecific binding at high concentrations of TFs may well cause situations that can no longer be considered dilute, in the sense that a considerable part of the DNA is occupied by the TFs, which to no extent can be considered as pointlike. The only difference between this case and the previous one is to not consider the full lengths of the DNA, but only the reduced lengths, corresponding to the overall space that TFs have for their motion. This length is Lred = L − Nλ = (M − Nμ)b, so that we obtain
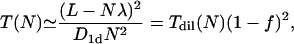 |
(2) |
where n0 is the initial concentration of the TFs. In Fig. 6, we compare the dilute case with the excluded volume expression in Eq. 2.
FIGURE 6.

Behavior of the mean first passage time T(N) as a function of the number N of TFs attached to a DNA of length 1000b according to Eq. 2, for the dilute case (solid line), TF-size λ = b (long-dashed line) and λ = 10b (short-dashed line). Excluded volume effects reduce the target search time T(N).
CONTINUUM APPROXIMATION
In this section, we use a continuum approach to verify the scaling result 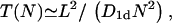 but also allow for explicit adsorption and desorption effects with constant rates k0 and k1, respectively. Although these latter effects apparently do not come into play for the experimental results reported in Experimental Evidence, above, we include them for completeness: adsorption and desorption are expected to be relevant to the search dynamics of other binding proteins such as transcription factors or other SSBs. We also note that although some of the results below are known per se for the case of one-particle diffusion or for phantom particles (26,27), in the present case they are based on a mapping of the case of impenetrable particles, a problem that, to our knowledge, has not been studied so far. In that sense, our analysis is new, and provides a detailed tool for the interpretation of experiments on protein motion coupled to DNA molecules.
but also allow for explicit adsorption and desorption effects with constant rates k0 and k1, respectively. Although these latter effects apparently do not come into play for the experimental results reported in Experimental Evidence, above, we include them for completeness: adsorption and desorption are expected to be relevant to the search dynamics of other binding proteins such as transcription factors or other SSBs. We also note that although some of the results below are known per se for the case of one-particle diffusion or for phantom particles (26,27), in the present case they are based on a mapping of the case of impenetrable particles, a problem that, to our knowledge, has not been studied so far. In that sense, our analysis is new, and provides a detailed tool for the interpretation of experiments on protein motion coupled to DNA molecules.
The continuous approximation corresponds to concentrations much smaller than unity (i.e., f ≪ 1), and to rather large systems consisting of many searching proteins (N ≫ 1). In other words, we consider large, dilute systems in the sense that the diffusion time through the whole system, T1 ≃ L2/D1d, is much larger than the typical first passage time corresponding to the characteristic target search time, being of the order of T ≃ 1/(f2D1d). Finite size effects can be incorporated into the model. However, this is beyond the scope of the present work, and we refer to a forthcoming study (I. M. Sokolov, R. Metzler, K. Pant, and M. C. Williams, unpublished). According to these results, the diluteness condition should hold for most in vitro experiments involving only a small number of different species of binding proteins, at concentrations that are not significantly higher than in vivo.
Let us first consider a one-sided problem (one target site at x = 0 of a semi-infinite DNA). The time evolution of the number concentration ν(x, t) (of dimension 1/cm, in contrast to the dimensionless quantity n0; ν can be made dimensionless by n(x, t) = ν/b) at position x at time t on a semi-infinite interval is given by the equation
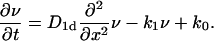 |
(3) |
Apart from diffusion, in this equation we take into account adsorption (with rate k0) and desorption (with rate k1) of the TFs, which, apart from real physical absorption/desorption processes, might mimic other nonlocal processes such as macrohops and intersegmental transfer in a mean field sense. Following Smoluchowski's approach to diffusion-controlled reactions, we represent the target site by an absorbing boundary condition at x = 0; i.e., when a diffusing particle hits this site, it will be removed. The possibility of double occupation of sites is disregarded, as it represents a higher order effect proportional to f2. Moreover, the fact that particles are impenetrable to each other does not change the behavior at low concentrations, since, neglecting the excluded volume on encounter of two particles, it does not matter whether the right particle always stays to the right of the other particle (impenetrable particles), or they change roles and the right particle becomes the left one (phantom particles), as long as the particles are indistinguishable, in contrast to the case of distinguishable particles (addressed in I. M. Sokolov, R. Metzler, K. Pant, and M. C. Williams, unpublished).
Finding the target corresponds to the event when the first particle hits the target site. Mathematically, this is equivalent to the first passage time of a particle from a site x > 0 to x = 0, given by the particle flux into the reaction center, j(t) = D1d ∂ν/∂x|x=0. The survival probability 𝒮(t) of the target site (i.e., the probability of not yet having been hit by a TF) is consequently given by the first-order kinetic equation
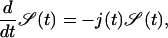 |
(4) |
whose formal solution reads
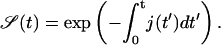 |
(5) |
In what follows we use the notation 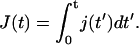 The first passage time density is then given by
The first passage time density is then given by
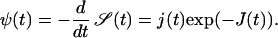 |
(6) |
In our one-sided problem, the mean first passage time becomes 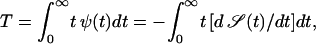 i.e.,
i.e.,
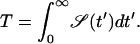 |
(7) |
To obtain an explicit expression for 𝒮(t), we solve the reaction-diffusion Eq. 3 by Laplace transformation techniques. With the initial condition ν(x, 0) = ν0Θ(x), where Θ(x) is the Heaviside jump function, we obtain for all x > 0 for the Laplace transform ñ(x,u),
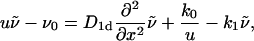 |
(8) |
i.e., a linear inhomogeneous differential equation of the form
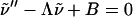 |
(9) |
with Λ = (k1 + u)/D1d > 0 and B = (k0/u + ν0)/D1d > 0. The boundary conditions we impose are of the absorbing Dirichlet type ν(0, u) = 0 at the target site placed at the origin, and the natural boundary condition ν(x, u) < ∞ for x → ∞. The corresponding solution reads
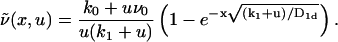 |
(10) |
From this expression, we find for the flux j(t) in Laplace space
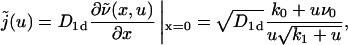 |
(11) |
an expression whose inverse Laplace transform can be calculated explicitly, yielding
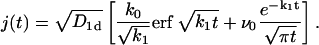 |
(12) |
The survival probability of the target site is then given by 𝒮(t) = exp(−J(t)) with
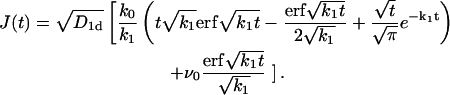 |
(13) |
Without adsorption and desorption (i.e., k0 = k1 = 0), we obtain the survival probability
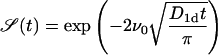 |
(14) |
and first passage time density
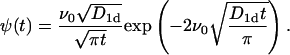 |
(15) |
We thus find for the mean first passage time 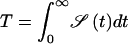 the simple form
the simple form
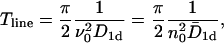 |
(16) |
showing the typical 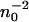 dependence on the initial concentration.
dependence on the initial concentration.
The first passage time distribution for the general case with nonvanishing rates k0 and k1 becomes
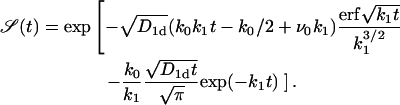 |
(17) |
In the case of no adsorption k0 = 0 but nonvanishing desorption k1 ≠ 0 that corresponds to a situation with vanishing concentration of TFs in the free volume, the function
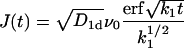 |
(18) |
is bounded from above, by 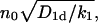 and the survival probability 𝒮(t) never reaches zero (all particles desorb with a nonzero probability without ever reaching the target site x = 0), and the probability density ψ(t) is a nonproper one, corresponding to a diverging mean first passage time. In all other cases ψ(t) is a proper probability density, and the mean target search time T is finite.
and the survival probability 𝒮(t) never reaches zero (all particles desorb with a nonzero probability without ever reaching the target site x = 0), and the probability density ψ(t) is a nonproper one, corresponding to a diverging mean first passage time. In all other cases ψ(t) is a proper probability density, and the mean target search time T is finite.
Performing an expansion in powers of t (the corresponding series contains only the half-integer powers), we find for the function J(t) in the general case with finite k0, k1,
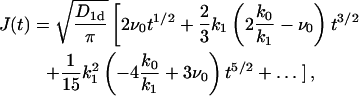 |
(19) |
so that the ith term of the expansion has a structure 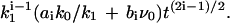 Thus, in essence, this expansion corresponds to an expansion in powers of k1. Note that k0/k1 = ns is a steady-state concentration of proteins in the absence of the absorbing target site. As long as both k0 and k1 are small, the overall behavior given by Eq. 16 is preserved, provided the initial concentration n0 is not too small. In the case without desorption (k1 → 0) we get
Thus, in essence, this expansion corresponds to an expansion in powers of k1. Note that k0/k1 = ns is a steady-state concentration of proteins in the absence of the absorbing target site. As long as both k0 and k1 are small, the overall behavior given by Eq. 16 is preserved, provided the initial concentration n0 is not too small. In the case without desorption (k1 → 0) we get
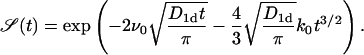 |
(20) |
This equation is important when finite-size effects are considered (I. M. Sokolov, R. Metzler, K. Pant, and M. C. Williams, unpublished).
The two-sided problem (a ring geometry with a perimeter that is much larger than the typical interparticle distance) corresponds to the situation where two competing processes occur, i.e., the survival probability of having an empty target site changes in time through the influx of TFs from both sides. This practically corresponds to using twice the probability current j in Eq. 4 due to symmetry, and therefore to
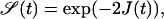 |
(21) |
with J(t) given by Eq. 13. The corresponding mean first passage time for the case k0 = k1 = 0 is then given by
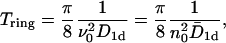 |
(22) |
that is, by a factor of 4 smaller than in the one-sided case. This result (Eq. 22) is also confirmed by numerical simulations. We note that the reduction by a factor 4 can be easily understood by mapping the circle with one absorbing site onto a line with both ends as absorbing boundaries. It then corresponds to two one-sided geometries as considered above, but with an effective length of L/2. With ν0 = N/L, this reproduces the factor 4.
DISCUSSION AND CONCLUSIONS
The problem of target search of the specific binding site on a genome by a TF is a complex stochastic process including three-dimensional volume exchange (macrohops); local detachment, displacement, and reattachment (microhops); intersegmental jumps mediated by DNA-looping; and one-dimensional sliding along the genome. The relative importance of the various contributions to the targeting problem is determined by the corresponding rates (binding, unbinding) and diffusivities. At normal salt conditions, the combination of sliding and volume exchange in the classical Berg-von Hippel model reduces the characteristic time of the targeting process considerably, thereby guaranteeing the surprisingly efficient control of gene expression.
To investigate the individual processes in more detail, variation of salt conditions (low salt will highly favor binding to the DNA) or volume diffusivity (e.g., by adding sugar to the solution to decrease the mobility) will bias the relative contributions and make it possible, for instance, to observe an almost exclusive combination of sliding and intersegmental jumps. Moreover, by suppressing DNA-looping (e.g., by stretching the DNA using optical tweezers), as we have shown here, it is possible to solely investigate sliding. We have demonstrated that for *I, a truncate of T4 gene 32 protein, the one-dimensional sliding mechanism determines the observed protein binding rate under a wide variety of solution conditions, including under physiological salt concentrations. In principle, it should be possible to experimentally reach a situation with pure sliding at very low salt concentrations for double-stranded DNA binding TFs as well. Given these perspectives, we provide here the framework for studying the dependence of the characteristic target search time on the number of proteins N, or, by knowledge of the Gibbs free energy for nonspecific binding, the concentration C of proteins in the solution. We distinguish some of the standard geometries used in the in vitro setups. In particular, we demonstrated by comparison of experimental and simulations data and analytical results that this approach is quantitative.
Finally, a few words concerning potential anomalous transport features are in order. As mentioned in the Introduction, there exist possible scenarios that, due to the heteropolymer character of DNA, the sliding motion of TFs can become subdiffusive (25), i.e., the mean-squared displacement of the diffusing TF grows sublinearly in time: 〈(Δx(t))2〉 ≃ Dαtα, with 0 < α < 1 and the anomalous diffusion constant with dimension [Dα] = cm2/sα (28–31). This corresponds to an infinite system producing a waiting time density of the inverse power-law form ψ(t) ≃ τα/t1+α, and, according to Slutsky and Mirny (19), can be overcome by a semidetached sliding mode of TFs. For the system we had in mind in this study, we thus assumed a normal-diffusive sliding. Moreover, the typical length covered by a single TF before one of the N TFs hit the target sequence, is relatively short, and the heteropolymer character of the DNA is not expected to produce fully pronounced subdiffusion. It has to be seen whether subdiffusion can be observed for sliding TFs, an interesting question that may be approached by single DNA imaging methods. Conversely, one expects the occurrence of Lévy flights in chemical coordinates due to DNA-looping. The typical distance covered by a sliding TF is expected to scale like p(l) ≃ l−c, where c < 3, such that, statistically, the mean-squared displacement diverges (compare the discussions in Refs. 13–15). This phenomenon, which is expected to contribute to the overall target search, will be discussed elsewhere.
Acknowledgments
We thank Ulrich Gerland and Oleg Krichevsky for helpful discussions. The *I protein used in the experiments presented here was provided by Richard L. Karpel.
I.M.S. gratefully acknowledges the Fonds der Chemischen Industrie for partial financial support. This research was supported by the National Science Foundation under grant No. MCB-0238190, and the Research Corporation.
References
- 1.Orphanides, G., and D. Reinberg. 2002. A unified theory of gene expression. Cell. 108:439–451. [DOI] [PubMed] [Google Scholar]
- 2.Ptashne, M., and A. Gunn. 2002. Genes and Signals. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 3.Kornberg, A., and T. A. Baker. 1992. DNA Replication. W. H. Freeman and Company, New York, NY.
- 4.Bakk, A., and R. Metzler. 2004a. In vivo non-specific binding of λ-CI and Cro repressors is significant. FEBS Lett. 563:66–68. [DOI] [PubMed] [Google Scholar]
- 5.Bakk, A., and R. Metzler. 2004b. Nonspecific binding of the OR repressors CI and Cro of bacteriophage λ. J. Theor. Biol. 231:525. [DOI] [PubMed] [Google Scholar]
- 6.Aurell, E., S. Brown, J. Johanson, and K. Sneppen. 2002. Stability puzzles in phage-λ. Phys. Rev. E. 65: art. no. 051914. [DOI] [PubMed]
- 7.Bakk, A., R. Metzler, and K. Sneppen. 2004. Sensitivity of OR in phage-λ. Biophys. J. 86:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Little, J. W., D. P. Shepley, and D. W. Wert. 1999. Robustness of a gene regulatory circuit. EMBO J. 18:4299–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adam, G., and M. Delbrück. 1968. Structural Chemistry and Molecular Biology. A. Rich, and N. Davidson, editors. W. H. Freeman, San Francisco, CA.
- 10.Richter, P. H., and M. Eigen. 1974. Diffusion controlled reaction rates in spheroidal geometry. Applications to repressor-operator association and membrane bound enzymes. Biophys. Chem. 2:255–263. [DOI] [PubMed] [Google Scholar]
- 11.Berg, O. G., R. B. Winter, and P. H. von Hippel. 1981. Diffusion-driven mechanisms of protein translocation on nucleic acids. I. Models and theory. Biochemistry. 20:6929–6948. [DOI] [PubMed] [Google Scholar]
- 12.Winter, R. B., O. G. Berg, and P. H. von Hippel. 1981. Diffusion-driven mechanisms of protein translocation on nucleic acids. III. The Escherichia coli lac repressor-operator interaction: kinetic measurements and conclusions. Biochemistry. 20:6961–6977. [DOI] [PubMed] [Google Scholar]
- 13.Hanke, A., and R. Metzler. 2003. Entropy loss in long-distance DNA looping. Biophys. J. 85:167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sokolov, I. M., J. Mai, and A. Blumen. 1997. Paradoxical diffusion in chemical space for nearest-neighbor walks over polymer chains. Phys. Rev. Lett. 79:857–860. [Google Scholar]
- 15.Sokolov, I. M., J. Mai, and A. Blumen. 1998. Energy transfer in polymers in dilute solutions. J. Luminesc. 76–77:377–380. [Google Scholar]
- 16.Coppey, M., O. Benichou, R. Voituriez, and M. Moreau. 2004. Kinetics of target site localization of a protein on DNA: a stochastic approach. Biophys. J. 87:1640–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerland, U., J. D. Moroz, and T. Hwa. 2002. Physical constraints and functional characteristics of transcription factor-DNA interaction. Proc. Natl. Acad. Sci. USA. 99:12015–12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halford, S. E., and J. F. Marko. 2003. How do site-specific DNA binding proteins find their targets? Nucleic Acids Res. 32:3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slutsky, M., and L. A. Mirny. 2004. Kinetics of protein-DNA interaction: facilitated target location in sequence-dependent potential. Biophys. J. 87:4021–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grillo, A. O., M. P. Brown, and C. A. Royer. 1999. Probing the physical basis for Trp repressor-operator recognition. J. Mol. Biol. 287:539–554. [DOI] [PubMed] [Google Scholar]
- 21.Spolar, R. S., and M. T. Record. 1994. Coupling of local folding to site-specific binding of proteins to DNA. Science. 263:777–784. [DOI] [PubMed] [Google Scholar]
- 22.Pant, K., R. L. Karpel, I. Rouzina, and M. C. Williams. 2004. Mechanical measurement of single-molecule binding rates: kinetics of DNA helix-destabilization by T4 gene 32 protein. J. Mol. Biol. 336:851–870. [DOI] [PubMed] [Google Scholar]
- 23.Pant, K., R. L. Karpel, I. Rouzina, and M. C. Williams. 2005. Salt dependent binding of T4 gene 32 protein to single and double-stranded DNA: single molecule force spectroscopy measurements. J. Mol. Biol. 349:317–330. [DOI] [PubMed] [Google Scholar]
- 24.Shimamoto, N. 1999. One-dimensional diffusion of proteins along DNA: its biological and chemical significance revealed by single-molecule measurements. J. Biol. Chem. 274:15293–15296. [DOI] [PubMed] [Google Scholar]
- 25.Slutsky, M., M. Kardar, and L. A. Mirny. 2004. Diffusion in correlated random potentials, with applications to DNA. Phys. Rev. E. 69: art. no. 061903. [DOI] [PubMed]
- 26.Redner, S. 2001. A Guide to First-Passage Processes. Cambridge University Press, Cambridge, UK.
- 27.Rice, S. A. 1985. Diffusion-limited reactions. In Comprehensive Chemical Kinetics, Vol. 25. C. H. Bamford, C. F. H. Tipper, and R. G. Compton, editors. Elsevier, Amsterdam, The Netherlands.
- 28.Metzler, R., and J. Klafter. 2004. The restaurant at the end of the random walk: recent developments in the description of anomalous transport by fractional dynamics. J. Phys. A. 37:R161–R208. [Google Scholar]
- 29.Metzler, R., and J. Klafter. 2000. The random walk's guide to anomalous diffusion: a fractional dynamics approach. Phys. Rep. 339:1–77. [Google Scholar]
- 30.Sokolov, I. M., J. Klafter, and A. Blumen. 2002. Fractional kinetics. Phys. Today. 55:48–54. [Google Scholar]
- 31.Williams, M. C. 2002. Optical Tweezers: Measuring PicoNewton Forces. Biophysics Textbook Online: http://www.biophysics.org/btol/.