

Abstract
Mammalian ClC-type chloride channels have large cytoplasmic carboxy-terminal domains whose function is still insufficiently understood. We investigated the role of the distal part of the carboxy-terminus of the muscle isoform ClC-1 by constructing and functionally evaluating two truncation mutants, R894X and K875X. Truncated channels exhibit normal unitary conductances and anion selectivities but altered apparent anion binding affinities in the open and in the closed state. Since voltage-dependent gating is strictly coupled to ion permeation in ClC-1 channels, the changed pore properties result in different fast and slow gating. Full length and truncated channels also differed in methanethiosulphonate (MTS) modification rate constants of an engineered cysteine at position 231 near the selectivity filter. Our data demonstrate that the carboxy-terminus of ClC channels modifies the conformation of the outer pore vestibule.
INTRODUCTION
ClC-type anion channels and transporters are expressed in prokaryotic and eukaryotic cells and fulfill diverse physiological functions. Nine human ClC channels (hClC-1 to hClC-7, hClC-Ka, and hClC-Kb) play roles in the regulation of cell excitability (1–3), transepithelial chloride transport (4,5), and pH adjustment in certain cell compartments (6–8). Bacterial homologs function as secondary active, proton-coupled chloride transporters (9) and were proposed to provide an electrical shunt for an outwardly directed proton pump linked to amino acid decarboxylation (10).
Prokaryotic and eukaryotic ClC isoforms exhibit a unique, evolutionarily conserved double-barreled architecture with two ion conduction pathways, each formed by a single subunit (11–13). In contrast to prokaryotic isofoms, all eukaryotic channels have large carboxy-termini that reside in the cytoplasm and contain two cystathionine-β-synthase (CBS) domains (14,15). For ClC-0 and ClC-1, complete removal of the carboxy-terminus has been demonstrated to render the corresponding protopore nonfunctional (16–20). On the other hand, point mutations or deletions within the region distal to the second CBS result in functional channels with altered voltage-dependent gating (18,21). Since ClC channels are thought to be gated by the permeant anion and to lack an endogenous voltage sensor (22–24), modification of gating properties by a cytoplasmic domain is intriguing. Mutations within the distal carboxy-terminus might alter ClC channel gating either by affecting intrinsic pore properties or, alternatively, by modifying inherently voltage-dependent conformational changes of the channel protein that were not identified before.
We generated and functionally characterized two truncation mutants of hClC-1 (R894X and K875X) (Fig. 1 A). R894X represents a naturally occurring mutation identified in patients with recessive myotonia congenita (25), and K875X hClC-1 was engineered to remove the whole region distal to the second CBS domain. Both mutations affect the interaction between the permeating anion and its binding sites within the ion conduction pathway, and these alterations result in changed channel gating. Our results demonstrate that the carboxy-terminus modifies the outer pore vestibule of ClC-type chloride channels and further support the concept of ClC channel gating by permeant anions.
FIGURE 1.

Carboxy-terminal truncations alter gating properties of hClC-1 channels. (A) Topology and (B–D) representative current recordings of WT and mutant channels.
MATERIAL AND METHODS
Expression of WT and mutant hClC-1
Truncations were introduced into the plasmid pRc/CMV-hClC-1 containing the full length wild-type (WT) hClC-1 cDNA (26) by polymerase chain reaction-based strategies. For all constructs, polymerase chain reaction-amplified sequences were verified by direct sequencing, and two independent mutant clones were used for expression studies. Some of the experiments were performed with WT and mutant pSVL-hClC-1 constructs obtained by subcloning the respective cDNA into the pSVL vector (Amersham Biosciences, Piscataway, NJ). Mutant and WT hClC-1 channels were transiently expressed in tsA201 cells with passage numbers between 3 and 20 and examined typically 2 days after transfection with a calcium phosphate precipitation method (27). To identify cells with a high probability of expressing recombinant ion channels, cells were cotransfected with a plasmid encoding the CD8 antigen and incubated 5 min before use with polystyrene microbeads precoated with anti-CD8 antibodies (Dynabeads M-450 CD 8, Dynal, Great Neck, NY) (28). For some experiments examining WT hClC-1, stably transfected human embryonic kidney (HEK) 293 cells (29) were used. Functional properties of channels stably expressed in HEK 293 cells and those transiently expressed in HEK 293 or tsA201 cells were indistinguishable from one another.
Electrophysiology
Standard whole-cell, excised inside-out or outside-out patch clamp recordings were performed as previously described (19,30). The standard extracellular solution contained (in mM): NaCl (140), KCl (4), CaCl2 (2), MgCl2 (1), HEPES (5), pH 7.4; the standard intracellular (in mM): NaCl (120), MgCl2 (2), EGTA (5), HEPES (10), pH 7.4. Cells were clamped to 0 mV between two test sweeps. To exchange the external or the internal solution, cells/excised patches were moved into the stream of solutions containing external or internal solutions whose composition was altered by substituting NaCl equimolarly with sodium glutamate, NaI, or NaNO3. Fast opening rate constants of K875X hClC-1 channels (see Figs. 3 C and 4 C) were determined using an intracellular solution containing 230 mM NaCl and an external solution with 250 mM NaCl that was partly substituted with Naglutamate to change [Cl−]o. To study block of chloride currents by I− or 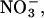 instantaneous current amplitudes were determined after a 200 ms prepulse to +75 mV before and after application of a given anion concentration. Agar bridges were used to connect the amplifier and the bath solution. Junction potentials calculated using the JPCalc software (Dr. P. Barry, University of South Wales, Sydney, Australia (31)) and offset potentials measured at the end of each experiment were used to correct results. Modification by 2-(trimethylammonium)ethyl-methanethiosulphonate (MTSET) and 2-aminoethyl-methanethiosulphonate (MTSEA) was performed as described (32). Modification rates were determined at holding potentials of −80 mV, 0 mV, and +75 mV.
instantaneous current amplitudes were determined after a 200 ms prepulse to +75 mV before and after application of a given anion concentration. Agar bridges were used to connect the amplifier and the bath solution. Junction potentials calculated using the JPCalc software (Dr. P. Barry, University of South Wales, Sydney, Australia (31)) and offset potentials measured at the end of each experiment were used to correct results. Modification by 2-(trimethylammonium)ethyl-methanethiosulphonate (MTSET) and 2-aminoethyl-methanethiosulphonate (MTSEA) was performed as described (32). Modification rates were determined at holding potentials of −80 mV, 0 mV, and +75 mV.
FIGURE 3.

Voltage and chloride dependence of opening rate constants for the fast (A, C, and E) and the slow (B and D) gate of WT and truncated hClC-1. Solid lines represent fits to Eq. 2. Mean ± SE (n > 5).
FIGURE 4.

Opening rate constants for the fast and the slow gate of WT and truncated hClC-1. (A, C, and E) Dependence of the opening rate constants of the fast gate of WT (A), K875X (C), and R894X (E) hClC-1 channels on [Cl−]o. (B and D) Dependence of the opening rate constants of the slow gate of WT (B) and K875X (D) channels on [Cl−]o. Solid lines represent fits to Eq. 2. Mean ± SE (n > 5).
Data analysis
Data were analyzed by a combination of PulseFit and PulseTools (HEKA Electronics, Lambrecht, Germany), Clampfit (Axon Instruments, Foster City, CA), and SigmaPlot (Jandel Scientific, San Rafael, CA) programs. Instantaneous current amplitudes were calculated by extrapolating deactivation time courses with biexponential functions to the moment of the voltage step. To determine the unitary current amplitude and the open probabilities of mutant hClC-1 channels, nonstationary noise analysis was performed as described (19,30). To determine the time course of current deactivation, a sum of two exponentials and a time-independent value (I(t) = a1exp(−t/τfast) + a2exp(−t/τslow) + c) were fit to data recorded during a series of hyperpolarizing voltage steps from a holding potential of 0 mV. To obtain the voltage dependence of the relative open probability, the instantaneous current amplitudes after a voltage step to −125 mV were normalized by their maximum value and plotted versus the preceding potential. The absolute open probability at 0 mV determined by noise analysis was then used to calculate the voltage dependence of the open probability (19) (Fig. 2 A). The voltage dependences of the opening of the fast and the slow gate were distinguished as described (33) (Fig. 2, C and D). The relative open probability of the slow gate is determined by measuring normalized tail current amplitudes after 200-ms prepulses to various potentials and a short fixed pulse to positive voltages inserted before the test step (Fig. 2 C, inset). For a separation of fast and slow gating, the short pulse has to be long enough to fully activate the fast gate without significantly affecting the slow gate. Time constants of fast and slow activation were determined at +180 mV for [Cl−]o = 10 mM or 150 mM using envelope protocols (33) (data not shown), demonstrating that a pulse duration of 500 μs satisfies these requirements for all constructs. Activation curves for the fast and for the slow gate were fit with the sum of a voltage-independent minimum open probability (Pmin) and a voltage-dependent term: I(V) = Amp/(1 + e(V− V0.5)/kV) + Pmin.
FIGURE 2.

Carboxy-terminal truncations modify the voltage dependence of fast and slow gating. (A) Voltage dependence of the open probability. (B) Voltage dependence of the fast (solid symbols) and slow (open symbols) deactivation time constants for WT (•), R894X (▾), and K875X (▪) hClC-1 channels. Mean ± SE (n > 58). The voltage dependence of the fast deactivation time constants was fitted with τ = 1/(αoeaeoV/kT + βoe−bzoV/kT), giving an equivalent gating charge of 1 for the opening rate constant and 0.75 for the closing time constants. Slow time constants were fitted with single exponentials. (C and D) Voltage dependence of the slow (C) and the fast (D) gate of WT (•) and mutant (R894X, ▾, and K875X, ▪) channels. (E and F) Plot of the midpoint of the voltage dependence of slow (E) and fast (F) gating versus [Cl−]o. Mean ± SE (n > 8).
The voltage dependences of the fast deactivation time constants (Fig. 2 B) were fitted in two steps. First, the τ-V curve of R894X hClC-1 was fitted with τ = 1/(αoeaeoV/kT + βoe−bzoV/kT), with αo and βo as opening/closing rate constants at 0 mV, and a and b as equivalent gating charges defining the voltage dependences of these rate constants. The voltage dependence of the deactivation time constants of WT and K875X hClC-1 was then fitted to the same equation with fixed equivalent gating charges. Slow time constants were fitted with single exponentials (Fig. 2 B). Because of the limited range of values, the voltage dependence of opening and closing rate constants cannot be deduced from these values.
We calculated opening rate constants of the fast and the slow gate for various voltages by dividing the open probability of the particular gate by the fast or the slow deactivation time constant, respectively (34):
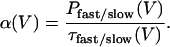 |
(1) |
Fast and slow gating was then further analyzed using the gating scheme developed by Chen and Miller for the fast gate of ClC-0 (24):
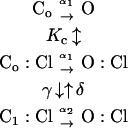 |
Opening rate constants measured at a given potential were plotted versus [Cl−]o (Fig. 4) and fit with
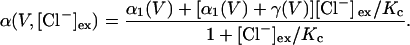 |
(2) |
These fits provide the opening rate constants in the absence of external Cl− (α1), the rate constants describing a conformational change of the Cl−-liganded channel that permits rapid channel opening (γ), and the Cl− dissociation constant of the closed channel (KC) (24). α1, γ, and KC were determined at various voltages, and the voltage dependences of these parameters were fit with the following: α1(V) = α1(0) ezαeoV/kT, γ(V) = γ(0) ezγeoV/kT, and KC(V) = KC(0) ezKCeoV/kT, respectively. The so-determined amplitudes and the voltage dependences of α1 and γ were then optimized by fitting Eq. 2 to the voltage dependence of opening rate constants at different [Cl−]o (Table 1). For these fits, the KCs obtained from the concentration dependences of opening rate constants were used as fixed values. We tested different voltage dependences of KC and obtained best results with voltage-independent KCs.
TABLE 1.
Gating properties of WT and truncated ClC-1 channels
|
Popen under standard conditions V0.5 (mV)
|
Fast gate
|
|||||||
|---|---|---|---|---|---|---|---|---|
| V0.5 fast under standard conditions (mV) | Pmin fast under standard conditions | α1 fast(0) (s−1) | z (α1 fast) | γfast(0) (s−1) | I (γfast) | KC fast(Cl−) at −35 mV (mM) | ||
| hClC-1 | −75.9 ± 1.7 | −90.5 ± 0.8 | 0.18 ± 0.03 | 0.69 | −0.61 | 677.9 | 0.42 | 54.1 ± 3.1 |
| n = 79 | n = 13 | n = 13 | n = 9 | |||||
| R894X hClC-1 | −96.2 ± 0.8 | −107.8 ± 1.8 | 0.23 ± 0.02 | 0.42 | −0.55 | 1088.7 | 0.50 | 33.0 ± 1.5 |
| n = 58 | n = 8 | n = 8 | n = 10 | |||||
| p < 0.01 | p < 0.01 | p = 0.51 | p < 0.01 | |||||
| K875X hClC-1 | −21.4 ± 1.1 | −59.0 ± 1.5 | 0.27 ± 0.05 | 0.52 | −0.47 | 258.4 | 0.33 | 155.0 ± 20.5 |
| n = 71 | n = 13 | n = 13 | n = 5 | |||||
| p < 0.01 | p < 0.01 | p = 0.25 | p < 0.01 | |||||
| Slow gate
|
||||||||
| V0.5 slow under standard conditions (mV) | Pmin slow under standard conditions | α1 slow(0) (s−1) | z (α1 slow) | γslow(0) (s−1) | z (γslow) | KC slow(Cl−) at −35 mV (mM) | ||
| hClC-1 | −74.7 ± 2.2 | 0.54 ± 0.02 | 0.46 | −0.61 | 16.43 | 0.54 | 16.7 ± 0.8 | |
| n = 14 | n = 14 | n = 6 | ||||||
| R894X hClC-1 | −98.4 ± 2.0 | 0.77 ± 0.08 | n.d. | n.d. | n.d. | n.d. | n.d. | |
| n = 8 | n = 8 | |||||||
| p < 0.01 | p < 0.01 | |||||||
| K875X hClC-1
|
−25.5 ± 1.8 | 0.23 ± 0.01 | 0.55
|
−0.60
|
13.08
|
0.51
|
45.3 ± 6.7 | |
| n = 13 | n = 13 | n = 7 | ||||||
| p < 0.01 | p < 0.01 | P < 0.01 | ||||||
n.d.: not determined.
To determine rate constants of MTS modification, the time course of modification was fit with a single exponential giving the time constant of modification. Pseudo-first order rate constants were calculated as the inverse of the modification time constant, and dividing by the concentration of the MTS reagents provided the second order rate constants (see Fig. 7).
FIGURE 7.

Truncation mutants alter cysteine accessibility of K231C. (A) Time-dependent reduction of K231C/K875X hClC-1 channels after application of 40 μM MTSET. (Inset) Current recordings during modification by MTSET. The solid line represents the fit to a single exponential. (B) Concentration dependence of inverse modification time constants for various [MTSET] concentrations at 125 mV for K231C (•), K231C/R894X (▾), and K231C/K875X (▪) hClC-1 channels. Data were fit with linear regressions. (C) MTSET second order modification rates for K231C, K231C/R894X, and K231C/K875X hClC-1 channels. Mean ± SE (n > 8). (D) Voltage dependence of the relative open probability of K231C (•), K231C/R894X (▾), and K231C/K875X (▪) channels. Solid lines represent fits to the Boltzman equation (K231C: V0.5 = −70.7 ± 2.6 mV (n = 10), K231C/R894X: V0.5 = −69.8 ± 0.9 mV (n = 14, p > 0.05); K231C/K875X: V0.5 = −75.4 ± 2.8 mV (n = 9, p > 0.05). (Inset) voltage dependence of MTSET second order modification rates for K231C hClC-1. Mean ± SE (n > 7).
All data are shown as mean ± SE from at least three cells and at least two distinct transfections. In general, mean ± SE are small and, in the figures, often smaller than the symbols. For statistical evaluation, Student's t-test was applied.
RESULTS
Functional expression of WT and C-terminally truncated hClC-1 channels
Fig. 1 shows representative current recordings from tsA201 cells expressing WT (Fig. 1 B), R894X (Fig. 1 C), and K875X (Fig. 1 D) hClC-1 channels. Cells were held at 0 mV, and voltage steps between −155 mV and +125 mV were applied, each followed by a fixed test step to −125 mV. WT and mutant currents increased instantaneously upon voltage steps in the positive as well as in the negative direction. At positive potentials, the current amplitudes were time independent; upon membrane hyperpolarization the instantaneous rise was followed by a slower decrease of the current amplitude due to channel deactivation. WT and mutant channels displayed comparable macroscopic current amplitudes but differed in the time course and degree of deactivation (Fig. 1).
To determine the voltage dependence of the open probability for WT and mutant channels, instantaneous current amplitudes were measured at a voltage step to −125 mV after prepulses to different voltages, plotted versus the preceding potential and normalized to the open probability at 0 mV obtained by nonstationary noise analysis (Fig. 2 A). Both C-terminal truncations change the voltage dependence of activation but leave the maximum absolute open probability unchanged (Fig. 2 A). The activation curve of R894X hClC-1 channels was shifted to more negative potentials and the voltage dependence of K875X hClC-1 to more positive potentials compared to WT hClC-1 (Table 1).
ClC-1 channels are known to exhibit two different gating processes. Fast gating opens and closes individual protopores, whereas slow cooperative gating steps act on both protopores together (35). Time constants of fast and slow deactivation were determined through fitting a sum of two exponentials and a time-independent value to current relaxations at negative potentials. Both deactivation time constants are voltage dependent, and carboxy-terminal truncations cause a shift of these voltage dependences (Fig. 2 B). To separate the open probabilities of the fast and the slow gate, we used the method developed by Accardi and Pusch (33). A short pulse to +180 mV, inserted before the test step into the pulse protocol given in Fig. 1 (Fig. 2 C, inset), fully activates the fast gate and permits acquisition of the relative open probability of the slow gate (Fig. 2 C). Dividing the channel open probability by the relative slow gate activation curve yields the open probability of the fast gate (Fig. 2 D). Since maximum absolute open probabilities are close to 1 for all tested conditions, absolute open probabilities are equal to relative open probabilities in all cases. WT and mutant channels differ in the voltage dependence of the fast gate and in the voltage dependence and minimum open probability of the slow gate (Fig. 2, C and D, Table 1).
Gating was investigated for various external chloride concentrations, and under all conditions tested, a linear relationship between midpoint of activation and the logarithm of the [Cl−]o was observed (Fig. 2, E and F). This is in agreement with the notion that the voltage dependence of fast and slow gating is conferred entirely by the permeant anion and further supports the notion that the stimulus for ClC-1 activation is a change of the electrochemical gradient for Cl−, rather than one of the transmembrane voltage (23).
We next determined opening rate constants for both gates from open probabilities and relaxation time constants under various [Cl−]o and voltages (Fig. 3). Such an analysis is restricted to voltages, where relaxation time constants can be reliably determined, i.e., only in a voltage range between −155 mV and −25 mV for the fast gate and −115 mV and −25 mV for the slow gate. For the slow gate of R894X hClC-1 channels, relative current components and voltage-dependent changes of absolute open probabilities are very small, precluding accurate determination of the open probability and the deactivation time constants for several [Cl−]o. We therefore did not calculate slow opening rate constants for this mutant.
Augmenting the [Cl−]o increases fast and slow opening rate constants at more positive voltages, leaving rate constants at very negative voltages unaffected. Opening rate constants at different [Cl−]o converge at negative voltages but do not cross over. All these results demonstrate that fast and slow gating of ClC-1 can be adequately described by the gating scheme developed by Chen and Miller (24) for the Torpedo ClC isoform, ClC-0.
Within this gating scheme, opening rate constants are a function of three different parameters: the dissociation constant of anion binding sites accessible in the closed state (KC), the opening rate constant in the absence of external Cl− (α1), and the rate constant (γ) describing a conformational change of the Cl−-liganded channel that permits rapid channel opening (24). These parameters were determined for fast and slow gating of WT and truncated ClC-1 channels in two steps. We first plotted opening rate constants of the fast and the slow gate versus [Cl−]o and fitted the dependences with Eq. 2 (Fig. 4). These fits provide values for KC in a voltage range of 30 mV (Fig. 5, A and B). α1 and γ exhibit a significant voltage dependence that cannot be reliably determined from these fits because of the limited voltage range. We therefore computed these values by fitting the voltage dependence of fast and slow opening rate constants for different [Cl−]o (Figs. 3 and 5, Table 1).
FIGURE 5.

Alteration of gating parameters by carboxy-terminal truncations. (A and B) Voltage dependence for the chloride dissociation constants (KCs) of the gating binding site for the fast (A) and the slow (B) gate. Mean ± SE (n > 5). (C and D) Voltage dependence of α1 and γ for the fast (C) and the slow (D) gate obtained from fitting the voltage dependence of the opening rate constants with Eq. 2.
For all constructs, we obtained best fits with voltage-independent KCs, similar to the voltage-independent anion binding observed in ClC-0 (24). KCs associated with fast gating were larger than the corresponding values of slow gating, whereas values of γ were larger for fast than for slow gating (Fig. 5, C and D, Table 1). The α1 rate constants are similar for the fast and the slow gate. They increase with membrane hyperpolarization, whereas the voltage dependence of γ is opposite (Table 1). The voltage dependence of α1fast and γfast differs from fast gating of the Torpedo isoform (Table 1). γfast is less voltage dependent, whereas α1fast exhibits a steeper voltage dependence than the corresponding values of ClC-0.
Our results demonstrate that fast and slow activation gating of ClC-1 fits to a model in which anions bind voltage independently and their subsequent translocation to the electric field fully confers the voltage dependence of channel opening. The truncation-induced differences in hClC-1 gating (Fig. 1) are caused by altered anion binding to the closed states. K875X hClC-1 channels exhibit the largest KCs, whereas those for WT and R894X hClC-1 channels are significantly smaller (KC(Cl−), Table 1). Both mutant channels also exhibit altered γfast rate constants. These results demonstrate that carboxy-terminal truncations modify the apparent affinity for anions and, in the case of fast gating, anion translocation. These variations result in altered voltage dependences of fast and slow gating.
Truncation mutations exhibit altered anion binding to the open channel
We next studied anion binding to WT and mutant chloride channels in the open state. Various permeant anions, such as I− and 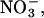 block chloride current through hClC-1 by binding to sites within the ion conduction pathway with higher affinity than Cl− (27,36,37). The block of chloride currents by anions applied to the internal or the external membrane side allows one to distinguish between two anion binding sites of the open channel, one accessible from the inside and one from the outside. The two anion binding sites differ in their apparent binding affinity and in their effect on gating properties (27,36,37). Fig. 6 A shows the concentration dependence of external I− block measured at −115 mV for WT and mutant channels. In all cases a Michaelis-Menten relationship with a Hill coefficient of 1 adequately described the data, providing the Michaelis constants (KMs) for WT and mutant channels. WT, R894X, and K875X hClC-1 channels display different KMs for external I− (Fig. 6 B, Table 2), and K875X differ from WT and R894X hClC-1 in block by external
block chloride current through hClC-1 by binding to sites within the ion conduction pathway with higher affinity than Cl− (27,36,37). The block of chloride currents by anions applied to the internal or the external membrane side allows one to distinguish between two anion binding sites of the open channel, one accessible from the inside and one from the outside. The two anion binding sites differ in their apparent binding affinity and in their effect on gating properties (27,36,37). Fig. 6 A shows the concentration dependence of external I− block measured at −115 mV for WT and mutant channels. In all cases a Michaelis-Menten relationship with a Hill coefficient of 1 adequately described the data, providing the Michaelis constants (KMs) for WT and mutant channels. WT, R894X, and K875X hClC-1 channels display different KMs for external I− (Fig. 6 B, Table 2), and K875X differ from WT and R894X hClC-1 in block by external 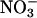 (Fig. 6 C, Table 2). In contrast, no differences were observed for internal I− (Fig. 6 D, Table 2).
(Fig. 6 C, Table 2). In contrast, no differences were observed for internal I− (Fig. 6 D, Table 2).
FIGURE 6.

Anion current block by external and internal anions. (A) Concentration dependence of current block by external I− at −115 mV for WT (•), R894X (▾), and K875X (▪) hClC-1 channels. The solid lines represent a fit to the Michaelis-Menten function. (Inset) Representative whole-cell recordings from a cell expressing WT hClC-1 channels at 0 (black line) and 20 mM external I− (shaded line). (B–D) Voltage dependence of the KMs determined by fits shown in (A) for external I− (B), external 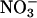 (C), and internal I− (D).
(C), and internal I− (D).
TABLE 2.
Pore properties of WT and truncated ClC-1 channels
| i at −160 mV (pA) | KM(I−)o at −115 mV (mM) |
KM(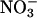 )o at −115 mV (mM) )o at −115 mV (mM) |
KM(I−)i at −115 mV (mM) | MTSET modification of K231C (s−1M−1) | |
|---|---|---|---|---|---|
| hClC-1 | 0.28 ± 0.01 | 8.7 ± 0.4 | 28.6 ± 3.3 | 4.8 ± 0.7 | 1692 ± 126 |
| n = 11 | n = 6 | n = 10 | n = 6 | n = 42 | |
| R894X hClC-1 | 0.24 ± 0.01 | 10.4 ± 0.4 | 27.7 ± 2.5 | 4.8 ± 0.9 | 2227 ± 90 |
| n = 8 | n = 7 | n = 8 | n = 5 | n = 43 | |
| p = 0.38 | p < 0.01 | p = 0.83 | p = 0.91 | p < 0.01 | |
| K875X hClC-1 | 0.29 ± 0.02 | 16.5 ± 1.3 | 53.2 ± 6.0 | 4.9 ± 1.3 | 1088 ± 57 |
| n = 7 | n = 8 | n = 8 | n = 5 | n = 42 | |
| p = 0.17 | p < 0.01 | p < 0.05 | p = 0.62 | p < 0.01 |
The unitary current amplitude (single channel current at −160 mV determined by nonstationary noise analysis (Table 2)) as well as the selectivity among anions (anion permeability ratios determined from reversal potential measurements (32): PSCN ∼ PCl > PBr > PNO3 > PI > PF) remained unaffected in mutant channels. The observed functional alterations, i.e., the changed apparent affinities for anion binding to the closed state and to the external binding site of the open channel, together with the unaltered selectivity, unitary conductance, and block by internal anions, suggest that the outer vestibule of the ion conduction pathway is altered in truncated hClC-1 channels.
Accessibility of a substituted cysteine within the ClC channel pore
Using site-directed mutagenesis, a conserved fixed charge close to the ClC channel selectivity filter was identified as playing a role in defining the anion binding affinity for hClC-1 (32). Neutralizing K231 (located in the N-terminus of the F helix in the atomic structure of a prokaryotic ClC isoform (13)) by substitution of a cysteine causes an inversion of the Cl > I permeability of WT hClC-1, and adding the positive charge back to C231 by reacting a MTSEA reagent restores WT anion selectivity (32). These results suggest that the positively charged side chain at position 231 is a major determinant of the electrostatic potential of the outer vestibule. We studied whether an altered relative position of this positively charged side chain is responsible for the observed differences in anion binding. MTS modification rate constants were measured for K231C, K231C/R894X, and K231C/K875X hClC-1 channels (Fig. 7). hClC-1 channels without substituted cysteines exhibit no functional change upon exposure to MTS reagents (32). Fig. 7 A (inset) shows current recordings from a cell expressing K231C/K875X hClC-1 during modification by 40 μM MTSET. Fig. 7 A gives the time course of isochronal current amplitudes determined at the end of a voltage step to −125 mV. Current amplitudes decayed in a monoexponential time course (Fig. 7 A). Time constants of current amplitude decay were determined for various [MTSET], and for all constructs a linear relationship between the inverse time constants and [MTSET] was observed (Fig. 7 B). This demonstrates that the obtained time course does indeed report modification of an engineered cysteine and not merely channel closure by a slow gating process. The different slopes were used to calculate second order rate constants of modification (Fig. 7 C). Constructs with truncated C-termini exhibit significantly different modification rate constants: K231C/R894X hClC-1 is modified with a higher, K231C/K875X hClC-1 with a lower second order rate constant (Fig. 7, A and C, Table 2).
The reaction of an MTS reagent with a thiol side chain of a channel protein occurs in two consecutive steps. The MTS reagent first has to diffuse from the aqueous solution to the side chain and then reacts with it in a second step. The experimentally determined reaction rates give information about the rate-limiting step in the cascade. All measured rate constants were well below the values observed for the modification of thiols in aqueous solution (38). This observation, together with the linear relationship between [MTS] and pseudo first order reaction constants, indicates that reagent access from the aqueous solution is the rate-limiting step. The voltage dependences of the relative open probability of full length and truncated K231C channels virtually superimpose (Fig. 7 D), and modification rates of full length K231C are voltage independent between −80 mV and +75 mV (Fig. 7 D, inset). These data indicate that the different modification rates are not due to an altered gating but rather to different arrangements of the access pathway to the thiol side chain. We conclude that the differences in rate constants between WT, R894X, and K875X channels demonstrate altered outer pore vestibules in the three constructs.
DISCUSSION
We investigated the functional effects of two carboxy-terminal truncations, R894X and K875X, of the human muscle chloride channel, hClC-1. Truncated hClC-1 channels exhibit unaltered unitary current amplitudes and relative anion permeabilities for SCN−, 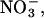 and I−. They differ, however, in anion binding to open and closed channels demonstrating a role of the carboxy-terminus in defining pore properties.
and I−. They differ, however, in anion binding to open and closed channels demonstrating a role of the carboxy-terminus in defining pore properties.
It is well established that ClC channels are gated by the permeant anion (22–24), in marked contrast to voltage-gated cation channels. However, one report appeared to be incompatible with this concept. A point mutation in the distal carboxy-terminus (A885P) shifts the activation curve of muscle chloride channels to more positive values and causes muscle hyperexcitability and muscle stiffness in an animal model of myotonia, the myotonic goat (21). Since the distal carboxy-terminus does not contribute to the formation of the ion conduction pathway, the gating alteration by A885P could have been interpreted as indicative for inherently voltage-dependent gating transitions. Our results remove this remaining doubt. The distal carboxy-terminus modifies anion binding to the closed pore, thus explaining the changed voltage dependence by the A885P substitution within this region. Moreover, testing fast and slow gating of WT and mutant channels over a broad range of external anion concentrations, no deviation from the expectations of anion-dependent gating could be seen (Fig. 2).
Surprisingly, the truncations cause similar alterations of fast and slow gating of hClC-1 channels. So far, these two gating transitions have been conceived as being two independent and structurally unrelated processes, with the slow gate as a structure closing both protopores together. Our experiments now demonstrate that both gating processes can be adequately described by the Chen-Miller model (24). In this model, the permeant anion binds voltage independently to the closed channel and subsequently translocates through the electric field, conferring the majority of the voltage dependence of channel opening. These findings suggest that opening of the fast as well as of the slow common gate requires anion translocation through an individual protopore. The KCs associated with fast gating are larger than those obtained for slow gating. This implies that hClC-1 anion conduction pathways exist in two closed states of differing apparent anion affinity, one with lower affinity corresponding to fast gating and one with higher affinity responsible for slow gating. Whereas the two gates dramatically differ in the translocation of the anion through the electric field, opening rate constants in the absence of external Cl− are quite similar (Table 1). A simple way of explaining our results is by proposing that fast and slow gating are resting and activated states of an allosteric gating process. A protopore might exist in two conformations, in one anion binding and translocation causes a slow cooperative opening of both of them, and in the other conformation fast opening of one protopore. Carboxy-terminal truncations modify anion binding in both conformations and thus alter fast and slow gating.
How does the distal carboxy-terminus modify anion binding to the pore of hClC-1? The cytoplasmic channel regions might extend the ion conduction pathway by forming a cytoplasmic pore, as observed in inwardly rectifying potassium channels (39,40) or, alternatively, affect the conformation of the ion conduction pathway. At present, no structural information about the ClC carboxy-terminus is available, but several lines of evidence support the idea of a changed conformation of the pore of truncated hClC-1 channels. Carboxy-terminal deletions have a pronounced effect on anion binding from the external side but no measurable effect on internal anion binding (Fig. 6). Moreover, anion binding is affected in the open (Fig. 6) as well as in the closed (Fig. 5) state. Lastly, MTS modification of an engineered cysteine demonstrates that there are discrete conformational differences between WT and truncated hClC-1 channels (Fig. 7).
There are several earlier reports that carboxy-terminal modifications change gating of ClC channels. Fong et al. (41) reported that carboxy-terminal truncations and chimeras affect slow gating of ClC-0. A disease-causing point mutation (G715E) in human ClC-2 alters the anion dependence of gating (3), and splice variants of a Caenorhabditis elegans homolog, Clh-3, with modified N- and C-terminus exhibit altered voltage-dependent gating (42). Our findings provide a possible explanation for all these findings. The carboxy-terminus likely determines anion binding and thus anion-dependent gating of various ClC isoforms.
The carboxy-terminus does not only modify functional properties, but is also required for proper targeting of several ClC isoforms into the respective cell compartment (18–20,43,44). The functional effect of the carboxy-terminus is not restricted to the time period immediately after translation in the endoplasmic reticulum and therefore is not involved in correct folding of the hydrophobic core domain. Maduke et al. (18) demonstrated that injection of a purified protein containing the last 154 amino acids of ClC-0 fully restores the function of truncated ClC-0 channels when injected into Xenopus oocytes, even under conditions abolishing protein synthesis. This demonstrates that the carboxy-terminus interacts with other protein domains and that modification of the carboxy-terminal domain by intracellular signals permits adjustment of ClC channel pore conformations. The physiological relevance of such a regulation of pore properties arises from the unique gating mechanisms of ClC channels. In voltage-gated cation channels, an endogenous voltage sensor moves across the membrane, and this movement can be modified by electrostatic or other interactions with regulatory pathways inside the cell, resulting in altered channel gating (45,46). Since ClC channels are gated by the permeant anion, changes of the voltage dependence of channel gating require altered anion binding to the closed pore. The functional interaction of the carboxy-terminus with the pore of hClC-1 might make voltage-dependent gating processes accessible to intracellular signal cascades, permitting modification of the macroscopic ClC anion conductance by regulatory pathways.
Acknowledgments
We thank Drs. Klaus Benndorf, Jennie Garcia-Olivares, Patrica Hidalgo, J. P. Johnson, Heider Lindner, and Reinhard Seifert for helpful discussions, and Hannelore Horstkott and Barbara Poser for excellent technical assistance.
This work was supported by the Deutsche Forschungsgemeinschaft (to Ch.F.) and the Muscular Dystrophy Association (to Ch.F.).
References
- 1.Koch, M. C., K. Steinmeyer, C. Lorenz, K. Ricker, F. Wolf, M. Otto, B. Zoll, F. Lehmann-Horn, K. H. Grzeschik, and T. J. Jentsch. 1992. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 257:797–800. [DOI] [PubMed] [Google Scholar]
- 2.George, A. L., M. A. Crackower, J. A. Abdalla, A. J. Hudson, and G. C. Ebers. 1993. Molecular basis of Thomsen's disease (autosomal dominant myotonia congenita). Nat. Genet. 3:305–310. [DOI] [PubMed] [Google Scholar]
- 3.Haug, K., M. Warnstedt, A. Alekov, T. Sander, A. Ramirez, B. Poser, S. Maljevic, S. Hebeisen, C. Kubisch, J. Rebstock, S. Horvath, K. Hallmann, J. Dullinger, B. Rau, F. Haverkamp, S. Beyenbrug, H. Schulz, D. Janz, B. Giese, G. Müller-Newen, P. Propping, C. Elger, Ch. Fahlke, H. Lerche, and A. Heils. 2003. Mutations in the voltage-gated chloride channel ClC-2 are associated with idiopathic generalized epilepsies. Nat. Genet. 33:527–533. [DOI] [PubMed] [Google Scholar]
- 4.Simon, D. B., R. S. Bindra, T. A. Mansfield, C. Nilson-Williams, E. Mendonca, R. Stone, S. Schurman, A. Nayir, H. Alpay, A. Bakkaloglu, J. Rodriguez-Soriano, J. M. Morales, S. A. Sanjad, C. M. Taylor, D. Pilz, A. Brem, H. Trachtman, W. Griswold, G. A. Richard, E. John, and R. P. Lifton. 1997. Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nat. Genet. 17:171–178. [DOI] [PubMed] [Google Scholar]
- 5.Matsumura, Y., S. Uchida, Y. Kondo, H. Miyazaki, S. B. Ko, A. Hayama, T. Morimoto, W. Liu, M. Arisawa, S. Sasaki, and F. Marumo. 1999. Overt nephrogenic diabetes insipidus in mice lacking the CLC-K1 chloride channel. Nat. Genet. 21:95–98. [DOI] [PubMed] [Google Scholar]
- 6.Stobrawa, S. M., T. Breiderhoff, S. Takamori, D. Engel, M. Schweizer, A. A. Zdebik, M. R. Bosl, K. Ruether, H. Jahn, A. Draguhn, R. Jahn, and T. J. Jentsch. 2001. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron. 29:185–196. [DOI] [PubMed] [Google Scholar]
- 7.Lloyd, S. E., S. H. S. Pearce, S. E. Fisher, K. Steinmeyer, B. Schwappach, S. J. Scheinman, B. Harding, B. Alessandra, M. Devota, P. Goodyear, S. P. A. Rigden, O. Wrong, T. J. Jentsch, I. W. Craig, and R. V. Thakker. 1996. A common molecular basis for three inherited kidney stone diseases. Nature. 379:445–449. [DOI] [PubMed] [Google Scholar]
- 8.Kornak, U., D. Kasper, M. R. Boesl, E. Kaiser, M. Schweizer, A. Schulz, W. Friedrich, G. Delling, and T. J. Jentsch. 2001. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 104:205–215. [DOI] [PubMed] [Google Scholar]
- 9.Accardi, A., and C. Miller. 2004. Secondary active transport mediated by a prokaryotic homologue of ClC Cl- channels. Nature. 427:803–807. [DOI] [PubMed] [Google Scholar]
- 10.Iyer, R., T. M. Iverson, A. Accardi, and C. Miller. 2002. A biological role for prokaryotic ClC chloride channels. Nature. 419:715–718. [DOI] [PubMed] [Google Scholar]
- 11.Miller, C. 1982. Open-state substructure of single chloride channels from Torpedo electroplax. Philos. Trans. R. Soc. Lond. B Biol. Sci. 299:401–411. [DOI] [PubMed] [Google Scholar]
- 12.Mindell, J. A., M. Maduke, C. Miller, and N. Grigorieff. 2001. Projection structure of a ClC-type chloride channel at 6.5 Å resolution. Nature. 409:219–223. [DOI] [PubMed] [Google Scholar]
- 13.Dutzler, R., E. D. Campbell, M. Cadene, M. B. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- 14.Ponting, C. P. 1997. CBS domains in CIC chloride channels implicated in myotonia and nephrolithiasis (kidney stones). J. Mol. Med. 75:160–163. [PubMed] [Google Scholar]
- 15.Bateman, A. 2003. The structure of a domain common to archeabacteria and the homocysteine disease protein. Trends Biochem. Sci. 22:12–13. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt-Rose, T., and T. J. Jentsch. 1997. Reconstitution of functional voltage-gated chloride channels from complementary fragments of CLC-1. J. Biol. Chem. 272:20515–20521. [DOI] [PubMed] [Google Scholar]
- 17.Hryciw, D. H., G. Y. Rychkov, B. P. Hughes, and A. H. Bretag. 1998. Relevance of the D13 region to the function of the skeletal muscle chloride channel, ClC-1. J. Biol. Chem. 273:4304–4307. [DOI] [PubMed] [Google Scholar]
- 18.Maduke, M., C. Williams, and C. Miller. 1998. Formation of CLC-0 chloride channels from separated transmembrane and cytoplasmic domains. Biochemistry. 37:1315–1321. [DOI] [PubMed] [Google Scholar]
- 19.Hebeisen, S., A. Biela, B. Giese, G. Müller-Newen, P. Hidalgo, and Ch. Fahlke. 2004. The role of the carboxyl terminus in ClC chloride channel function. J. Biol. Chem. 279:13140–13147. [DOI] [PubMed] [Google Scholar]
- 20.Estevez, R., M. Pusch, C. Ferrer-Costa, M. Orozco, and T. J. Jentsch. 2004. Functional and structural conservation of CBS domains from CLC chloride channels. J. Physiol. (Lond.). 557:363–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck, C. L., Ch. Fahlke, and A. L. George Jr. 1996. Molecular basis for decreased muscle chloride conductance in the myotonic goat. Proc. Natl. Acad. Sci. USA. 93:11248–11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pusch, M., U. Ludewig, A. Rehfeldt, and T. J. Jentsch. 1995. Gating of the voltage-dependent chloride channel ClC-0 by the permeant anion. Nature. 373:527–530. [DOI] [PubMed] [Google Scholar]
- 23.Rychkov, G. Y., M. Pusch, D. S. Astill, M. L. Roberts, T. J. Jentsch, and A. H. Bretag. 1996. Concentration and pH dependence of skeletal muscle chloride channel ClC-1. J. Physiol. (Lond.). 497:423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen, T. Y., and C. Miller. 1996. Nonequilibrium gating and voltage dependence of the ClC-0 Cl− channel. J. Gen. Physiol. 108:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer-Kleine, C., K. Steinmeyer, K. Ricker, T. J. Jentsch, and M. C. Koch. 1995. Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am. J. Hum. Genet. 57:1325–1334. [PMC free article] [PubMed] [Google Scholar]
- 26.Fahlke, Ch., R. Rüdel, N. Mitrovic, M. Zhou, and A. L. George Jr. 1995. An aspartic acid residue important for voltage-dependent gating of human muscle chloride channels. Neuron. 15:463–472. [DOI] [PubMed] [Google Scholar]
- 27.Fahlke, Ch., C. L. Beck, and A. L. George, Jr. 1997. A mutation in autosomal dominant myotonia congenita affects pore properties of the muscle chloride channel. Proc. Natl. Acad. Sci. USA. 94:2729–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jurman, M. E., L. M. Boland, Y. Liu, and G. Yellen. 1994. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 17:876–881. [PubMed] [Google Scholar]
- 29.Fahlke, Ch., A. Rosenbohm, N. Mitrovic, A. L. George Jr., and R. Rüdel. 1996. Mechanism of voltage-dependent gating in skeletal muscle chloride channels. Biophys. J. 71:695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hebeisen, S., L. Heidtmann, D. Cosmelli, C. Gonzalez, B. Poser, R. Latorre, O. Alvarez, and Ch. Fahlke. 2003. Anion permeation in human ClC-4 channels. Biophys. J. 84:2306–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barry, P. H. 1994. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J. Neurosci. Methods. 51:107–116. [DOI] [PubMed] [Google Scholar]
- 32.Fahlke, Ch., H. T. Yu, C. L. Beck, T. H. Rhodes, and A. L. George Jr. 1997. Pore-forming segments in voltage-gated chloride channels. Nature. 390:529–532. [DOI] [PubMed] [Google Scholar]
- 33.Accardi, A., and M. Pusch. 2000. Fast and slow gating relaxations in the muscle chloride channel CLC-1. J. Gen. Physiol. 116:433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen, M. F., and T. Y. Chen. 2001. Different fast-gate regulation by external Cl− and H+ of the muscle-type ClC chloride channels. J. Gen. Physiol. 118:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saviane, C., F. Conti, and M. Pusch. 1999. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 113:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fahlke, Ch., C. Dürr, and A. L. George Jr. 1997. Mechanism of ion permeation in skeletal muscle chloride channels. J. Gen. Physiol. 110:551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rychkov, G. Y., M. Pusch, M. L. Roberts, T. J. Jentsch, and A. H. Bretag. 1998. Permeation and block of the skeletal muscle chloride channel, ClC-1, by foreign anions. J. Gen. Physiol. 111:653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheung, M., and M. H. Akabas. 1997. Locating the anion-selectivity filter of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel. J. Gen. Physiol. 109:289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishida, M., and R. MacKinnon. 2002. Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1at 1.8 Å resolution. Cell. 111:957–965. [DOI] [PubMed] [Google Scholar]
- 40.Kuo, A., J. M. Gulbis, J. F. Antcliff, T. Rahman, E. D. Loewe, J. Zimmer, J. Cuthbertson, F. M. Ashcroft, and D. A. Doyle. 2003. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 300:1922–1926. [DOI] [PubMed] [Google Scholar]
- 41.Fong, P. Y., A. Rehfeldt, and T. J. Jentsch. 1998. Determinants of slow gating in ClC-0, the voltage-gated chloride channel of Torpedo marmorata. Am. J. Physiol. Cell Physiol. 274:C966–C973. [DOI] [PubMed] [Google Scholar]
- 42.Denton, J., K. Nehrke, E. Rutledge, R. Morrison, and K. Strange. 2004. Alternative splicing of N- and C-termini of a C. elegans ClC channel alters gating and sensitivity to external Cl− and H+. J. Physiol. (Lond.). 555:97–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwappach, B., S. Stobrawa, M. Hechenberger, K. Steinmeyer, and T. J. Jentsch. 1998. Golgi localization and functionally important domains in the NH2 and COOH terminus of the yeast CLC putative chloride channel Gef1p. J. Biol. Chem. 273:15110–15118. [DOI] [PubMed] [Google Scholar]
- 44.Carr, G., N. Simmons, and J. Sayer. 2003. A role for CBS domain 2 in trafficking of chloride channel CLC-5. Biochem. Biophys. Res. Commun. 310:600–605. [DOI] [PubMed] [Google Scholar]
- 45.Hille, B. 1992. Ionic Channels of Excitable Membranes. Sinauer Associates, Sunderland, MA.
- 46.Perozo, E., and F. Bezanilla. 1990. Phosphorylation affects voltage gating of the delayed rectifier K+ channel by electrostatic interactions. Neuron. 5:685–690. [DOI] [PubMed] [Google Scholar]